
Abstract
Introduction
Exosomes are important mediators of intercellular communication. Previously, we characterized circulating exosomal miR-425-3p as a non-invasive prognostic marker for predicting clinical response to platinum-based chemotherapy in patients with non-small cell lung cancer (NSCLC).
Methods
Circulating exosomal miR-425-3p was validated by qRT-PCR in paired serum samples from NSCLC patients during the course of platinum-based chemotherapy. Cell coculture was performed to examine the effects of exosomal miR-425-3p on the sensitivity of recipient A549 cells to cisplatin. Using bioinformatics, ChIP and luciferase reporter assays, the transcription factor essential for miR-425-3p expression was identified. Autophagic activity in the recipient cells was determined by Western blot and fluorescence microscopy.
Results
Higher levels of exosomal miR-425-3p were found in serum samples from the patients in tolerance versus those at baseline. An upward trend in the expression of circulating exosomal miR-425-3p was revealed during chemotherapy. Furthermore, the expression of exosomal miR-425-3p could be induced by cisplatin in NSCLC cells. Exosomes isolated from either cisplatin-treated or cisplatin-resistant NSCLC cells conferred chemoresistance to sensitive A549 cells in a miR-425-3p-dependent manner. Cisplatin-induced c-Myc was found to directly bind the miR-425-3p promoter and transactivated its expression. Exosomal miR-425-3p facilitated autophagic activation in the recipient cells by targeting AKT1, eventually leading to chemoresistance.
Discussion
Our results suggest that apart from a prognostic marker of treatment response, exosomal miR-425-3p might be a potential dynamic biomarker to tailor cisplatin resistance in NSCLC patients during the treatment and represent a promising therapeutic target for therapy-resistant NSCLC.
Introduction
Chemotherapy resistance, whether intrinsic or acquired, is one of the most fundamental problems in cancer and gives rise to local and distant tumor recurrence and disease progression. Although platinum-based schemes are a cornerstone for broad-based treatments of patients with non-small cell lung cancer (NSCLC), 5-year survival rates remain poor largely due to the emergence of resistance prior to and during the course of treatment.Citation1,Citation2 Non-coding single-stranded microRNAs (miRNAs), important epigenetic regulators of protein coding genes, have been described relevant to platinum-based chemotherapy in NSCLC.Citation3–Citation5 This class of RNA negatively regulates gene expression by binding to the 3ʹ-UTR regions of their target mRNAs involving in epithelial-mesenchymal transition, cell proliferation and apoptosis, and eventually contributes to the regulation of chemoresistance in NSCLC.Citation6,Citation7
Exosomes are 30–100 nm membrane vesicles that are actively released into the extracellular environment by many cell types, including cancer cells.Citation8 Exosomes contain proteins and nucleic acids, such as mRNA and miRNAs, that allow these structures to operate as signaling platforms for short-range or long-range delivery of molecular information to the recipient cells.Citation9 The miRNA contents of exosomes frequently reflect those of the cells they originated from.Citation10 Extensive attention is now focusing on exosomes partly because of their capability to confer chemoresistance in many cancer types, in particular through transfer of miRNAs.Citation11–Citation13 In addition, exosomal miRNAs can be obtained from the peripheral blood and easily measured, making them attractive biomarkers to predict response to therapy or monitor disease progression.
Our recent study has demonstrated that circulating exosomal miR-425-3p acts as a prognostic biomarker for improved predictions of the clinical response to platinum-based chemotherapy in NSCLC patients, and high level of miR-425-3p leads to the increase of basal autophagic activity in NSCLC cells and lung tissues and impairs therapeutic response.Citation3 Given the role of exosomes in intercellular communication,Citation9,Citation14 we therefore hypothesized that miR-425-3p might be transported among cancer cells via exosomes during the course of platinum-based treatment and promote chemotherapeutic drug resistance. In this study, our results showed that cisplatin treatment induced the expression of miR-425-3p in cells and exosomes through c-Myc-mediated transactivation. More importantly, coculture with exosomes isolated from either cisplatin-treated or cisplatin-resistant NSCLC cells impaired the sensitivity of the recipient A549 cells in a miR-425-3p-dependent manner. Exosomal miR-425-3p facilitated autophagic activation in the recipient cells by targeting AKT1, eventually leading to their resistance to drug stress.
Materials And Methods
Patients And Serum Samples
NSCLC patients, diagnosed at Nanjing First Hospital between 2012 and 2016, were uniformly followed and received standard first-line platinum-based chemotherapy. Based on progression-free survival (PFS), the patients with PFS ≤ 6 months were diagnosed platinum-resistant. Paired serum samples were collected from these patients at the start of treatment (non-resistant, n=19) and the date of disease progression (resistant, n=19), or after the first cycle and the last cycle of trials (n=15), respectively. Each pair was from the same patient. Patients provided written informed consent in accordance with the Declaration of Helsinki. The study was approved by the ethics committee on Human Research of Nanjing First Hospital.
Isolation, Quantification And Labeling Of Exosomes
Exosomes in serum samples were isolated and purified using the ExoQuick serum exosome precipitation solution (SBI, Mountain View, CA) according to the manufacturer’s instruction. Exosomes from cultured cells were purified by differential centrifugation and confirmed by transmission electron microscopy and NanoSight analyses as previously described.Citation3 Exosomes were quantified in bicinchoninic acid (BCA) method in which exosomal protein was measured by BCA protein assay kit (Beyotime Biotechnology, China). Purified exosomes were labeled with CM-Dil (Invitrogen, Carlsbad, CA) as described previously.Citation15 The internalization of CM-Dil-labeled exosomes by A549 cells was analyzed quantitatively by flow cytrometry on a FACScan (Becton Dickinson, Franklin Lakes, NJ).
Cell Culture
The human NSCLC cell line A549 and its cisplatin-resistant A549/DDP were purchased from the Shanghai Institute of Cell Biology (Shanghai). We developed 3 cisplatin-resistant variants by exposure of A549/DDP cells to stepwise increase in cisplatin concentrations and maintained them at a final concentration of 500 (A549/DDP-500), 1000 (A549/DDP-1000) and 2000 ng/mL (A549/DDP-2000), respectively.Citation16 All the cells were cultured in DMEM medium (Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS, Life Technologies). For resistant variants, cisplatin was withdrawn from the culture medium 3 days before use. In cell coculture experiments, purified exosomes (10 μg/mL) were added into the fresh media containing 0.5% FBS.
Quantitative Real-Time PCR (qRT-PCR)
Exosomal RNAs were isolated using a miRNeasy Serum/Plasma Kit (Qiagen, Hilden, Germany) and cDNA was synthesized using a miScript II Reverse Transcription Kit (Qiagen). Quantification of miRNAs was performed using a miScript SYBR Green PCR Kit (Qiagen) on the Bio-Rad CFX96 TouchTM Real-Time PCR Detection System (Bio-Rad, Hercules, CA). Quantitative real-time PCR analyses of hsa-miR-425-3p and AKT1 mRNA expression were conducted as previously reported.Citation3 The absolute concentrations of hsa-miR-425-3p in exosomes were calculated from calibration curves developed with synthetic miRNA oligonucleotides and normalized the miRNA concentration to the exosomal protein content. U6 snRNA was used as an endogenous control to normalize miR-425-3p expression in cells, and β-actin was used as the endogenous control to normalize AKT1 expression.
Transient miR-425-3p Inhibitor Transfection And siRNA Treatment
A549 cells were transfected with either miR-425-3p inhibitor or c-Myc siRNA, which were synthesized by GenePharma Co. Ltd. (Shanghai), using Lipofectamine™ 2000 transfection reagent (Invitrogen) as previously reported.Citation3 The sequence of miR-425-3p inhibitor and c-Myc siRNA are 5′-GGGCGGACACGACAUUCCCGAU-3′ and 5′-CGTCCAAGCAGAGGAGCAA-3′, respectively.
Cell Viability And Apoptosis Assays
Cell proliferation was determined using the MTT assay in triplicate as previously described.Citation3 The dose-response curves were fitted by a nonlinear regression function in GraphPad Prism 7.0 (La Jolla, CA). Cell apoptosis was determined by annexin V/PI staining.Citation17 Samples were analyzed by flow cytrometry on a FACScan.
Western Blot
Western blot was performed as previously described.Citation3 The anti-PARP, anti-p-β-catenin (S33/37/T41), anti-β-catenin, anti-c-Myc, anti-AKT1, anti-p-AKT1 (S473), anti-p-mTOR (S2448), anti-mTOR and anti-LC3B antibodies were purchased from Cell Signaling Technology (Beverly, MA). The anti-β-actin antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The anti-SQSTM1/p62 antibody was from Abcam (Cambridge, UK). The densitometry of the immunoblots was performed with Image J software (NIH, Bethesda, MD)
Chromatin Immunoprecipitation (ChIP)
ChIP was conducted using Pierce Magnetic ChIP Kit (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s protocol. Immunoprecipitation of proteins, after ChIP with the antibody against c-Myc, was confirmed by Western blot analysis before the ChIP-real time PCR analysis for the fragments of the miR-425-3p promoter with the appropriated promoter primers. Relative quantification of the target was normalized to input control. The primer sequences used in ChIP-PCR were as follows: miR-425-3p forward, 5ʹ-CGAGCGTGGATTGGCTTCTC-3ʹ; miR-425-3p reverse, 5ʹ-GTGAACGAGGACAAGGTGATGC-3ʹ.
Construction Of Luciferase Plasmids And Reporter Assay
Based on bioinformatics analysis, 2000 bp before transcriptional start site was selected as miR-425-3p promoter. Then the promoter sequence was amplified and cloned into pGL-Basic plasmids containing a luciferase reporter. Since there were 5 predicted c-Myc binding sites in miR-425-3p promoter regions, a mutant luciferase plasmid was constructed by deleting 2 c-Myc binding sites (−1173~-1183, −1307~-1317) in the promoter regions. For luciferase reporter assay, the WT or Mut luciferase plasmids were transfected into the cells using Lipofectamine™ 2000. Transfected cells were lysed 24 h after transfection, and luciferase activities were assayed following the instruction of the Luciferase Assay System (Promega). The activity of the product of the β-galactosidase gene under the control of a constitutive β-actin promoter was used to normalize the transfection efficiency.
Autophagy Analysis
A549 cells transfected with GFP-LC3 plasmids were treated with purified exosomes for 24 h. Then cells were fixed with 4% paraformaldehyde (40 min, room temperature) and permeabilized with methanol and nuclei were stained with DAPI. The formation of vacuoles containing GFP-LC3 (dots) was examined by fluorescence microscopy (BX51TRF, Olympus).
Statistical Analysis
All statistical analyses were performed using the GraphPad Prism 7.0 software. The significance of the differences between groups was estimated using the Student’s t-test and Mann–Whitney U-test. For the MTT assay, statistical analyses were performed by extra sum-of-squares F-test. The data are shown as the mean ± SD. A P value of < 0.05 was considered statistically significant.
Results
The Expression Of Circulating Exosomal miR-425-3p Is Up-Regulated During The Course Of Platinum-Based Chemotherapy
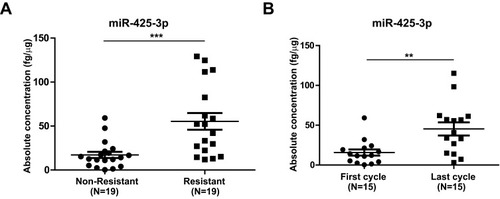
We compared the expression of miR-425-3p in circulating exosomes of paired serum samples that were collected from NSCLC patients (n = 19, Supplementary Table S1) at the start of platinum-based treatment (non-resistant) and at the date of disease progression (resistant), respectively. The levels of miR-425-3p expression were significantly higher from the patients in tolerance versus those at baseline (). When compared between paired serum samples from resistant NSCLC patients (n = 15) collected after the first cycle and the last cycle of trials, respectively, higher levels of miR-425-3p expression were detected in the circulating exosomes from the patients after the last cycle than after the first cycle of trials ().
Figure 1 Increased miR-425-3p expression in serum exosomes from NSCLC patients during the course of platinum-based chemotherapy. Circulating exosomes were derived from paired serum samples of NSCLC patients (A) at the start of treatment (non-resistant) and at the date of disease progression (resistant) (n = 38 samples from 19 patients) or (B) of NSCLC patients receiving the first and last cycle of treatment before diagnosed as drug-resistant (n = 30 samples from 15 patients), respectively. The absolute concentrations of miR-425-3p in exosomes were evaluated by qRT-PCR. Each point represents one sample. **P < 0.01; ***P < 0.001.
Cisplatin Promotes Exosome Release And Its miR-425-3p Expression In NSCLC Cells
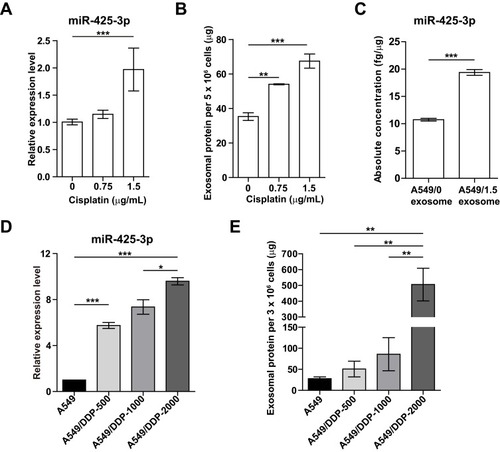
To investigate whether platinum-based chemotherapy, mainly composed of cisplatin treatment, induces the expression of NSCLC cell-derived exosomal miR-425-3p, we treated A549 cells with various concentrations of cisplatin for 24 h. As shown in , cellular miR-425-3p expression was significantly increased by 1.5 μg/mL of cisplatin, which is around the IC50 value (1.15 ± 0.04 μg/mL) of cisplatin in A549 cells. When exosomes were isolated and quantified using BCA protein assay, we found that cisplatin promoted exosome release from NSCLC cells in a concentration-dependent manner (). In addition, the absolute concentration of miR-425-3p was dramatically higher in the exosomes from A549 cells treated with 1.5 μg/mL of cisplatin (A549/1.5 exosome) than those from untreated A549 cells (A549/0 exosome, ). In agreement with our previous study,Citation3 greatly higher level of exosomal miR-425-3p was observed in cisplatin-resistant A549/DDP cells than in cisplatin-sensitive A549 cells (data not shown). To monitor the change of miR-425-3p expression in the development of cisplatin resistance, we compared the parental A549 cells and three cisplatin-resistant variant cells that were developed by long-term exposure of A549/DDP cells to cisplatin and maintained at various final concentrations (500–2000 ng/mL) as previously described.Citation16 Compared with A549 cells, the increase of both cellular miR-425-3p expression and exosome release accompanied the development of cisplatin resistance in these variants (). Taken together, these findings suggest that cisplatin could enhance miR-425-3p expression in cells and exosomes, as well as exosome release during the short- or long-term treatment, which might be linked with the development of cisplatin resistance.
Figure 2 Cisplatin stimulates exosome release and its miR-425-3p expression in NSCLC cell lines. (A-C) A549 cells were exposed to various concentrations of cisplatin for 48 h. (A) Intracellular miR-425-3p expression were evaluated by qRT-PCR. (B) Quantity of exosomes from cultured cells were determined using BCA protein assay and normalized to cell numbers. (C) The absolute concentrations of miR-425-3p in exosomes were evaluated by qRT-PCR. (D, E) Cisplatin-resistant variant cells were maintained at a final concentration of cisplatin (500–2000 ng/mL). (D) Intracellular miR-425-3p expression in the variant cells. (E) Quantity of exosomes from the cultured variant cells. *P < 0.05; **P < 0.01; ***P < 0.001.
Exosomal miR-425-3p Impairs The Sensitivity Of NSCLC Cells To Cisplatin
To determine whether cisplatin-induced miR-425-3p can be directly transfered to neighboring or distant recipient cells via exosomes, we treated A549 cells with CM-Dil-labeled A549/1.5 exosome or exosomes purified from A549/DDP-1000 cells (A549/DDP-1000 exosome). After 3 hr, confocal microscopy detected fluorescently labeled signals in the treated cells (). The efficacy of exosomal transfer depended on the amounts of exosomes added into the medium (Supplementary Figure S1).
Figure 3 Exosomal miR-425-3p decreases the sensitivity of A549 cells to cisplatin. (A) Representative confocal microscopy image showing the internalization of CM-Dil-labeled exosomes (red) by A549 cells. A549 cells were incubated with exosomes isolated from A549 cells exposed to the indicated concentration of cisplatin or from cisplatin-resistant variant cells for 6 h. Then the resulting cells were treated with various concentrations of cisplatin for 72 h. (B, E) Cell viability was determined by MTT assay in triplicate. An extra sum-of-squares F-test was performed to test whether dose-response curves statistically differ from PBS control. P < 0.05, (B) A549/1.5 exosome vs PBS; (E) A549/DDP-1000 or −2000 exosome vs PBS. (C, F) Intracellular miR-425-3p expression were evaluated by qRT-PCR. (D, G) Enhanced sensitivity to cisplatin in the treated cells after transfection of the miR-425-3p inhibitor. An extra sum-of-squares F-test was performed to test whether dose-response curves statistically differ from NC control. P < 0.05, (D) NC + A549/1.5 exosome vs NC; (G) NC + A549/DDP-1000 exosome vs NC. (H) Analysis of cell apoptosis by annexin V/PI double staining. Left, exosome-treated A549 cells were exposed to 2 μg/mL of cisplatin for 24 h. FACS analyses were performed. Right, statistical graph analysis. (I) Enhanced apoptosis in the treated cells after transfection of the miR-425-3p inhibitor. Upper, representative data of FACS. Lower, statistical graph analysis. (J) Enhanced cleavage of PARP in the treated cells after transfection of the miR-425-3p inhibitor was assessed using Western blotting. β-actin was used as a loading control. The densitometry of the immunoblots was performed with image J software and is presented in the histograms. The data are shown as the mean ± SD of three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001.
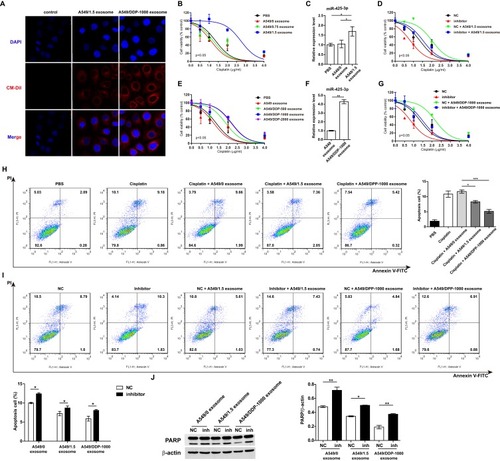
Next, we determined the impact of exosomal miR-425-3p in regulating chemoresistance. A549 cells were cocultured with the same amount of exsomes isolated from A549 cells treated with various concentrations of cisplatin. A549/1.5 exosome remarkably decreased the sensitivity of recipient A549 cells to cisplatin (). Accordingly, higher level of miR-425-3p expression was detected in the cells cocultured with A549/1.5 exosome than those with PBS or A549/0 exosome (). In contrast, the miR-425-3p inhibitor resensitized the A549/1.5 exosome-treated cells (). Similar results were obtained in A549 cells cocultured with exosomes purified from cisplatin-resistant variant cells. Impaired sensitivity was induced by these resistant variants-derived exosomes compared with PBS or A549 exosome (). Increase in cellular miR-425-3p expression and inhibition of the chemoresistance by the miR-425-3p inhibitor were also detected in A549/DDP-1000 exosome-treated cells (). Apoptosis is the main mechanism for cell death related to cisplatin. Annexin V/PI double staining confirmed that both A549/1.5 exosome and A549/DDP-1000 exosome reduced cisplatin-induced apoptosis in A549 cells (Fig. H). Such cell apoptosis was reversed by the miR-425-3p inhibitor (). Accordingly, the miR-425-3p inhibitor enhanced the cleavage of PARP in the cells cocultured with A549/1.5 exosome or A549/DDP-1000 exosome, while both the exosomes reduced the cleavage of PARP compared with A549/0 exosome ().
Cisplatin Up-Regulates miR-425-3p Through The β-Catenin Signaling Pathway In NSCLC Cells
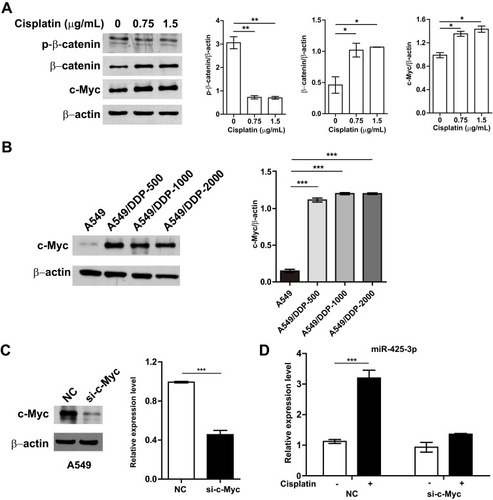
To investigate molecular mechanisms by which cisplatin enhances miR-425-3p expression in NSCLC cells, we examined possible regulation via transcription factors. JASPAR online analysis (http://jaspar.genereg.net) identified five predicted c-Myc binding sites in putative miR-425-3p promoter regions (Supplementary Table S2). It has been reported that cisplatin induces DNA-protein cross-linking damage that activates the Wnt/β-catenin signaling and c-Myc is one of its main downstream target genes.Citation18–Citation20 As respected, cisplatin decreased the phosphorylation of β-catenin at Ser33/37/Thr41 while increasing the expression of total β-catenin and c-Myc in A549 cells, indicative of the activation of the β-catenin pathway (). All the levels of c-Myc expression in three cisplatin-resistant variant cells were remarkably higher than that in A549 cells (). Notably, knocking down c-Myc completely abolished the up-regulation of miR-425-3p induced by cisplatin (), implying that c-Myc might promote the transcription activity of miR-425-3p during cisplatin treatment.
Figure 4 Cisplatin enhances miR-425-3p expression by activating the β-catenin pathway. (A) A549 cells were exposed to various concentrations of cisplatin for 24 h. The protein levels of phosphorylated β-catenin (p-β-catenin), β-catenin and c-Myc were assessed using Western blotting. β-actin was used as a loading control. (B) Western blotting analysis showing protein expression of c-Myc in the cisplatin-resistant variant cells. (C) Knockdown of c-Myc in A549 cells transfected with c-Myc-targeting siRNA. Left, the protein level of c-Myc. Right, the mRNA level of c-Myc. (D) A549 cells transfected with c-Myc siRNA were treated with 1.5 μg/mL of cisplatin for 24 h. Then intracellular miR-425-3p expression were evaluated by qRT-PCR. The data are shown as the mean ± SD of three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001.
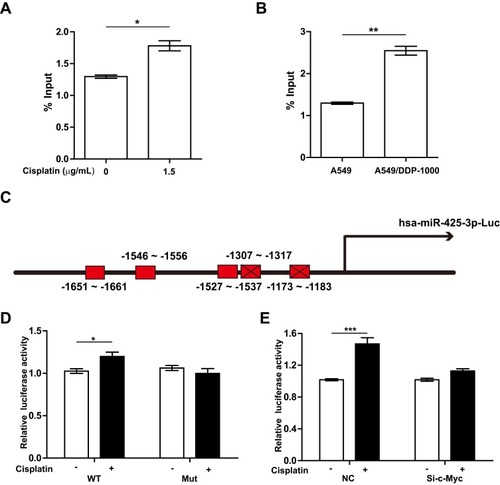
Further, we performed ChIP assay with anti-c-Myc antibody and real-time PCR analysis. 1.5 μg/mL of cisplatin enhanced the recruitment of c-Myc onto the miR-425-3p promoter regions in A549 cells (). An enhanced binding activity was also detected in A549/DDP-1000 cells compared with A549 cells (). Two luciferase reporter vectors containing the WT or mutant c-Myc binding sequences in the miR-425-3p promoter were constructed (). Cisplatin-induced transcription of miR-425-3p was confirmed by the observation of increased luciferase activity when the WT reporter was transfected in A549 cells (). In contrast, no changes in the luciferase activity was observed in the case of the mutant reporter. In addition, cisplatin-increased luciferase activity of the WT miR-425-3p reporter was inhibited by c-Myc siRNA ().
Figure 5 c-Myc transactivates miR-425-3p via its binding to the miR-425-3p promoter. (A) A549 cells were treated with 1.5 μg/mL of cisplatin for 24 h. ChIP assays were performed using an antibody against c-Myc, followed by qRT-PCR with primers designed for c-Myc-binding sites in miR-425-3p promoter regions. (B) ChIP analysis showing the binding of c-Myc to the miR-425-3p promoter in A549 and A549/DDP-1000 cells. (C) Schematic of a luciferase reporter of the miR-425-3p promoter with 5 predicted c-Myc binding sites (wide type). Mutated luciferase plasmids were designed with depletion of 2 predicted binding sites. (D) A549 cells transfected with the wide-type (WT) or mutated (Mut) plasmids were treated with or without 1.5 μg/mL of cisplatin for 24 h. Luciferase activity was detected. (E) A549 cells co-transfected with the WT plasmids or NC and c-Myc siRNA were treated with or without 1.5 μg/mL of cisplatin for 24 h. Luciferase activity was detected. The data are shown as the mean ± SD of three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001.
Exosomal miR-425-3p Enhances Autophagy Via Targeting AKT1 And Reduces Cisplatin Sensitivity
Our previous study identified AKT1 as the direct target gene of miR-425-3p.Citation3 To examine whether cisplatin-induced exosomal miR-425-3p confers cisplatin resistance in NSCLC through targeting AKT1, we treated A549 cells with A549/DDP-1000 exosome. The addition of the exosomes reduced AKT1 mRNA expression by about 2-fold compared with the untreated control, while the miR-425-3p inhibitor reversed such a reduction (). Consistently, A549/DDP-1000 exosome markedly down-regulated both total and phosphorylated AKT1, as well as its downstream p-mTOR at protein levels (). In addition, A549/DDP-1000 exosome enhanced the LC3B-I to LC3B-II conversion and reduced the expression of SQSTM1/p62, indicative of autophagy activation in the recipient A549 cells. Co-treatment of the miR-425-3p inhibitor reversed the changes in these protein levels. Moreover, the formation of GFP-fused LC3 puncta was increased by adding A549/DDP-1000 exosome, while greatly decreased after the combined treatment with the miR-425-3p inhibitor ().
Figure 6 Exosomal miR-425-3p-activated autophagy contributes to apoptosis resistance in the recipient A549 cells. (A, B) A549 cells transfected with NC or the miR-425-3p inhibitor were incubated with or without A549/DDP-1000 exosome for 24 h. (A) qRT-PCR analysis for AKT1 mRNA expression. (B) Western blotting analysis showing protein expression of AKT1, p-AKT1, p-mTOR, mTOR, LC3B and SQSTM1/p62. β-actin was used as a loading control. (C) A549 cells co-transfected with the GFP-LC3 plasmid and NC or the miR-425-3p inhibitor were incubated with or without A549/DDP-1000 exosome for 24 h. Representative images show GFP-LC3 localization. Scale bar: 5 μm. (D) A549 cells were pretreated with BafA1 (100 nM) for 2 h and incubated with indicated exosomes 6 h. The resulting cells were exposed to 2 μg/mL of cisplatin for another 24 h. The protein levels of PARP were assessed using Western blotting. β-actin was used as a loading control. (E) Schematic representation of the mechanism by which cisplatin-induced miR-425-3p confers chemoresistance via exosome transfer in NSCLC. The data are shown as the mean ± SD of three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001.
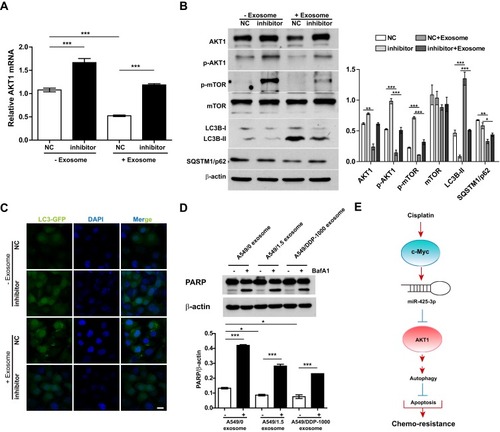
Given the role of autophagy activation in chemotherapy resistance,Citation16,Citation21,Citation22 we examined the effect of exosomal miR-425-3p on cisplatin-induced apoptosis in the presence of autophagy inhibitor BafA1. Both A549/1.5 exosome and A549/DDP-1000 exosome reduced cisplatin-induced PARP cleavage in A549 cells (). But the reduction was remarkably reversed by BafA1. These results suggest that exosomal miR-425-3p can target AKT1 and activate autophagy by negatively regulating the AKT/mTOR pathway, eventually leading to resistance to cisplatin-induced apoptosis ().
Discussion
Circulating miRNAs are produced through two main mechanisms: cell death via apoptosis or necrosis, which lead to the release of AGO protein-bound miRNAs, and a process by active section of exosomes containing mature miRNAs.Citation23,Citation24 Exosomal miRNAs that truly reflect the biological changes occurring in cancer cells and play important roles in cell-to-cell communication may represent more specific molecular biomarkers than cell-free miRNAs.Citation25 Our previous study has shown the negative relationship between circulating exosomal miR-425-3p and clinical responsiveness in NSCLC patients treated with platinum-based chemotherapy, and identified exosomal miR-425-3p as a valid prognostic biomarker for predicting the therapeutic response.Citation3 In this study, by monitoring circulating exosomal miR-425-3p from NSCLC patients, we revealed an upward trend in its expression levels during the course of platinum-based chemotherapy. The result needs to be further confirmed in a large series of NSCLC patients following treatment. In fact, recent studies have showed high levels of miR-425-3p in IgA nephropathy and early-stage lung adenocarcinoma,Citation26,Citation27 although its role in disease progression remains determined. MiR-425-3p levels also predicts response to sorafenib therapy in patients with hepatocellular carcinoma.Citation28 Assessment of circulating exosomal miR-425-3p could help in stratifying patients in NSCLC for platinum-based chemotherapy.
It is accepted that cancer cells are able to actively adapt to stressors within the environment, including chemotherapy-induced toxicities, for survival. In the present study, we found that once exposure to the DNA-damaging agent cisplatin, miR-425-3p expression was induced in both cells and exosomes, supporting that the exosomal miRNA profiles resemble those of the original cells and reflect their biologic state in the environment.Citation29 Cisplatin treatment, whether short-time or long-time, enhanced miR-425-3p expression. More importantly, these tumor-derived exosomal miR-425-3p conferred chemoresistance to the recipient A549 cells, evidenced by the decrease in cisplatin-induced apoptosis after incubation with A549/1.5 exosome or A549/DDP-1000 exosome. Such chemoresistance was almost completely reversed by miR-425-3p specific inhibitor. In line with this finding, some previous studies also reported that NSCLC-derived exosomes decreased the sensitivity of the recipient NSCLC cells to cisplatin.Citation12,Citation30,Citation31 Therefore, exosomal miR-425-3p may function not only as a prognostic biomarker for predicting for the responsiveness in NSCLC,Citation3 but also as a potential molecule to deliver chemoresistance information among NSCLC cells.
Cisplatin-induced DNA damage activates multiple pathways, including the Wnt-β-catenin pathway, in NSCLC.Citation18–Citation20 Consistently, cisplatin dramatically up-regulated c-Myc expression, accompanied by increased β-catenin levels in A549 cells. All three cisplatin-resistant variant cells displayed higher c-Myc levels than their parental A549 cells. Combined bioinformatics analysis with ChIP and luciferase reporter assays, c-Myc was found to directly bind the miR-425-3p promoter and positively regulate its transcription. Emerging evidence has demonstrated the role of c-Myc in chemoresistance in lung cancer.Citation19,Citation32,Citation33 Xie et al reported that c-Myc participates in β-catenin–mediated cisplatin resistance in A549/DDP cells.Citation19 A recent study showed that miR-296-3p-PRKCA-FAK-Ras-c-Myc feedback loop modulated by HDGF/DDX5/β-catenin complex attenuates cisplatin resistance in lung adenocarcinoma,Citation34 suggesting that c-Myc functions as a key determinant of chemoresistance. Cisplatin-upregulated c-Myc could transactive miR-425-3p and promote its transfer via exosomes among NSCLC cells during the treatment.
Given stress adaptation of cancer cells, we revealed that exosomal miR-425-3p enhanced autophagic activity in the recipient cells by targeting AKT1 that activates the mTOR complex, exerting an inhibitory role on autophagy.Citation35 In fact, autophagy can be activated by stressful conditions such as drug stress, facilitating the survival of tumor cells and increasing resistance to chemotherapy.Citation22 Previously, we showed that basal autophagy is progressively increased during the development of cisplatin resistance in NSCLC cells and that inhibiting basal autophagy sensitizes NSCLC cells to cisplatin-induced apoptosis.Citation16,Citation36 The present study showed that autophagy inhibitor BafA1 indeed greatly promoted cisplatin-induced apoptosis in the recipient A549 cells incubated with A549/1.5 exosome or A549/DDP-1000 exosome. Interestingly, Qin et al reported that cisplatin-resistant NSCLC cell-derived exosomes are characterized by low expression of miR-100-5p, which negatively regulates mTOR expression, and that higher mTOR expression contributes to cisplatin resistance in recipient cells.Citation12 However, we failed to find any change of total mTOR expression after A549 cells were incubated with A549/DDP-1000 exosome. Conversely, the p-mTOR level was greatly induced by miR-425-3p inhibitor possibly due to loss of AKT1 inhibition.
Conclusion
In summary, our study verified that exosomal transfer of miR-425-3p could promote the development of chemoresistance in NSCLC during the treatment of cisplatin. Moreover, circulating exosomal miR-425-3p might be a potential dynamic biomarker to tailor platinum-based chemotherapy and to facilitate adjustment in NSCLC therapeutic regiment.
Disclosure
The authors report no conflicts of interest in this work.
Acknowledgments
This study was supported by National Natural Science Foundation of China (Nos. 81573446, 81802297) and The Drug Innovation Major Project (2018ZX09711001-003-007).
References
- MacDonagh L, Gray SG, Breen E, et al. Lung cancer stem cells: the root of resistance. Cancer Lett. 2016;372(2):147–156. doi:10.1016/j.canlet.2016.01.01226797015
- Rosell R, Karachaliou N. Lung cancer in 2014: optimizing lung cancer treatment approaches. Nat Rev Clin Oncol. 2015;12(2):75–76. doi:10.1038/nrclinonc.2014.22525533943
- Yuwen D, Ma Y, Wang D, et al. Prognostic role of circulating exosomal miR-425-3p for the response of NSCLC to platinum-based chemotherapy. Cancer Epidemiol Biomarkers Prev. 2019;28(1):163–173. doi:10.1158/1055-9965.EPI-18-056930228154
- Wang X, Chen X, Meng Q, et al. MiR-181b regulates cisplatin chemosensitivity and metastasis by targeting TGFbetaR1/Smad signaling pathway in NSCLC. Sci Rep. 2015;5:17618. doi:10.1038/srep1761826620926
- Zhao J, Fu W, Liao H, et al. The regulatory and predictive functions of miR-17 and miR-92 families on cisplatin resistance of non-small cell lung cancer. BMC Cancer. 2015;15:731. doi:10.1186/s12885-015-1584-326482648
- Zang H, Wang W, Fan S. The role of microRNAs in resistance to targeted treatments of non-small cell lung cancer. Cancer Chemother Pharmacol. 2017;79(2):227–231. doi:10.1007/s00280-016-3130-727515517
- Zang H, Peng J, Wang W, Fan S. Roles of microRNAs in the resistance to platinum based chemotherapy in the non-small cell lung cancer. J Cancer. 2017;8(18):3856–3861. doi:10.7150/jca.2126729151973
- Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2(8):569–579. doi:10.1038/nri85512154376
- Martins VR, Dias MS, Hainaut P. Tumor-cell-derived microvesicles as carriers of molecular information in cancer. Curr Opin Oncol. 2013;25(1):66–75. doi:10.1097/CCO.0b013e32835b7c8123165142
- Skog J, Wurdinger T, van Rijn S, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10(12):1470–1476. doi:10.1038/ncb180019011622
- Kong JN, He Q, Wang G, et al. Guggulsterone and bexarotene induce secretion of exosome-associated breast cancer resistance protein and reduce doxorubicin resistance in MDA-MB-231 cells. Int J Cancer. 2015;137(7):1610–1620. doi:10.1002/ijc.2954225833198
- Qin X, Yu S, Zhou L, et al. Cisplatin-resistant lung cancer cell-derived exosomes increase cisplatin resistance of recipient cells in exosomal miR-100-5p-dependent manner. Int J Nanomedicine. 2017;12:3721–3733. doi:10.2147/IJN.S13151628553110
- Huang X, Yuan T, Liang M, et al. Exosomal miR-1290 and miR-375 as prognostic markers in castration-resistant prostate cancer. Eur Urol. 2015;67(1):33–41. doi:10.1016/j.eururo.2014.07.03525129854
- Li L, Li C, Wang S, et al. Exosomes derived from hypoxic oral squamous cell carcinoma cells deliver miR-21 to normoxic cells to elicit a prometastatic phenotype. Cancer Res. 2016;76(7):1770–1780. doi:10.1158/0008-5472.CAN-15-162526992424
- Wang M, Zhao C, Shi H, et al. Deregulated microRNAs in gastric cancer tissue-derived mesenchymal stem cells: novel biomarkers and a mechanism for gastric cancer. Br J Cancer. 2014;110(5):1199–1210. doi:10.1038/bjc.2014.1424473397
- Mi S, Xiang G, Yuwen D, et al. Inhibition of autophagy by andrographolide resensitizes cisplatin-resistant non-small cell lung carcinoma cells via activation of the Akt/mTOR pathway. Toxicol Appl Pharmacol. 2016;310:78–86. doi:10.1016/j.taap.2016.09.00927639426
- Li X, Pang J, Xue W, et al. Inducible SHP-2 activation confers resistance to imatinib in drug-tolerant chronic myeloid leukemia cells. Toxicol Appl Pharmacol. 2018;360:249–256. doi:10.1016/j.taap.2018.09.04430290167
- Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;740:364–378. doi:10.1016/j.ejphar.2014.07.02525058905
- Xie C, Pan Y, Hao F, et al. C-Myc participates in beta-catenin-mediated drug resistance in A549/DDP lung adenocarcinoma cells. APMIS. 2014;122(12):1251–1258. doi:10.1111/apm.1229625131138
- Vesel M, Rapp J, Feller D, et al. ABCB1 and ABCG2 drug transporters are differentially expressed in non-small cell lung cancers (NSCLC) and expression is modified by cisplatin treatment via altered Wnt signaling. Respir Res. 2017;18(1):52. doi:10.1186/s12931-017-0537-628340578
- Zhou J, Hu SE, Tan SH, et al. Andrographolide sensitizes cisplatin-induced apoptosis via suppression of autophagosome-lysosome fusion in human cancer cells. Autophagy. 2012;8(3):338–349. doi:10.4161/auto.1872122302005
- White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12(6):401–410. doi:10.1038/nrc326222534666
- Cortez MA, Bueso-Ramos C, Ferdin J, Lopez-Berestein G, Sood AK, Calin GA. MicroRNAs in body fluids–the mix of hormones and biomarkers. Nat Rev Clin Oncol. 2011;8(8):467–477. doi:10.1038/nrclinonc.2011.7621647195
- Schwarzenbach H. The clinical relevance of circulating, exosomal miRNAs as biomarkers for cancer. Expert Rev Mol Diagn. 2015;15(9):1159–1169. doi:10.1586/14737159.2015.106918326202667
- Melo SA, Sugimoto H, O’Connell JT, et al. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell. 2014;26(5):707–721. doi:10.1016/j.ccell.2014.09.00525446899
- Wu J, Zhang H, Wang W, et al. Plasma microRNA signature of patients with IgA nephropathy. Gene. 2018;649:80–86. doi:10.1016/j.gene.2018.01.05029459010
- Wang Y, Zhao H, Gao X, et al. Identification of a three-miRNA signature as a blood-borne diagnostic marker for early diagnosis of lung adenocarcinoma. Oncotarget. 2016;7(18):26070–26086. doi:10.18632/oncotarget.842927036025
- Vaira V, Roncalli M, Carnaghi C, et al. MicroRNA-425-3p predicts response to sorafenib therapy in patients with hepatocellular carcinoma. Liver Int. 2015;35(3):1077–1086. doi:10.1111/liv.1263625040368
- Liao J, Liu R, Yin L, Pu Y. Expression profiling of exosomal miRNAs derived from human esophageal cancer cells by Solexa high-throughput sequencing. Int J Mol Sci. 2014;15(9):15530–15551. doi:10.3390/ijms15091553025184951
- Dong C, Liu X, Wang H, et al. Hypoxic non-small-cell lung cancer cell-derived exosomal miR-21 promotes resistance of normoxic cell to cisplatin. Onco Targets Ther. 2019;12:1947–1956. doi:10.2147/OTT.S18692230881046
- Xiao X, Yu S, Li S, et al. Exosomes: decreased sensitivity of lung cancer A549 cells to cisplatin. PLoS One. 2014;9(2):e89534. doi:10.1371/journal.pone.008953424586853
- Jia X, Zhang Z, Luo K, et al. TCRP1 transcriptionally regulated by c-Myc confers cancer chemoresistance in tongue and lung cancer. Sci Rep. 2017;7(1):3744. doi:10.1038/s41598-017-03763-028623290
- Chen D, Huang J, Zhang K, et al. MicroRNA-451 induces epithelial-mesenchymal transition in docetaxel-resistant lung adenocarcinoma cells by targeting proto-oncogene c-Myc. Eur J Cancer. 2014;50(17):3050–3067. doi:10.1016/j.ejca.2014.09.00825310895
- Fu Q, Song X, Liu Z, et al. miRomics and proteomics reveal a miR-296-3p/PRKCA/FAK/Ras/c-Myc lated by HDGF/DDX5/beta-catenin complex in lung adenocarcinoma. Clin Cancer Res. 2017;23(20):6336–6350. doi:10.1158/1078-0432.CCR-16-281328751441
- Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15(2):81–94. doi:10.1038/nrm373524401948
- Yuwen D, Mi S, Ma Y, et al. Andrographolide enhances cisplatin-mediated anticancer effects in lung cancer cells through blockade of autophagy. Anticancer Drugs. 2017;28(9):967–976. doi:10.1097/CAD.000000000000053728692436