 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Background
Genistein, one of the major isoflavones, has received great attention as a phytoestrogen and potential cancer chemoprevention agent. However, the dissolution and bioavailability of genistein from solid oral preparations is low due to its poor water solubility.
Methods
In order to improve the oral bioavailability of genistein, genistein nanoparticles were prepared by the nanoprecipitation technique using Eudragit® E100 as carriers and an optimized formulation of mass ratio (genistein:Eudragit E100, 1:10). The mean particle size of genistein nanoparticles was approximately 120 nm when diluted 100 times with distilled water. The drug-loaded nanoparticles were spherical on observation by transmission electric microscopy.
Results
Encapsulation efficiency and drug loading of the genistein nanoparticles were approximately 50.61% and 5.02%, respectively. Release of drug from the genistein nanoparticles was two times greater than that from the conventional capsules. After administration of genistein suspension or genistein nanoparticles at a single dose of 100 mg/kg to fasted rats, the relative bioavailability of genistein from the nanoparticles compared with the reference suspension was 241.8%.
Conclusion
These results suggested that a nanoparticle system is a potentially promising formulation for the efficient delivery of poorly water-soluble drugs by oral administration.
Introduction
Genistein (5, 7, 4′-triatomic isoflavone, see ) is a primary active component of the soybean, kudzuvine root, scoparius, and other leguminous plants. As one of the most extensively studied isoflavones, it is generally recognized as a phytoestrogen.Citation1 With its weak estrogenic and antiestrogenic properties, genistein is not only used as an antioxidant, ie, an inhibitor of protein tyrosine kinase or topoisomerase II, but is also used to induce cell differentiation.Citation2 Asiatic women with a high intake of isoflavones from soy products have been shown to have a decreased risk of osteoporosis, cardiovascular disease, breast and uterine cancer, and climacteric syndrome.Citation3,Citation4 Although frequent reports demonstrate positive influences of isoflavones on human health, the clinical application of genistein is hindered by its disadvantages, including poor solubility in water and low serum levels after oral administration.Citation5
Figure 1 Structure of genistein.
Various drug delivery systems, including self-nanoemulsified systems, superparamagnetic systems, and chitosan microspheres, have been used to increase the dissolution and bioavailability of genistein. It is reported that the optimized formulation dissolved more than 90% of genistein over five minutes in a self-nanoemulsified drug delivery system.Citation6 Genistein encapsulated in Fe3O4-carboxymethylated chitosan nanoparticles shows greater water solubility than the free genistein.Citation5
The Eudragit® E cationic copolymer has been widely used to improve the solubility of poorly water-soluble drugs.Citation7 It has a basic site containing tertiary amine groups which are ionized in gastric fluid.Citation8 Therefore, Eudragit E is easy to dissolve in the gastric environment. Eudragit E is one type of high polymer material, and is nontoxic, easily absorbed orally, and is widely used in coating and film forming. In recent years, it has been used to prepare microcapsules and nanoformulations to improve the solubility of poorly water-soluble drugs. It is reported that some new nanoparticle formulations of naringenin-Eudragit E nanoparticles and quercetin-Eudragit E nanoparticles have been prepared for improving solubility.Citation9,Citation10
In this study, a drug delivery system for genistein was designed based on Eudragit E nanoparticles. It was expected that this system would improve the water solubility of the drug, and hence improve its bioavailability by oral administration. This novel nanoparticle system was characterized by its particle size, morphology, encapsulation efficiency, drug loading, and dissolution. Furthermore, the oral bioavailability of genistein nanoparticles was assessed and compared with that of a genistein suspension.
Materials and methods
Materials
Genistein was purchased from Shanxi Huike Botanical Development Co Ltd (Xi’an, Shanxi, China). Standard phenacetin was supplied by the National Institute for Control of Pharmaceutical and Biological Products (Beijing, China). Sodium dodecyl sulfate was obtained from Tianjin Chemical Reagent Co Ltd (Tianjin, China). Pluronic® F68 (Poloxamer 188) was kindly provided by BASF Corporation (Ludwigshafen, Germany). Filters (0.22 μm and 0.8 μm) were supplied by Millipore Corporation (Billerica, MA). Eudragit E100 was supplied by Röhm Pharma as a gift (Darmstadt, Germany). Sulfatase extracted from Helix pomatia was a product of Sigma-Aldrich Chemicals Co Ltd (St Louis, MO). All other reagents were of chromatographic grade. Distilled water, prepared from deionized water, was used throughout the study.
Preparations of genistein nanoparticles
The genistein nanoparticles were prepared by a nanoprecipitation method.Citation9,Citation11,Citation12 Genistein 5 mg and Eudragit E 50 mg were dissolved in 2 mL of ethanol. The internal organic phase solutions were slowly injected into 10 mL of the external aqueous solution (1% Poloxamer 188 in water), and the mixtures were then stirred at 500 rpm for 50 minutes. Internal organic phase solutions are always composed of solvents, making the drug and Eudragit soluble completely, and the external aqueous phase comprises aqueous solution, sometimes with surfactant in it. The surfactant can penetrate into the genistein nanoparticles during the nanoprecipitation process to form a stable nanoparticle delivery system. Ethanol was completely removed by rotary vacuum evaporation under a water bath at 32°C. The nanoparticles formed were filtered using a 0.8 μm filter and centrifuged at 11,000 × g for 60 minutes. The supernatant was separated and analyzed by high-pressure liquid chromatography (HPLC) for free drug content. The genistein nanoparticles formed were isolated, washed three times with distilled water, and freeze-dried. Lactose was chosen as a cryoprotectant, which had a good freeze-drying effect. Blank nanoparticles were prepared following the above method without genistein. All the nanoparticles were prepared in triplicate.
Particle size analysis
The mean size of the genistein nanoparticles was determined using a LB-500 dynamic light scattering particle size analyzer (Horiba, Kyoto, Japan). Each sample was appropriately diluted 100 times with distilled water for analysis. The particle size analyzer could measure sizes ranging from 3 nm to 6000 nm.
Morphological analysis
A drop of the fresh nanoparticle sample was placed onto a carbon-coated copper grid, forming a thin liquid film. The films on the grid were negatively stained by addition of a drop of 1% (w/v) phosphotungstic acid. The excess staining solution was removed with filter paper, then air-dried. The stained films were observed on a Tecnai G2 transmission electron microscope (Philips, Eindhoven, The Netherlands) and photographed.
Encapsulation efficiency and drug loading
The supernatant was separated by ultracentrifugation,Citation13 and the encapsulation efficiency was determined by HPLC.Citation14 The HPLC system consisted of a 600 pump (Waters, Milford, MA), a PDA-996 ultraviolet detector (Waters), and a 717 autosampler (Waters). A reversed-phase column (DiamonsilTM C18, 250 mm × 4.6 mm, 5 μm, Beijing, China) was utilized for drug separation at 25°C. The mobile phase comprised a degassed mixture of methanol and water (85:15, v/v). The flow rate of the mobile phase was maintained at 1.0 mL/minute and detection was performed at 261 nm. A 10 μL sample was injected into the HPLC column for analysis. The genistein concentration in the sample was determined using a calibration curve. The encapsulation efficiency and drug loading were determined using the following formulae:
The method showed excellent reproducibility, with intraday and interday precision of less than 2% (relative standard deviation), as well as excellent accuracy between 91.7% and 100.0% for genistein. The lower limit of quantitation for genistein was 0.02 μg/mL.
Dissolution test
The dissolution test was performed using a drug intelligence dissolution tester according to the second method of the Chinese Pharmacopoeia dissolution procedure. Briefly, lyophilized genistein nanoparticles in capsules and common capsules (drug filled in blank capsules) containing genistein approximately 1.6 mg were put into different sinkers. Each sinker was loaded into 400 mL of phosphate buffer (pH 1.2) with 0.5% sodium dodecyl sulfate at 37° ± 0.5°C and a paddle speed of 100 rpm. Each sample (4 mL) was withdrawn at 5, 10, 20, 30, 45, and 60 minutes, and an equal volume of temperature-equilibrated blank media was then added to the beaker. The samples were filtered using a 0.22 μm filter, and 10 μL was used for determination of genistein. The concentrations of drug were determined by HPLC analysis as already mentioned.
Animals and drug administration
The experimental protocols were approved by the animal care committee of Harbin Medical University, Heilongjiang, China. Twelve male Sprague-Dawley rats weighing 180–220 g were provided by the experimental animal center at our institution. They were housed in an air-conditioned room with temperature maintained at 25°C ± 1°C and humidity at 55% ± 5% under a regular 12:12 hour light/dark cycle for one week prior to treatment. All animals were fed with standard rodent chow and water ad libitum. They were randomly divided into two groups, ie, one to receive genistein nanoparticles and the other to receive genistein suspension. The genistein suspension was prepared by suspending genistein in a 1% sodium carboxymethyl cellulose solution. All groups were fasted and given genistein suspension or genistein nanoparticles at a dose of 100 mg/kg by intragastric administration. Blood samples (approximately 0.25 mL) were collected and put into heparinized tubes at hours 0.17, 0.5, 1, 2, 3, 4, 6, 8 and 12 after administration. Plasma samples were immediately separated by centrifugation at 3000 × g for 10 minutes and stored at −20°C until analysis.
Sample preparation
The 100 μL plasma samples were incubated with 100 U of sulfatase at 37°C for eight hours, and phenacetin 10 μg/mL in mobile phase and 3 mL of methyl tert-butyl ether were then added to the samples in 5 mL Eppendorf tubes. After vortex-mixing for three minutes and centrifugation at 3000 × g for 10 minutes, the supernatant was transferred to another tube and evaporated to dryness under a gentle stream of nitrogen at 40°C. The residue was reconstituted with 100 μL of mobile phase followed by vortex-mixing. A 20 μL aliquot of the supernatant was injected into the HPLC system.
HPLC assay
The concentration of genistein was determined by HPLC using a 600 pump (Waters, Milford MA), a PDA-996 UV detector (Waters), and a 717 autosampler (Waters). A reversed-phase column (Diamonsil TM C18, 250 mm × 4.6 mm, 5 μm, Beijing, China) was utilized for drug separation at 30°C. The mobile phase comprised a degassed mixture of methanol and water (60:40, v/v). The flow rate of the mobile phase was maintained at 1.0 mL/minute. Detection was performed at 261 nm. The method showed excellent reproducibility, with an intraday and interday precision of less than 12.1% (relative standard deviation), as well as an excellent accuracy of between 91.2% and 106.8% for genistein. The lower limit of quantitation for genistein was 0.15 μg/mL. The extraction recovery of genistein and the internal standard was 55.55% ± 6.65% and 52.88% ± 2.96%, respectively.
Genistein was stable in rat plasma samples after three freeze-thaw cycles, following benchtop storage at room temperature for 12 hours, and in reconstituted solution after being kept at room temperature for 12 hours. When stored at −20°C, the genistein added to the plasma samples was found to be stable for at least 30 days.
Pharmacokinetic analysis
The chromatographic data were automatically processed to obtain the peak-area ratio of compound to internal standard. The maximum plasma concentration (Cmax) and the time to reach this peak concentration (Tmax) were determined by visual inspection of the experimental data. The elimination rate constant (Ke) was calculated by applying the least-squares regression technique to the data for the last three or four points of the plasma concentration-time curve, and the half-life (t1/2) of the drug was obtained by 0.693/Ke. The area under the curve (AUC) was calculated by the trapezoid method. The relative bioavailability of genistein nanoparticles (test formulation) to the genistein suspension (reference formulation) was calculated using the following equation:
The data were presented as the mean ± standard error of the mean for the individual groups. The unpaired Student’s t-test was used to determine any statistically significant differences. Differences were considered to be statistically significant at P < 0.05.
Results
Characterization of genistein nanoparticles
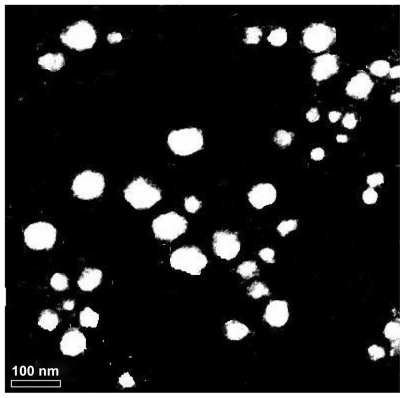
Genistein nanoparticles prepared by the nanoprecipitation technique showed a blue opalescent and uniform appearance. The optimal encapsulation efficiency and drug loading of the genistein nanoparticle formulation were 50.61% ± 0.41% and 5.02% ± 0.04%, respectively. The size of the genistein nanoparticles was approximately 120.0 ± 9.25 nm when diluted 100 times with distilled water (). The drug-loaded nanoparticles were small, spherical, and uniform, and there was no adhesion between particles seen on transmission electron microscopy ().
Figure 2 Size distribution of genistein nanoparticles.
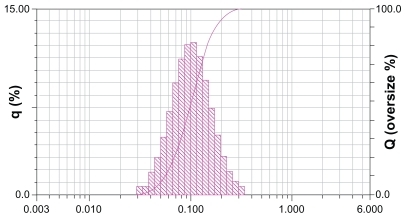
Figure 3 Transmission electron micrograph of genistein nanoparticles.
In vitro dissolution study
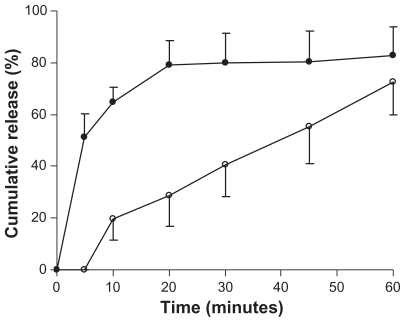
Dissolution studies were performed for lyophilized genistein nanoparticles filled in capsules and for conventional genistein capsules. The dissolution rates of the drug from these preparations were evaluated in pH 1.2 phosphate buffer with 0.5% sodium dodecyl sulfate. The release percentages of the drug from genistein nanoparticles were significantly higher than those from conventional genistein capsules (). The release of genistein nanoparticles was more than 80% within 20 minutes. In contrast, less than 30% of genistein from the conventional genistein capsules dissolved within 20 minutes.
Figure 4 Dissolution profiles of genistein from genistein nanoparticles (●) and genistein capsules (○). Tests were conducted in 400 mL of pH 1.2 phosphate buffer with 0.5% sodium dodecyl sulfate at 37°C ± 0.5°C with a rotation speed of 100 rpm. Each value is the mean ± standard deviation (n = 3).
Bioavailability studies
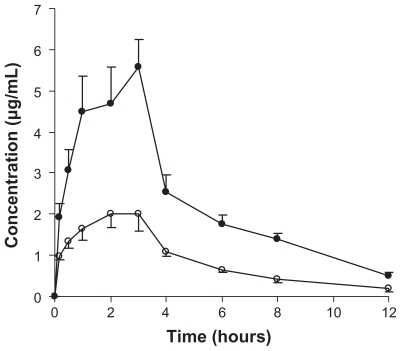
The plasma profile of genistein in rats following oral administration of a single 100 mg/kg dose of genistein was investigated in the two groups. The plasma concentration profiles for genistein in the nanoparticles showed significantly greater improvement on oral absorption than did the genistein suspension (). The main pharmacokinetic parameters for genistein in the two formulations are summarized in . The results show pronounced differences in the pharmacokinetics of genistein following oral administration in rats. The difference in Cmax value of 5.32 ± 0.71 μg/mL in the formulation group and 2.17 ± 0.87 μg/mL in the control group was statistically significant (P < 0.05). Marked differences were also observed for the AUC. The AUC0→12 for the genistein suspension was 10.8 μg/mLμhour and that for the genistein nanoparticles was 26.1 μg/mLμhour (P < 0.05). The relative bioavailability of genistein from the nanoparticles compared with the suspension was 241.8%, suggesting that a nanoparticle formulation could improve the oral absorption of genistein.
Table 1 Main pharmacokinetic parameters of genistein after a single 100 mg/kg oral dose of genistein in two groups, ie, a nanoparticle group and a suspension group
Figure 5 Genistein plasma concentration-time profiles of either genistein nanoparticles (●) or genistein suspension (○) following oral administration of a single dose of 100 mg/kg genistein in rats. Each value is the mean ± standard error (n = 6).
Discussion
In previous studies, various methods have been used to prepare nanoparticles, including a water-in oil-in-water (w1/o/w2) double emulsion method which is only used for hydrophilic drugs, and a film ultrasonic dispersion process which is complex, time-consuming, and difficult to control. Due to the limitations of these methods and the conditions of this experiment, the nanoprecipitation technique was used, based on its advantages of being relatively simple and rapid to perform. Moreover, it has been demonstrated in the literature that this nanoparticle delivery system can substantially transform the original physicochemical properties of drugs and greatly improve their bioavailability.Citation16,Citation17
For this experiment, a series of solvents, ie, ethanol, acetone, and dichloromethane, were used to investigate their influence on the preparation of genistein nanoparticles. Of these, the ethanol solvent was able to dissolve the drug and the polymer well and was quite clear, while acetone was more toxic than ethanol, and dichloromethane could not dissolve the drug or polymer completely. Therefore, ethanol was selected as the organic solvent for further study.
There were several reasons for enhancement of the drug dissolution rate. First, both genistein and Eudragit E are hydrophobic substances, which implies that a stronger affinity will be generated between them when blended with the organic ethanol solvent during the process of nanoparticle preparation. Second, the enhancement of drug dissolution could be attributed to the reduction of drug particle size, the enhanced hydrophilic properties of the drug when encapsulated in Eudragit E polymer, and enhanced wettability at the acidic pH provided by the dissolved Eudragit E. Finally, the hydrophilic and hydrophobic portion of Poloxamer 188 can penetrate into genistein nanoparticles during the nanoprecipitation process to form a stable nanoparticle delivery system. Similar observations have been reported elsewhere. Citation9,Citation18
Nanoparticle size is an important factor in gastrointestinal absorption, because only nanoparticles of appropriate size can be absorbed significantly. Several mechanisms for gastrointestinal absorption of nanoparticles have been reported, ie, cell bypass channel transport, intestinal epithelial cells cross-cell uptake, and collection of lymph nodes in the ileum (Peyer’s patches) of microfold cells.Citation19,Citation20 Studies have documented that when rats are given PLGA particles sized 100 nm, 500 nm, 1 μm, and 10 μm by oral administration, the amount of 100 nm particles captured by Peyer’s patches is 10–250 times greater than that of the other particle sizes.Citation21 Further studies have shown a range of particle sizes to be suitable for gastrointestinal absorption,Citation22 and that nanoparticles of around 100 nm are absorbed several times more efficiently than those of larger size.Citation21 The nanoparticles prepared in the present experiment were 120 nm, and could be absorbed well by the gastrointestinal tract, and thus would play a key role in efficacy.
There has been some research reporting on different genistein doses being administered to rats. For example, genistein 40 mg/kg was orally administered in a pharmacokinetic rat model,Citation23 and a dose of 480 mg/kg was used in another study to investigate toxicity.Citation24 In the present study, an intermediate dose of 100 mg/kg was administered.
There has been a report of genistein being administered orally in healthy premenopausal women to investigate the pharmacokinetics of genistein. Pharmacokinetic analysis of the plasma concentration-time curves showed that the t1/2 of genistein was 7.77 hours.Citation25 Similar findings have been reported by other investigators. Pharmacokinetic analysis showed that the serum half-life of genistein was 4.53 ± 1.40 hours in rats.Citation23 In the present study, the t1/2 of genistein was 4.60 ± 0.59 hours.
The faster absorption of genistein nanoparticles than that of genistein alone is mainly due to the higher hydrophilicity and dispersion of the nanoparticles. There were differences in the plasma concentration-time profiles and pharmacokinetic parameters of genistein in individual rats after oral administration in the two groups. This can be attributed to the fact that the absorption of genistein and activity of sulfatase can be very different from each other.
Sulfatase is from Helix pomatia, and has deglucuronidation and desulfation activities. Glucoside conjugates and sulfate conjugates of genistein are the major naturally occurring isoflavones in soybean and soybean-based food products. After being absorbed, genistein exists in the systemic circulation in the form of multiple molecules, such as glucuronide and sulfate conjugates.Citation26 These conjugates of genistein are converted to the aglycone form by sulfatase. Genistein glucuronides become active in vivo by hydrolysis, and only in this way can genistein have strong physiological functions. Sulfatase treatment is necessary because it plays an important role in the measurement of genistein concentration in plasma samples. A similar observation has been reported by other investigators.Citation23,Citation27
In conclusion, genistein, the principal soy isoflavone, is a molecule of great interest as a lead compound in anticancer drug design and an innovative chemotherapeutic agent. However, genistein is poorly soluble in water, and the dissolution and bioavailability of the drug from the solid oral preparations are different and not reproducible. In this study, the use of nanoparticles improved the dissolution of genistein. Furthermore, nanoparticles showed better oral absorption than the genistein suspension based on in vivo bioavailability studies. The relative bioavailability of genistein-loaded nanoparticles was 241.8% compared with the genistein suspension. In summary, Eudragit E nanoparticles offer a promising means to improve the solubility and bioavailability of genistein after oral administration.
Disclosure
The authors report no conflicts of interest in this work.
References
- RusinAKrawczykZGrynkiewiczGGoglerAZawisza-PuchałkaJSzejaWSynthetic derivatives of genistein, their properties and possible applicationsActa Biochim Pol201057233420216977
- LamartiniereCAMurrillWBManzolilloPAGenistein alters the ontogeny of mammary cancer development and protects against mammary cancer in ratsProc Soc Exp Biol Med19982173583649492348
- UsuiTPharmaceutical prospects of phytoestrogensEndocr J20065372016543667
- CassidyAAlbertazziPLise NielsenICritical review of health effects of soyabean phyto-oestrogens in post-menopausal womenProc Nutr Soc200665769216441947
- SiHYLiDPWangTMImproving the anti-tumor effect of genistein with a biocompatible superparamagnetic drug delivery systemJ Nanosci Nanotechnol2010102325233120355429
- ZhuSHongMLiuCPeiYApplication of Box-Behnken design in understanding the quality of genistein self-nanoemulsified drug delivery systems and optimizing its formulationPharm Dev Technol20091464264919883253
- JungJYYooSDLeeSHKimKHYoonDSLeeKHEnhanced solubility and dissolution rate of itraconazole by a solid dispersion techniqueInt J Pharm199918720921810502627
- WangSLLinSYChenTFChengWTEudragit E accelerated the diketopiperazine formation of enalapril maleate determined by thermal FTIR microspectroscopic techniquePharm Res2004112127213215587937
- YenFLWuTHLinLTChamTMLinCCNaringenin-loaded nanoparticles improve the physicochemical properties and the hepatoprotective effects of naringenin in orally-administered rats with CCl4-induced acute liver failurePharm Res20082689390219034626
- WuTHYenFLLinLTTsaiTRLinCCChamTMPreparation, physicochemical characterization, and antioxidant effects of quercetin nanoparticlesInt J Pharm200834616016817689897
- UbrichNSchmidtCBodmeierRHoffmanMMaincentPOral evaluation in rabbits of cyclosporin-loaded Eudragit RS or RL nanoparticlesInt J Pharm200528816917515607269
- ZiliZSfarSFessiHPreparation and characterization of poly-epsilon-caprolactone nanoparticles containing griseofulvinInt J Pharm200529426126715814249
- DevarajanPVSonavaneGSPreparation and in vitro/in vivo evaluation of gliclazide loaded Eudragit nanoparticles as a sustained release carriersDrug Dev Ind Pharm20073310111117454041
- SarmentoBRibeiroAVeigaFSampaioPNeufeldRFerreiraDAlginate/chitosan nanoparticles are effective for oral insulin deliveryPharm Res2007242198220617577641
- GovenderTStolnikSGarnettMCIllumLDavisSSPLGA nanoparticles prepared by nanoprecipitation: drug loading and release studies of a water-soluble drugJ Control Rel199957171185
- RatnamDVAnkolaDDBhardwajVSahanaDKKumarMNRole of antioxidants in prophylaxis and therapy: A pharmaceutical perspectiveJ Control Release200611318920716790290
- DaiJNagaiTWangXZhangTMengMZhangQpH-sensitive nanoparticles for improving the oral bioavailability of cyclosporine AInt J Pharm200428022924015265562
- ShahPPMashruRCRaneYMThakkarADesign and optimization of mefloquine hydrochloride microparticles for bitter taste maskingAAPS Pharm Sci Tech20089377389
- HussainNJaitleyVFlorenceATRecent advances in the understanding of uptake of microparticulates across the gastrointestinal lymphaticsAdv Drug Deliv Rev20015010714211489336
- DesaiMPLabhasetwarVAmidonGLLevyRJGastrointestinal uptake of biodegradable microparticles: effect of particle sizePharm Res199613183818458987081
- WinKYFengSSEffects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugsBiomaterials2005262713272215585275
- FlorenceATHilleryAMHussainNJaniPUNanoparticles as carriers for oral peptide absorption: studies on particle uptake and fateJ Control Rel1995363946
- KwonSHKangMJHuhJSComparison of oral bioavailability of genistein and genistin in ratsInt J Pharm200733714815417280808
- XuJNWangQKCuiTLuoYSWangSWZhengYXStudy on reproductive toxicity of genistein I. General reproductive toxicityChinese Traditional and Herbal Drugs200334830832
- SetchellKDFaughnanMSAvadesTComparing the pharmacokinetics of daidzein and genistein with the use of 13C-labeled tracers in premenopausal womenAm J Clin Nutr20037741141912540402
- ShelnuttSRCiminoCOWigginsPARonisMJBadgerTMPharmacokinetics of the glucuronide and sulfate conjugates of genistein and daidzein in men and women after consumption of a soy beverageAm J Clin Nutr20027658859412198004
- ChenJLinHHuMAbsorption and metabolism of genistein and its five isoflavone analogs in the human intestinal Caco-2 modelCancer Chemother Pharmacol20055515916915455178