 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Fexofenadine (FEX) has high solubility and low permeability (BCS, Class III). In this work, novel FEX loaded water in oil microemulsion (w/o) was designed to improve bioavailability and compared with Fexofen® syrup in in vitro and in vivo studies. In addition, pharmacokinetic parameters in permeability studies were estimated by using WinNonLin software program. w/o microemulsion system was optimized using a pseudoternary phase diagram, composed of span 80/lutrol F 68 (9.5:0.5 w/w), oleic acide, isopropyl alcohol and water as surfactant mixture; oil and cosurfactant was developed for oral drug delivery. w/o microemulsion systems were characterized by phase behavior, particle size, viscosity and solubilization capacity. In vitro studies were studied using Caco-2 cell monolayer. Pharmacokinetic parameters of w/o microemulsion were investigated in rabbits and compared to Fexofen® syrup. Fexofen® syrup and microemulsion were administered by oral gavage at 6 mg/kg of the same concentration. The experimental results indicated that microemulsion (HLB = 5.53) formed nanometer sized droplets (33.29 ± 1.76) and had good physical stability. This microemulsion increased the oral bioavailability of FEX which was highly water-soluble but fairly impermeable. The relative bioavailability of FEX microemulsion was about 376.76% compared with commercial syrup in rabbits. In vitro experiments were further employed for the enhanced effect of the microemulsion for FEX. These results suggest that novel w/o microemulsion plays an important role in enhancing oral bioavailability of low permeability drugs.
Introduction
Allergic rhinitis is a common disorder associated with a high incidence of morbidity. Citation1 Several mediators are involved in the pathophysiology of allergic diseases, so a variety of drugs, including antihistamines, decongestants, intranasal corticosteroids, leukotriene receptor antagonists, topical anticholinergics, mast cell stabilizers, mucolytics, and anti-IgE antibodies are used in the clinical treatment of allergic rhinitis. However, among these mediators, antihistamine remains the principal one, and plays a fundamental role in the genesis of allergic rhinitis. Therefore, antihistamines have been the main class of medications used for the treatment of allergic rhinitis over the past 60 years.Citation2,Citation3
Fexofenadine hydrochloride, the active metabolite of terfenadine, a well known and effective H1 receptor antagonist, is administered by the oral route. It is a nonsedating antihistamine providing rapid, long-acting, and highly selective peripheral H1 receptor antagonist activity.Citation4–Citation6 The oral bioavailability of fexofenadine in humans is not established. Citation7 However, in other animals, it is known to be as low as 4.2% (in rats)Citation7 and 2.6% (in horses).Citation8 In a review of 16 human pharmacokinetic studies, Chen et al reported that the half-life of fexofenadine varies between 3 and 17 hours.Citation9 In our study, the half-life of fexofenadine w/o microemulsion was significantly (P < 0.05) decreased (by 2.04-fold) when compared with a commercial fexofenadine syrup (Fexofen®, Sanovel, Istanbul, Turkey) because of differences in clearance rates and distribution volumes for the two formulations. In addition, the enterohepatic circulation is involved in the prolonged plasma elimination half-life of fexofenadine due to increased oral absorption.
The efflux transporter, P glycoprotein, has been reported to transport fexofenadine in in vitro models. Thus, it is considered to be an important determinant of the pharmacokinetics of fexofenadine.Citation10 Fexofenadine is a substrate for P glycoprotein, and concomitant food and coadministration of drugs have significant effects on its oral bioavailability.Citation11 In addition, according to the biopharmaceutical classification system, fexofenadine belongs to the Class 3 drugs, which means it has high solubility and low permeability, so an effort to increase the permeability of fexofenadine is needed.Citation12
Microemulsions known to enhance bioavailability in the gastrointestinal tract are thermodynamically stable, isotropically clear dispersions of two immiscible liquids, such as oil and water, and are stabilized by an interfacial film of surfactant molecules.Citation13–Citation17 Further advantages of using microemulsions as drug delivery systems include better drug solubilization and protection against enzymatic hydrolysis, as well as the potential for enhanced absorption due to a surfactant-induced increment in permeability. In addition, microemulsions represent an interesting and potentially quite powerful alternative carrier system for drug delivery because of their high solubilization capacity, transparency, ease of preparation, and high diffusion and absorption rates when compared with solvents without the surfactant system.Citation18,Citation19
In this study, we designed and developed a novel microemulsion formulation to improve the oral absorption of fexofenadine. Absorption of fexofenadine in the intestine was compared with that of Fexofen syrup in vitro and in vivo in rabbits. In particular, Caco-2 cell permeability and the pharmacokinetics of the fexofenadine microemulsion were determined to evaluate the potential of the microemulsion to be an effective oral delivery carrier for drugs with bioavailability problems, like fexofenadine.
Materials and methods
Materials
Fexofenadine was provided by Basel Drug Company (Istanbul, Turkey). Diphenylhydramine was purchased from Sigma Chemical Co (St Louis, MO). Lutrol F 68 was purchased from BASF (Ludwigshafen, Germany). Span 80, oleic acid, and isopropyl alcohol were obtained from Sigma- Aldrich (St Louis, MO). Cell culture reagents and supplies were obtained from Gibco Invitrogen (Grand Island, NY). Sodium dodecyl sulfate was purchased from Sigma Chemical Co.
Preparation of microemulsion
Solubility studies
The solubility of fexofenadine in various oils and surfactants was determined by adding an excess amount of fexofenadine into 1 mL of each vehicle, followed by shaking (100 rpm) at 25°C for 24 hours. The samples were centrifuged at 10000 rpm for 10 minutes to remove excess fexofenadine, and the concentration of fexofenadine in the supernatant was measured by high-performance liquid chromatography (HPLC) after appropriate dilution with isopropyl alcohol.
Construction of pseudoternary phase diagram
Microemulsion fields formed by dilution and gentle agitation were identified from ternary phase diagrams of systems containing oil-surfactant (S)-cosurfactant (coS). Oleic acid was selected as the oil phase. The effects of surfactants (mixtures of span 80 and lutrol F 68 at w/w ratios of 9.5:0.5 with a hydrophilic-lipophilic balance of 5.53) and the cosurfactant (isopropyl alcohol) on the pseudoternary phase diagram were systematically observed at room temperature. Lutrol F 68 were melted at 50°C–60°C and blended with span 80 to make the surfactant mixture. Afterwards, the oil phase and the surfactant mixture were mixed. The boundaries of the microemulsion domains were determined for different values of the S/coS (w/w) ratios. The S/coS weight ratios were 1:1, 2:1, 3:1, 4:1, and 5:1. Distilled water was added dropwise to each clear oil and surfactant mixture with gentle stirring to allow equilibration. Following addition of aliquots in the water phase, the mixture was examined visually for transparency. Based on the results of the pseudoternary phase diagrams, one microemulsion was selected for further experiments (). Thereafter, the fexofenadine-loaded microemulsion was prepared by adding 6 mg of fexofenadine per 1 mL of microemulsion with vortexing.
Table 1 The solubility of FEX in various vehicles at 25°C
Characterization of microemulsion
Droplet size determination
The droplet size, polydispersity, and zeta potential of the microemulsions (with and without fexofenadine) were measured using a zeta sizer (3000 HSA, Malvern Instruments, Worcestershire, UK) with six measurements.
Viscosity
The viscosity of the microemulsions (with and without fexofenadine) was measured at 25°C ± 0.1°C using a Brookfield digital viscometer-III rheometer V 3.3 HB (Middleboro, MA) at 200 rpm (n = 6).
Conductivity measurements
The effect of the amount of water phase in the microemulsions was monitored quantitatively by measuring electrical conductivity. The water phase was added drop by drop into the mixture of oil phase and, after each drop of water, electrical conductivity was measured using a conductometer WPA CM 35 (Cambridge, UK), at 25°C ± 0.1°C. The conductivity of microemulsions with and without fexofenadine was measured (n = 6).
pH measurements
The pH values of the samples were measured by a pH meter (Jenway 3040 Ion Analyze, Combined Glass Electrode, Mettler- Toledo, Greifensee, Switzerland) at 25°C ± 0.1°C (n = 6).
HPLC analysis of fexofenadine
HPLC was used for the loading content and permeability studies of fexofenadine. Samples were appropriately diluted with isopropyl alcohol to 20 μL and injected directly into the HPLC system without further treatment. The HPLC system was equipped with a Waters 2487. A reverse phase C18 column (250 mm × 4 mm, 5 μm) LiChroCART® (Merck, Darmstadt, Germany) was used at room temperature. The excitation and emission wavelengths of the fluorescence detector were set as 220 nm and 290 nm, respectively. A mixture of acetonitrile and 0.05 M pH 3.2 potassium dihydrogen phosphate buffer solutions (60:40, v/v) was used as the mobile phase at a flow rate of 1.0 mL/min.
Content of fexofenadine
Petroleum ether 2.5 mL was added to 200 mL of microemulsion containing fexofenadine and mixed for three minutes at 1000 rpm, followed by addition of 2.5 mL 0.05 M pH 3.2 potassium dihydrogen phosphate buffer solution. The mixture was vortexed and centrifuged at 3000 rpm for 15 minutes. Finally, the upper liquid phase was constituted with 2.5 mL of mobile phase, and 20 μL was injected into the HPLC system (n = 6).
Stability test
To evaluate the stability of the optimized formulation of fexofenadine, the microemulsion was put into sealed glass vials, which were stored at 25°C and 40°C for 6 months. The clarity, concentration of fexofenadine, and droplet size were thereafter investigated at predetermined intervals (n = 6).
Permeability studies in Caco-2 cell monolayer
Cell cultures
The colonic adenocarcinoma cell line, Caco-2, was obtained from the American Type Culture Collection, and cultured in Dulbecco’s Modified Eagle Medium. The cell monolayers were prepared by seeding 4 × 105 cells/well on a 6-well Transwell® insert filter. Cell cultures were maintained at 37°C under 90% humidity and 5% CO2. Monolayers were used 19–22 days after seeding. The integrity of each cell monolayer was checked by measuring its transepithelial electrical resistance (TEER) with an epithelial Volt-Ohm meter (EVOM, World Precision Instrument, Sarasota, FL) before and after the experiments (n = 6).
Permeability of fexofenadine from microemulsion versus syrup
The in vitro permeability study was developed in Caco-2 cell monolayers grown in Transwell inserts with collagen-coated polycarbonate membranes having a pore size of 0.4 μm and a surface area of 4.7 cm2. The cells were maintained at 37°C in the atmospheric conditions described earlier. The medium was replaced every second day for 3 weeks. For the experiment with Caco-2 cell monolayers, w/o microemulsion (fexofenadine 6 mg/mL) and commercial syrup (6 mg/ mL) formulations were diluted up to 95% (v/v) with Hank’s Buffered Salt Solution (HBSS).
The permeability studies were performed in both directions, ie, from apical to basolateral and basolateral to apical. After washing the Caco-2 cell monolayer twice with prewarmed HBSS medium (pH 7.4), the fexofenadine microemulsion was diluted with HBSS (95% v/v). The transport experiments were done by adding the fexofenadine microemulsion diluted with HBSS to either the apical (2.2 mL) or basolateral side (3.2 mL), while the receiving chamber contained the corresponding volume of transport medium. After shaking at 50 rpm for 2 hours at 37°C in a water bath, samples were collected from both sides of the Caco-2 cell monolayer and immediately stored below −20°C for subsequent HPLC analysis. The same procedure was used for the syrup formulation.
Apparent permeability values (Papp) for each side were calculated according to the following equation:
where Papp is the apparent permeability (cm/sec), dQ/dt is the permeation rate, A is the diffusion area of monolayers (cm2 ), and Co is the initial concentration of drug in the donor compartment.Citation1
Pharmacokinetic studies in rabbits
Study design
Two groups of rabbits (weight 1.8–2.5 kg, provided by the Central Animal Laboratory of Ege University) were used to perform the in vivo pharmacokinetic study. Six rabbits were allocated to each group. All rabbits were maintained in a light-controlled room at a temperature of 25°C ± 0.5°C. The experimental protocols were approved by the Animal Care and Use Committee of the College of Pharmacy, Ege University. Before the experiment, the rabbits were fasted overnight for 12 hours with free access to water, and remained conscious throughout the experiment. For the oral administration, the fexofenadine syrup and w/o microemulsion formulations were administered at the same doses by oral gavage (6 mg/kg body weight). Blood samples (2 mL) were withdrawn from the ear vein at predetermined time intervals (hours 0, 1, 2, 3, 4, 5, 6, 7, and 8), and 75 μL/mL of heparin solution was used to maintain patency of the cannula between sampling times. Plasma samples were obtained by immediately centrifuging the blood samples at 10,000 rpm for 10 minutes, after which 500 μL of each plasma sample was stored at −20°C until analyzed by HPLC.
Plasma analysis
The fexofenadine concentration in the plasma samples was analyzed by HPLC.Citation20 Briefly, plasma samples (500 μL) were mixed with 2 μg/mL of internal standard (DFN) and 1500 μL of methanol. The mixture was vortexed for 10 minutes and centrifuged at 4000 rpm for 5 minutes at 5°C ± 0.5°C, and the supernatant was evaporated to dryness under nitrogen at 40°C. The residue was reconstituted with 200 μL of mobile phase and injected into the HPLC system. Chromatography was performed using 3 μm C18 columns (100 mm × 3 mm, Phenomenex, Torrance, CA) at a flow rate of 0.8 mL/min with acetonitrile 0.02 M potassium dihydrogen phosphate buffer solution mixture (25:75, v/v) as the mobile phase at room temperature. A calibration curve was constructed in the range of 0.5–30 μg/mL. The mean correlation coefficient (r2) for the calibration curve was at least 0.999.
Pharmacokinetic analysis
The pharmacokinetic parameters for fexofenadine after administration of the microemulsion and syrup were calculated for each rabbit using the WinNonlin® program (Version 3.1, Pharsight Co, Mountainview, CA). Noncompartmental analysis was used to calculate the peak plasma concentration, time taken to reach peak concentration, and the area under the curve (AUC). The relative bioavailability of the oral drug delivery system was calculated as follows:
Statistical analysis
A two-tailed unpaired Student’s t-test was performed, with P < 0.05 considered to be statistically significant.
Results
Solubility studies
The solubility of fexofenadine in the various vehicles is shown in . Isopropyl alcohol was selected as a cosurfactant for the study because it has the highest solubility for fexofenadine and has been reported to have a favorable enhancing effect on the oral bioavailability of fexofenadine. Citation9 Oleic acid was chosen as the oily phase for its good drug solubility (). Span 80 and lutrol F 68 were selected as surfactants due to their ability to augment drug solubilization.Citation7
Construction of pseudoternary phase diagrams
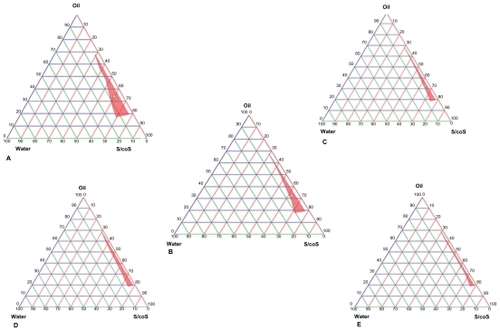
Investigation of the phase behavior of these systems demonstrated that our approach was suitable for determining the water phase, oil phase, surfactant concentration, and cosurfactant concentration at which the transparent phase microemulsion system was formed.Citation21 The construction of a phase diagram makes it easy to identify the concentration range of components in the microemulsions. shows the phase diagrams constructed to determine the optimum S/coS concentration ratio for the formulation of w/o microemulsion consisting of span 80, lutrol F 68, oleic acid, isopropyl alcohol, and water. The S/coS and surfactant mixture (span 80/lutrol F 68) ratios were found to be 1:1 and 9.5:0.5 for the optimized microemulsion. As shown in , the area of w/o microemulsion becomes enlarged and is highest at a S/coS ratio of 1:1. The exact composition according to oil, surfactant, cosurfactant, and aqueous phases is shown in .
Table 2 The contents of optimized microemulsion formulation
Figure 1 Pseudoternary phase diagrams of microemulsion system. (A) 1:1 S/coS ratio, (B) 2:1 S/coS ratio, (C) 3:1 S/coS ratio, (D) 4:1 S/coS ratio, (E) 5:1 S/coS ratio.
Characterization of microemulsion
The physicochemical characteristics of the microemulsions are shown in . Particle size analysis showed that the mean droplet size for the microemulsions with and without fexofenadine was below 100 nm. Microemulsions without fexofenadine were transparent colloidal dispersions with an average diameter of 39.05 nm and a polydispersity index of 0.044. When fexofenadine was loaded into the microemulsion system, the microemulsion did not change in transparency or polydispersity index value, but the mean droplet size decreased from 39.05 nm to 33.29 nm. A possible reason for this might be that the contents of the microemulsion and fexofenadine affect each other. When the fexofenadine molecule is dissolved and dispersed into the emulsifying membrane layer (composed of surfactant and cosurfactant) and oil phase, the chemical groups in the fexofenadine molecule can react with the other groups in the surfactant, cosurfactant, and oil phase, producing hydrogen bonds. This could be due to the decreased surface tension created by the presence of surfactant and cosurfactant.Citation22 In addition, the zeta potential was measured. When fexofenadine was loaded to the microemulsion system, the zeta potential of the microemulsion increased from 1.95 mV to 2 mV. This increment was not statistically significant (P < 0.05). It is clear from the physicochemical data () that microemulsions with and without fexofenadine had similar values for viscosity, conductivity, and pH (P > 0.05).
Table 3 Characterization of microemulsion
Fexofenadine content
shows that the fexofenadine content in the microemulsion system was approximately 5.98 mg/mL. Loading of fexofenadine into the w/o microemulsion system was obtained, with an encapsulation efficiency of 99.67%.
Stability test
After storage at 25°C and 40°C for 6 months, the optimized fexofenadine microemulsion remained clear and transparent without any phase separation. shows that the fexofenadine concentration and droplet size in the microemulsion was not changed significantly, suggesting that the fexofenadine-loaded microemulsion was stable under the above conditions.
Table 4 Stablity of optimum microemulsion containing FEX (A) and (B)
Permeability of fexofenadine in Caco-2 cell monolayer
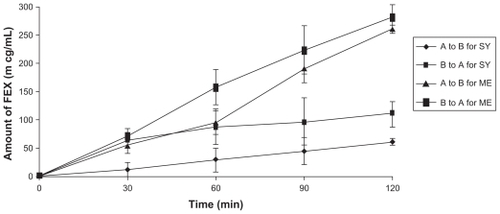
shows the permeability profiles for fexofenadine from the w/o microemulsion and from the commercial syrup across the Caco-2 cell monolayers. The permeability studies were performed at 37°C. The results indicate that permeability of fexofenadine from the w/o microemulsion was greater than that of the syrup formulation. The bidirectional Papp values for both formulations are summarized in .
Table 5 The permeability value of FEX with different formulations (n = 6 ± SD)
Figure 2 The amount of FEX permeability across Caco-2 cell (n = 6 ± SD).
Notes: A to B for SY = The amount of FEX from apical to basolateral direction of syrup, B to A for SY = The amount of FEX from basolateral to apical direction of syrup, A to B for ME = The amount of FEX from apical to basolateral direction of microemulsion, B to A for ME = The amount of FEX from basolateral to apical direction of microemulsion.
Abbreviations: A, apical; B, basolateral; FEX, fexofenadine; ME, microemulsion; SD, standard deviation; SY, syrup.
Generally, substances with an apparent permeability coefficient (Papp) < 1 × 10−6 cm/sec are classified as low permeability substances, and those with an apparent permeability coefficient > 1 × 10−5 cm/sec are classifed as high permeability substances.Citation7 The microemulsion and commercial syrup formulations had high permeability for both directions. Papp B to A (permeability from basolateral to apical direction) was higher than Papp A to B (permeability from apical to basolateral direction) for both formulations, consistent with the contribution of a secretion transporter, and fexofenadine has been demonstrated to be a P glycoprotein substrate.Citation8 When the fexofenadine microemulsion was applied to the Caco-2 cell, the Papp A to B was increased (2.55 × 10−5 ± 0.05 cm/sec) compared with the Papp A to B of the syrup (1.97 × 10−5 ± 1.23 cm/sec), and the Papp B to A was increased (2.61 × 10−5 ± 1.77 cm/sec) compared with the Papp B to A of the syrup (2.39 × 10−5 ± 1.33 cm/sec). These findings suggest that fexofenadine in the microemulsion has higher permeability than when in the commercial syrup form. This also indicates that the microemulsion is an enhancer of fexofenadine permeability.
Effect of fexofenadine on TEER across Caco-2 cell monolayer
TEER values across Caco-2 cells have been used as indicators of tight junction integrity in cell monolayers. Therefore, the effects of both formulations on the ability to modulate tight junctions could be monitored using this indicator. Experiments performed with the commercial syrup showed a change in TEER of < 20% for 2 hours in both directions (apical to basolateral and basolateral to apical). However, experiments with the microemulsion showed a reduction in TEER over the 2 hours, with TEER being 33.7% and 41.1% of control values at 2 hours in the apical to basolateral and basolateral to apical directions, respectively (). Changes in TEER after treatment with the microemulsion were statistically significant compared with the commercial syrup (P < 0.05). In general, the microemulsion-induced decrease in TEER was correlated with an increase in permeability of fexofenadine.
Table 6 The % TEER change of experiments across Caco-2 cell for both directions with ± SD (A → B: Apical to basolateral direction, B → A basolateral to apical direction)
Estimation of fexofenadine transport parameters
Fexofenadine permeability values for the microemulsion were higher than for the syrup in the basolateral to apical and apical to basolateral directions. This is consistent with the presence of surfactant and cosurfactant as permeability enhancers. The following model equations, including the passive diffusion component and the Michelis–Menten component, were fitted to the dataset for apical to basolateral and basolateral to apical permeabilities in order to estimate the passive component and Vm and Km.
where Peff is the experimental permeability value, Pdif is the passive diffusion component, and Vm and Km are the Michelis–Menten parameters. A second model was explored with the following equations:
in which A is a correction factor included to take into account the fact that the binding site of the secretion carrier is located inside the cells so it actually “sees” a different formulation than the donor chamber one. This factor helps to explain why permeability versus concentration is not symmetrical around Pdif in both directions. The change in apparent permeability is more evident in the basal to apical direction because the basolateral membrane has lower resistance, and when the drug is applied in the basolateral chamber, the extracellular and intracellular concentrations are more similar than when the drug is placed in the apical chamber.Citation23 The parameters obtained with both kinetic models for the commercial syrup and microemulsion are summarized in . In order to compare the two models, the sum of squared residuals and Akaike information criterion values were tabulated.
Table 7 Parameters of the fit of the models with passive component an Michaelis–Menten component and goodness of fit indexes
The residual variances from both fits (sum of squared/ degrees of freedom) were compared using the Snedecor’s F test and an alpha value of 0.05 with the following equation:
where SSR1 is the sum of the squared residual of the simplest models and SSR2 is the sum of the squared residuals of the more complex model, and df are the degrees of freedom of the fit (number of data points minus number of parameter estimated). The calculated F was higher than the tabulated F, indicating statistical significance of the more complex model, so the inclusion of parameter A improves the fit to the experimental data.
Pharmacokinetic studies in rabbits
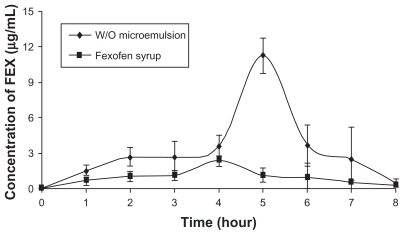
shows the plasma concentration-time profiles of fexofenadine after administration of the w/o microemulsion and the syrup to rabbits, each at a dose of 6 mg/kg body weight (n = 6). shows the pharmacokinetic parameters for fexofenadine that were evaluated by the noncompartmental method using the oral WinNonLin program. In this study, oral administration of w/o microemulsion in lower doses gave significantly higher peak plasma concentrations and AUC values compared with the syrup. The mean peak fexofenadine concentration in the group treated with the syrup was 1.97 μg/m; a higher mean peak value of 18.63 μg/mL was obtained in the group treated with the microemulsion. This difference was statistically significant (P < 0.05). In the case of oral administration, the mean residence time and time to peak plasma concentrations obtained from the w/o microemulsion were significantly prolonged (P < 0.05) when compared with those for the syrup.
Table 8 Pharmacokinetic results from curve fitting of in vivo plasma data after microemulsion and Fexofen® syrup administration (n = 6) with SD
Figure 3 Comparison of the plasma concentration values obtained after administration of FEX loaded microemulsion and Fexofen®syrup.
Abbreviation: FEX, fexofenadine; w/o, water in oil.
There have been many previous attempts at estimating the absolute oral bioavailability of fexofenadine, with values ranging widely from 10% to 90%.Citation24 For example, Chen found that the absolute bioavailability of fexofenadine was 35%.Citation9 In this study, the relative bioavailability related to the AUC0→8 of fexofenadine was found to be 376.76% in w/o microemulsion compared with the syrup. This may be because the microemulsion allows the drug to be retained long enough in the gastrointestinal system to be well permeated in the gastrointestinal mucosa. According to the relative bioavailability value, the amount of fexofenadine in the microemulsion can be reduced, enabling a lower-dose formulation. Furthermore, a higher relative bioavailability, delayed mean residence time, and extended time to peak concentration with the fexofenadine microemulsion suggested that this microemulsion could have extendedrelease characteristics, and might be useful for extendedrelease oral formulations of fexofenadine.
Discussion
The minimum absolute oral bioavailability of fexofenadine in this study was 10%–15%, based on the amount of unchanged drug appearing in the urine.Citation9 Extrapolation from animal data is unreliable, with oral bioavailability being 2%–4% in rodentsCitation25 and only marginally higher at 6.6% in monkeys.Citation26 An estimate of 30% bioavailability in man has been made based on two observations,Citation9 one being the 10% recovery in urine after oral fexofenadine and the other the finding that fexofenadine is formed by the metabolism of orally administered terfenadine (which is rapidly and extensively metabolized), and is assumed to occur systemically.
To improve its oral bioavailability, some absorption enhancers can be used to improve the oral absorption of fexofenadine, such as chitosan, sodium deoxycholate, and sodium dodecyl sulfate.Citation27 However, these enhancers can lead to irritation of the intestinal tract, resulting in substantial damage to the intestinal mucosa.Citation28 Thus, it is hoped that a suitable pharmaceutical formulation of fexofenadine can be developed to improve intestinal absorption without damaging the intestinal tissue. Our w/o microemulsion system with surfactant and cosurfactant has the ability to form microemulsions, with good biological acceptance, which is the main reason for their use.Citation29 Water-soluble drugs are solubilized in w/o microemulsion and incorporated mainly in the water-phase core and therefore are released rather slowly.Citation30 In this study, the viscosity, refractive index, particle size, zeta potential, and electric conductivity of the microemulsion were investigated. The results demonstrated successful formation of a w/o microemulsion containing oil, surfactant/cosurfactant, and an aqueous phase. The optimal w/o microemulsion, consisting of oleic acid, lutrol F-68, span 80, and isopropyl alcohol, is a promising formulation for increasing absorption of fexofenadine by the oral route and could also increase the oral bioavailability of other drugs with low permeability. There are several barriers to the oral delivery of such drugs, including luminal enzymatic hydrolysis, low solubility, and low membrane permeability.Citation31,Citation32 Fexofenadine has good stability against enzymatic hydrolysis and at different pH values.Citation33 For fexofenadine, low membrane permeability is the main barrier to its oral absorption. However, because of its good solubility, significantly improved absorption and bioavailability was found for fexofenadine after oral administration of the w/o microemulsion than with the commercial syrup formulation in rabbits.
Conclusion
A novel fexofenadine-loaded microemulsion was designed to improve the oral absorption of fexofenadine. The microemulsion system for fexofenadine was developed by construction of a pseudoternary phase diagram. The optimal formulation was as follows: 34.05% oil (oleic acid), 30% surfactant (span 80/lutrol F 68), 29.79% cosurfactant (isopropyl alcohol), and 6.16% distilled water. The optimized microemulsion was characterized according to viscosity, solubility, content of fexofenadine, conductivity, polydispersity, droplet size, and stability. In vitro and in vivo studies in Caco-2 cells and in rabbits showed that the microemulsion had significantly improved permeability across Caco-2 cells, a higher peak concentration and AUC, and shorter elimination half-life compared with the commercial syrup. Overall, the results indicate that the microemulsion was effective for enhancing the oral absorption of fexofenadine, and fexofenadine- loaded microemulsion demonstrates great potential for clinical application.
Disclosure
The authors report no conflicts of interest in this work.
References
- SusmanGMasonJComptonDStewartJRicardNThe efficacy and safety of fexofenadine HCl and pseudoephedrine, alone and in combination, in seasonal allergic rhinitisJ Allergy Clin Immunol199910410010610400846
- PiaoHMBalakrishnanPChoHJPreparation and evaluation of fexofenadine microemulsions for intranasal deliveryInt J Pharm201039530931620635476
- JohnHKAllergic rhinitis: current pharmacotherapyOtolaryngol Clin North Am20084134735818328373
- MarkhamAWagstaffAJFexofenadineDrugs1998552692769506246
- UjieKOdaMKobayashiMSaitohHRelative contribution of absorptive and secretory transporters to the intestinal absorption of fexofenadine in ratsInt J Pharm200836171118572335
- GarteizDAHookRHWalkerBJOkerholmRAPharmacokinetics and biotransformation studies of terfenadine in manArzneimittelforschung198232118511906817765
- KoganAKesselmanEDaninoDAserinAGartiNViability and permeability across Caco-2 cells of carbamazepine solubilized in fully dilutable microemulsionsColloids Surf B Biointerfaces20086611218599273
- ShimizuMUnoTSugawaraKTateishiTEffects of itraconazole and diltiazem on the pharmacokinetics of fexofenadine, a substrate of P-glycoproteinBr J Clin Pharmacol20066153854416669847
- ChenCSome pharmacokinetic aspects of the lipophilic terfenadine and zwitterionic fexofenadine in humansDrugs R D2007830131417767395
- DrescherSSchaeffelerEHitzlMMDR1 gene polymorphisms and disposition of the P-glycoprotein substrate fexofenadineBr J Clin Pharmacol20025352653411994059
- ShimizuTUnoKSugawaraKTateishiTEffects of itraconazole and diltiazem on the pharmacokinetics of fexofenadine, a substrate of P-glycoproteinBr J Clin Pharmacol20066153854416669847
- RitschelWAMicroemulsions for improved peptide absorption from the gastrointestinal tractMethods Find Exp Clin Pharmacol1991132052202051845
- SarciauxJMAcarLSadoPAUsing microemulsion formulations for oral delivery of therapeutic peptidesInt J Pharm1995120127136
- GaoZChoiHGShinHJPhysicochemical characterization and evaluation of a microemulsion system for oral delivery of cyclosporin AInt J Pharm19981617586
- EcclestonGMMicroemulsionsswarbrickSBoylanJCEncyclopedia of Pharmaceutical TechnologyNew York, NYmarcel Dekker1992
- ConstantinidesPPLipid microemulsions for improving drug dissolution and oral absorption: physical and biopharmaceutical aspectsPharm Res199512156115728592652
- LinHGebhardtMBianSEnhancing effect of surfactants on fexofenadine-HCl transport across the human nasal epithelial cell monolayerInt J Pharm2007330233116997520
- JadhavKRShaikhIMAmbadeKWKadamVJApplications of microemulsion based drug delivery systemCurr Drug Deliv2006326727316848728
- YinYMCuiFDMuCFDocetaxel microemulsion for enhanced oral bioavailability: preparation and in vitro and in vivo evaluationJ Control Release2009140869419709639
- KarakuşSKüçükgüzelIKüçükgüzelGSDevelopment and validation of a rapid RP-HPLC method for the determination of cetirizine or fexofenadine with pseudoephedrine in binary pharmaceutical dosage formsJ Pharm Biomed Anal20084629530218054459
- GhoshPKMajithiyaRJUmrethiaMLMurthyRSDesign and development of microemulsion drug delivery system of acyclovir for improvement of oral bioavailabilityAAPS Pharm Sci Tech2006777
- CuiJYuBZhaoYEnhancement of oral absorption of curcumin by self-microemulsifying drug delivery systemsInt J Pharm200937114815519124065
- Gonzalez-AlvarezIGonzalez-AlvarezMOltra-NogueraDUnique pharmacology of KAR–2, a potential anti-cancer agent: absorption modeling and selective mitotic spindle targetingEur J Pharm Sci200836911
- GolightlyLKGreosLSSecond-generation antihistamines: actions and efficacy in the management of allergic disordersDrugs20056534138415669879
- PengSXRitchieDMCousineauMDanserEDewireRFlodenJAltered oral bioavailability and pharmacokinetics of P-glycoprotein substrates by coadministration of biochanin AJ Pharm Sci2006951984199316850393
- OgasawaraAKumeTKazamaEEffect of oral ketoconazole on intestinal first-pass effect of midazolam and fexofenadine in cynomolgus monkeysDrug Metab Dispos20073541041817142564
- LiYZhangMWangJZhangSLiuJZhangQEffects of absorption enhancers on intestinal absorption of lumbrokinaseYao Xue Xue Bao200641939944 Chinese17184110
- YamamotoAUchiyamaTNishikawaRFujitaTMuranishiSEffectiveness and toxicity screening of various absorption enhancers in the rat small intestine: effects of absorption enhancers on the intestinal absorption of phenol red and the release of protein and phospholipids from the intestinal membraneJ Pharm Pharmacol199648128512899004192
- AlanyRGRadesTAgatonovic-KustrinSDaviesNMTuckerIGEffects of alcohols and diols on the phase behaviour of quaternary systemsInt J Pharm200019614114510699705
- PodlogarFGasperlinMTomsicMJamnikARogacMBStructural characterisation of water-Tween 40/Imwitor 308-isopropyl myristate microemulsions using different experimental methodsInt J Pharm200427611512815113620
- LangguthPBohnerVHeizmannJThe challenge of proteolytic enzymes in intestinal peptide deliveryJ Control Release1997463957
- LappinGShishikuraYJochemsenRPharmacokinetics of fexofenadine: evaluation of a microdose and assessment of absolute oral bioavailabilityEur J Pharm Sci20104012513120307657
- KumarLAlamMSMeenaCLJainRBansalAKFexofenadine hydrochlorideProfiles of Drug Substances, Excipients and Related Methodology200934153192