Abstract
Colchicinoids are very potent tubulin-binding compounds, which interfere with microtubule formation, giving them strong cytotoxic properties, such as cell mitosis inhibition and induction of microcytoskeleton depolymerization. While this makes them promising vascular disrupting agents (VDAs) in cancer therapy, their dose-limiting toxicity has prevented any clinical application for this purpose. Therefore, colchicinoids are considered attractive lead molecules for the development of novel vascular disrupting nanomedicine. In a previous study, a polymeric colchicinoid prodrug that showed favorable hydrolysis characteristics at physiological conditions was developed. In the current study, this polymeric colchicinoid prodrug was evaluated in vitro and in vivo for its toxicity and vascular disrupting potential. Cell viability studies with human umbilical vein endothelial cells, as an in vitro measure for colchicine activity, reflected the degradation kinetics of the prodrug accordingly. Upon intravenous treatment, in vivo, of B16F10 melanoma-bearing mice with colchicine or with the polymeric colchicinoid prodrug, apparent vascular disruption and consequent tumor necrosis was observed for the prodrug but not for free colchicine at an equivalent dose. Moreover, a five-times-higher dose of the prodrug was well tolerated, indicating reduced toxicity. These findings demonstrate that the polymeric colchicinoid prodrug has a substantially improved efficacy/toxicity ratio compared with that of colchicine, making it a promising VDA for cancer therapy.
Introduction
The extract of Colchicum autumnale, which is more commonly known as autumn crocus, wild saffron, naked lady, or any of several other names, has been used in the therapy of gout for more than 15 centuries.Citation1 At present, it is still in clinical use for the treatment of gout, as well as several other inflammatory diseases including familial Mediterranean fever and Behçet’s disease.Citation2,Citation3 Colchicine and its colchicinoid derivatives possess the ability to bind irreversibly to tubulin, forming tubulin-colchicine complexes, which hinder microtubule formation and inhibit cell mitosis.Citation2–Citation4 It has been described that colchicine possesses anti-inflammatory properties, mainly mediated by inhibition of leukocyte adhesion and activity.Citation2,Citation5 At higher doses, tubulin-colchicine complexes induce depolymerization of microtubules, resulting in destabilization of the tubulin cytoskeleton.Citation4,Citation6,Citation7 Whereas most cells rely on actin for their cell morphology, endothelial cells of angiogenic tumor vasculature are more dependent on tubulin to maintain their typically enlongated shape.Citation6,Citation8 Therefore, upon colchicinoid-induced microtubule depolymerization, the tumor endothelial cells lose their shape, thereby exposing the vascular basement membrane, which subsequently leads to coagulation, decreased perfusion, and hemostasis.Citation9,Citation10 This process, known as vascular disruption, deprives the surrounding (tumor) cells of oxygen and nutrients, leading to massive tissue necrosis. Currently, however, there is no use for colchicine and colchicinoids in cancer therapy due to their high systemic toxicity.Citation11 Although in preclinical cancer models doses of colchicine higher than 5 mg/kg induce a significant reduction in the perfusion of tumors, the maximum tolerated dose (MTD) of colchicine is limited to around 1 mg/kg.Citation12,Citation13 Even doses below 0.5 mg/kg, as used in the clinical management of gout and familial Mediterranean fever, are frequently accompanied by gastrointestinal comorbidity (eg, nausea, vomiting, and diarrhea) and hematologic disorders, such as thrombocytopenia. Citation14 Colchicine doses higher than 0.5 mg/kg are generally considered toxic, although lower doses may still cause significant side effects, illustrating its narrow therapeutic index. Overdosing of colchicine may eventually lead to multiple organ failure, including bone marrow suppression, hemolysis, liver failure, renal failure, convulsions, and cardiac arrest, and is often lethal.Citation14,Citation15
One strategy to limit the side effects caused by colchicinoid therapy is to design colchicinoid prodrugs, which possess pharmacological activity only upon conversion.Citation16 Colchicinoids have a partition coefficient (log P) of around 1 and a relatively high volume of distribution (±2 L/kg), which implies that upon intravenous injection they immediately redistribute into the tissues, explaining the high risk for side effects.Citation17–Citation19 Therefore, by creating a colchicinoid prodrug with improved aqueous solubility, its volume of distribution is expected to be reduced, confining its distribution to the circulation and extracellular compartment and lowering its off-target toxicity. Additionally, to keep the prodrug in the proximity of its target cells, that is, the angiogenic endothelial cells, the tissue penetration of the prodrug may be reduced by increasing its molecular weight. Previously, colchicinoid prodrugs based on glycopeptide dendrimers and cobalamin (vitamin B12) have been synthesized and characterized in vitro.Citation20,Citation21 However, to be converted to the active colchicinoid, both conjugates required cellular uptake in the tumor tissue. For exploiting the direct cytotoxic activity of colchicinoids – the inhibition of tumor cell mitosis – this is a rational approach. For colchicinoid-induced vascular disruption, however, a colchicinoid prodrug that is converted extracellularly, preferably in the proximity of the tumor vascular endothelium, is needed. This may be achieved by utilizing polymer-based colchicinoid prodrugs that are more readily transformed into the active colchicinoid, such as by hydrolysis of an ester bond which allows conversion in aqueous conditions. Previous work reported the synthesis of a hydrophilic colchicinoid prodrug, where colchicine was derivatized and conjugated to poly(ethylene glycol) (PEG) using a linker liable to hydrolysis.Citation22 The synthesis of nanomedicines by conjugating PEG-chains (PEGylation) to low-molecular-weight drugs increases the hydrophilicity and size of the construct, and shields them from interactions with plasma proteins.Citation23–Citation25 Upon intravenous injection, instantaneous and random diffusion of the colchicinoid prodrug into cells is impeded by the relatively large PEG moiety, thereby preventing the binding to tubulin and limiting its toxicity. However, due to the enhanced permeability of the imperfect angiogenic vasculature, the nanosized colchicinoid prodrug may be passively targeted to the tumor tissue, where, promoted by the reductive microenvironment in the tumor tissue, it hydrolyzes to the active colchicinoid.Citation26 In the present study, a polymeric colchicinoid prodrug containing a hydrolysable linker was studied in vitro and in vivo for its therapeutic potential and toxicity as vascular disrupting agent.
Materials and methods
Synthesis of polymeric colchicinoid prodrug
Colchicine was derived and conjugated to PEG5000 using methodology reported elsewhere ().Citation22 In brief, colchicine was hydroxyl-functionalized by substituting the N-acetyl moiety with an N-2-hydroxyacetyl moiety. Subsequently, the hydroxyl group was reacted with methoxy PEG-acetic acid to obtain the hydrolysable polymeric colchicinoid prodrug. The amount of colchicine derivative per milligram of material (ie, colchicine equivalents) was determined by means of ultra performance liquid chromatography (UPLC) using an Acquity UPLC® BEH C18 1.7 μm column (Waters, Milford, MA) and ultraviolet detection at 350 nm (Acquity UPLC® PDA; Waters). The mobile phase consisted of a gradient from 5%–95% methanol in water (v/v) and trifluoroacetic acid as modifier.
Figure 1 Synthesis and hydrolysis kinetics of colchicinoid prodrug. The synthesis of the colchicinoid prodrug is performed in three steps: (1) colchicine is deacetylated to obtain N-deacetylcolchicine; (2) N-deacetylcolchicine is acylated with glycolic acid resulting in a hydroxyl functionalized colchicinoid also known as colchifoline; and (3) the colchicinoid is coupled to methoxy PEG5000 to form the colchicinoid prodrug. By using esterification to conjugate PEG to the colchicinoid, a prodrug that is hydrolysable at physiological conditions is created: at 37°C, the prodrug is cleaved within a day (t1/2 5.4 hours), while at 4°C the hydrolysis rate is limited (calculated t1/2 14 days [zero-order kinetics]).
![Figure 1 Synthesis and hydrolysis kinetics of colchicinoid prodrug. The synthesis of the colchicinoid prodrug is performed in three steps: (1) colchicine is deacetylated to obtain N-deacetylcolchicine; (2) N-deacetylcolchicine is acylated with glycolic acid resulting in a hydroxyl functionalized colchicinoid also known as colchifoline; and (3) the colchicinoid is coupled to methoxy PEG5000 to form the colchicinoid prodrug. By using esterification to conjugate PEG to the colchicinoid, a prodrug that is hydrolysable at physiological conditions is created: at 37°C, the prodrug is cleaved within a day (t1/2 5.4 hours), while at 4°C the hydrolysis rate is limited (calculated t1/2 14 days [zero-order kinetics]).](/cms/asset/8845f498-0977-4c4b-a740-6a7caf40bd6e/dijn_a_24450_f0001_b.jpg)
In vitro hydrolysis study
The hydrolysis kinetics of the colchicinoid prodrug were determined at 4°C and 37°C in phosphate buffer (20 mM, pH 7.4). During 72 hours, samples were taken at regular time intervals, and stored at −20°C before analysis. For each time point, the concentration of colchicinoid prodrug and hydrolyzed prodrug were determined by UPLC, using the methodology described in the previous section.
In vitro cytotoxicity
Human umbilical vein endothelial cells (HUVECs) were grown at 37°C and 5% carbon dioxide in angiogenic growth factor rich EGM®-2 medium (Lonza Ltd, Basel, Switzerland). Cells were seeded in 96-well plates (1 × 104 cells/well) for 24 hours before further treatment. Subsequently, the cells were incubated with colchicine and colchicinoid prodrug at concentrations ranging from 0.025–2.5 μM colchicine equivalents. The cytotoxicity of each drug after 6 hours, 24 hours, and 48 hours incubation was determined by colorimetric XTT (2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide) cell viability assay.Citation27
In vivo vascular disrupting efficacy of colchicinoid prodrug
All animal experiments were conducted in agreement with the local applicable Dutch law, “Wet op de dierproeven” (1977),Citation28 and the European Convention for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes (1986).Citation29 The mice were housed in steel cages, and water and food were provided ad libitum. Female pathogen-free C57BL/6 inbred mice of 21–24 g (Charles River Laboratories International, Inc, Wilmington, MA) were subcutaneously inoculated with 1 × 106 B16F10 cells. Ten days after tumor cell inoculation, when tumor size reached >100 mm3, phosphate buffered saline, colchicine (1 mg/kg), and the colchicinoid prodrug (1 mg/kg and 5 mg/kg, colchicine equivalents) were administered intravenously in the tail vein. The mice were sacrificed at 4 and 24 hours after injection. The tumors were excised, snap frozen in liquid nitrogen, and stored at −80°C upon sectioning.
Histological evaluation
Frozen tumor samples (n = 3 per group) were sectioned (5 μm), acetone fixed, and hematoxylin and eosin stained. Images were taken with an inverted microscope (Nikon Eclipse TE2000U; Nikon Corporation, Tokyo, Japan) using NIS Elements software (Nikon Corporation). Small magnification (10×) overlapping images were taken of the complete tumor area and subsequently stitched together with PhotoFit (v 1.4; Tekmate, Inc, Anchorage, AK) software.
Results and discussion
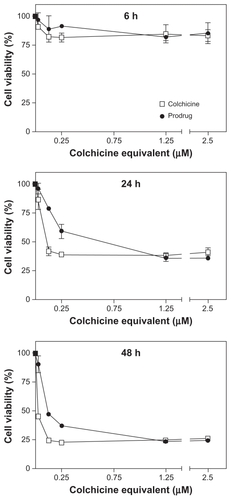
Although colchicine is widely recognized as a promising VDA for cancer therapy, its dose-limiting toxicity has prevented it from realising this potential.Citation11 Only by dosing colchicine well above its MTD, could significant vascular disruption and subsequent necrosis of tumor tissue be observed.Citation12,Citation13 In the present study, a PEG-based polymeric nanomedicine of colchicine was synthesized to attenuate systemic toxicity and to enhance its therapeutic index by improving its aqueous solubility. To this end, colchicine was derived and conjugated to PEG5000 via a hydrolysable linker (). The molecular structure of colchicine was modified at the acetamido moiety, which is not part of the pharmacophore, creating a colchicinoid also known as colchifoline, with similar anti-inflammatory and tubulin-binding activity.Citation7,Citation30,Citation31 Hydrolysis studies at physiological conditions (37°C, pH 7.4) showed that the half-life of prodrug conversion was approximately 5 hours, whereas, this was calculated by zero-order extrapolation at approximately 14 days at low temperature (). The conversion rate of the prodrug at physiological conditions correlated with its activity in endothelial cell viability experiments. To investigate the antimitotic tubulin-binding capacity as a measure of efficacy, colchicine and the colchicinoid prodrug were incubated at different concentrations (0.025–2.5 μM, colchicine equivalent) with primary HUVECs (). After 6 hours of incubation, few or no apparent effects on cell viability were measured for each treatment (two-way analysis of variance, P > 0.05), indicating that several hours of incubation are needed to allow colchicine to interfere with tubulin dynamics. However, after 24 hours and 48 hours of incubation, HUVEC viability was markedly decreased for both colchicine (dose ≥ 0.025 μM, P < 0.001) and the polymeric colchicinoid prodrug (dose ≥ 0.125 μM, P < 0.001). The prodrug, of which >95% is converted after 24 hours at 37°C, showed at the highest doses a similar cytotoxicity in comparison with colchicine. However, at lower concentrations the prodrug was less potent than colchicine after 24 hours and 48 hours incubation (P < 0.05, 0.125–0.25 μM at 24 hours; 0.025–0.25 μM at 48 hours), despite the fact that practically all prodrug has been converted at these time points. The lower activity of the prodrug can be explained by the delayed availability of the colchicinoid due to the time needed for conversion of the prodrug.
Figure 2 In vitro cytotoxicity of colchicine and colchicinoid prodrug. The endothelial cell toxicity of colchicine and the colchicinoid prodrug were determined as a measure of their ability to induce damage to angiogenic vasculature. Human umbilical vein endothelial cells were incubated with colchicine and colchicinoid prodrug at different equivalent concentrations during 6 hours, 24 hours, and 48 hours. Subsequently, the cell viability in respect to the untreated cells was determined by XTT assay. Whereas there was only low reduction in cell viability and no difference between the treatments after 6 hours of incubation, the colchicinoid prodrug was less cytotoxic than colchicine at 24 hours (0.125 μM and 0.25 μM, P < 0.05, two-way analysis of variance) and 48 hours (0.025–0.25 μM, P < 0.05).
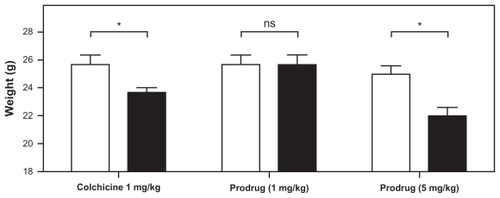
The in vivo efficacy and toxicity of colchicine and the colchicinoid prodrug as VDAs in solid tumors were assessed in mice bearing subcutaneous B16F10 melanoma tumors. To study the systemic toxicity, the weight of the mice was determined before and 24 hours after intravenous treatment with either colchicine or the prodrug. Approximately 8% of total body weight was lost 24 hours after administration of 1 mg/kg colchicine (P < 0.05 one-tailed paired t-test) (). The high loss of body weight at 24 hours after treatment with 1 mg/kg colchicine illustrates the high toxicity of colchicine, which limits the maximum dose to a level considered insufficient to result in VDA activity.Citation13 However, at 1 mg/kg colchicine equivalent dose, the polymeric prodrug did not induce significant weight loss, and only upon administration of a 5× higher dose (5 mg/kg), did it cause a drop in average body weight similar to that of colchicine at its MTD (12%, P < 0.05). This much higher tolerability of the prodrug compared with free colchicine may therefore allow for colchicinoid doses more likely to result in vascular disrupting activity.
Figure 3 Effect of in vivo toxicity of colchicine and colchicinoid prodrug on the body weight of mice. To study their in vivo toxicity, colchicine (1 mg/kg) and the colchicinoid prodrug (1 mg/kg and 5 mg/kg colchicine equivalents) were intravenously injected into B16F10 melanoma-bearing mice. The weight of the mice was measured upon injection (0 hours, white bars) and 24 hours (black bars) after injection.
Notes: Significant weight loss was observed for mice treated with 1 mg/kg colchicine (7.7%, P = 0.0371, one-tailed paired t-test) and 5 mg/kg colchicinoid prodrug (12.0%, P = 0.0175) (indicated by *), but not for mice treated with 1 mg/kg colchicinoid prodrug (0%, P > 0.05) (indicated by NS).
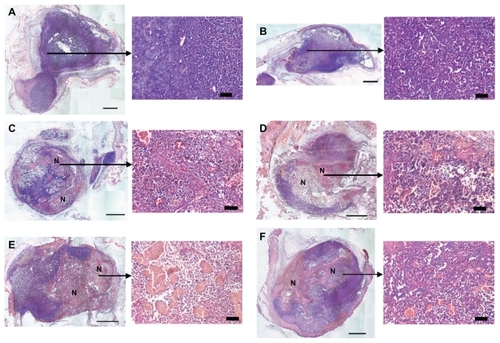
The mice were sacrificed at 4 and 24 hours after treatment and the tumors were excised, sectioned, and stained to examine vascular disruption-induced tissue necrosis. No tumor necrosis was observed at 4 hours () or 24 hours (data not shown) after intravenous injection of phosphate buffered saline or colchicine dosed at its MTD (1 mg/kg). The polymeric colchicinoid prodrug, however, induced tissue necrosis in multiple areas in the tumors 4 hours after administration at colchicine equivalent doses of 1 mg/kg and 5 mg/kg (, respectively). A similar extent of necrosis (approximately 50% of total tumor mass) was seen after 24 hours in the tumors of mice treated with colchicine equivalents of 1 mg/kg or 5 mg/kg of colchicinoid prodrug (). Although it has been shown previously for colchicine that intravenous doses of 5 mg/kg or higher are required to induce observable vascular disruption and subsequent necrosis in solid tumors,Citation12,Citation13 the polymeric colchicinoid prodrug exhibited vascular disrupting efficacy at a much lower dose (1 mg/kg), despite its reduced potency in vitro.
Figure 4 In vivo vascular disrupting activity of colchicine and colchicinoid prodrug. To investigate the vascular disrupting activity of colchicine and the colchicinoid prodrug, B16F10 melanoma-bearing mice were treated with phosphate buffered saline, colchicine (1 mg/kg), and colchicinoid prodrug (1 mg/kg and 5 mg/kg). The vascular disrupting activity of each treatment was evaluated by histological assessment of tumor tissue necrosis. Phosphate buffered saline-treated tumors did not show necrosis levels above background (A). Four hours after injection of 1 mg/kg colchicine tumor sections did not show tumor necrosis levels above control (B). Four hours after injection of 1 mg/kg (C) or 5 mg/kg (D) of colchicinoid prodrug, areas with congested blood vessels and necrotic cells were observed (as marked by N). Both 1 mg/kg (E) and 5 mg/kg (F) of the prodrug revealed considerable tumor necrosis 24 hours after injection (as marked by N).
Note: Scale bars of overview images: 1 mm; magnifications: 50 μm.
Polymer conjugation is a successful strategy in prodrug development that has been employed regularly for improving the aqueous solubility of the parent compound.Citation25 An improved aqueous solubility changes the tissue distribution, which might explain the potency of the colchicinoid prodrug in relation to colchicine.Citation16 By employing a PEG-chain larger than 35 kDa, or by utilizing colloidal drug delivery systems such as liposomes, a significant decrease in plasma clearance of the colchicinoid prodrug may be achieved, potentially enhancing its in vivo efficacy even more.Citation25,Citation32,Citation33 Nevertheless, the vascular disrupting efficacy at a low, nontoxic dose makes the polymeric colchicinoid prodrug presented here a promising VDA for cancer therapy. The observed favorable characteristics of the prodrug in vivo, on one hand, may be related to enhanced accumulation of the prodrug in the tumor tissue mediated by its improved aqueous solubility, limiting its distribution into other tissues and allowing it to penetrate via the “leaky” immature tumor vasculature.Citation34 On the other hand, the increased expression and activity of reductive enzymes, such as esterases and carboxylesterases, in tumor and endothelial cells may augment tumor-specific conversion of the prodrug into the active colchicinoid, and thus improve its efficacy at the target site, while the polymer conjugation, as such, limits its toxicity toward other healthy tissues.Citation26,Citation35–Citation39
Conclusion
The vascular disrupting efficacy and toxicity of a hydrolysable polymeric colchicinoid prodrug was studied in vitro and in vivo. The presented data convincingly demonstrate that the rate of hydrolysis of the prodrug at physiological conditions correlates with its reduced in vitro efficacy compared with colchicine. In vivo, the colchicinoid prodrug was found to be less toxic, while showing higher VDA efficacy than the parent compound, colchicine. Taken together, this study demonstrates the employment of a promising prodrug strategy using a polymeric nanomedicine for improving the vascular disrupting efficacy of colchicinoids while reducing their systemic toxicity, thereby opening the door for the application of these potent VDAs in cancer therapy.
Disclosure
This work was supported by MediTrans, an Integrated Project funded by the European Commission under the Nanotechnologies and Nano-Sciences, Knowledge-based Multifunctional Materials and New Production Processes and Devices (NMP) program, a thematic priority of the European Commission’s Sixth Framework Programme.
References
- HartungEFHistory of the use of colchicum and related medicaments in goutAnn Rheum Dis195413319020013198053
- TerkeltaubRAColchicine update: 2008Semin Arthritis Rheum200938641141918973929
- Ben-ChetritELevyMFamilial Mediterranean feverLancet199835191036596649500348
- RavelliRBGigantBCurmiPAInsight into tubulin regulation from a complex with colchicine and a stathmin-like domainNature2004428697919820215014504
- NielEScherrmannJMColchicine todayJoint Bone Spine200673667267817067838
- SchwartzELAntivascular actions of microtubule-binding drugsClin Cancer Res20091582594260119351751
- BhattacharyyaBPandaDGuptaSBanerjeeMAnti-mitotic activity of colchicine and the structural basis for its interaction with tubulinMed Res Rev200828115518317464966
- PasquierEAndréNBraguerDTargeting microtubules to inhibit angiogenesis and disrupt tumour vasculature: implications for cancer treatmentCurr Cancer Drug Targets20077656658117896922
- TozerGMKanthouCBaguleyBCDisrupting tumour blood vesselsNat Rev Cancer20055642343515928673
- KanthouCTozerGMTumour targeting by microtubule-depolymerizing vascular disrupting agentsExpert Opin Ther Targets200711111443145718028009
- JordanMAWilsonLMicrotubules as a target for anticancer drugsNat Rev Cancer20044425326515057285
- BaguleyBCHoldawayKMThomsenLLZhuangLZwiLJInhibition of growth of colon 38 adenocarcinoma by vinblastine and colchicine: evidence for a vascular mechanismEur J Cancer19912744824871827725
- NiheiYSuzukiMOkanoAEvaluation of antivascular and antimitotic effects of tubulin binding agents in solid tumor therapyJpn J Cancer Res199990121387139510665658
- FinkelsteinYAksSEHutsonJRColchicine poisoning: the dark side of an ancient drugClin Toxicol (Phila)201048540741420586571
- DickinsonMJunejaSHaematological toxicity of colchicineBr J Haematol2009146546519220282
- RautioJKumpulainenHHeimbachTProdrugs: design and clinical applicationsNat Rev Drug Discov20087325527018219308
- QuinnFRNeimanZBeislerJAToxicity and quantitative structure-activity relationships of colchicinesJ Med Chem19812456366397241524
- ZamoraJMPearceHLBeckWTPhysical-chemical properties shared by compounds that modulate multidrug resistance in human leukemic cellsMol Pharmacol19883344544623162758
- WallaceSLOmokokuBErtelNHColchicine plasma levels: implications as to pharmacology and mechanism of actionAm J Med19704844434485444299
- BagnatoJDEilersALHortonRAGrissomCBSynthesis and characterization of a cobalamin-colchicine conjugate as a novel tumor-targeted cytotoxinJ Org Chem200469268987899615609930
- LagnouxDDarbreTSchmitzMLReymondJLInhibition of mitosis by glycopeptide dendrimer conjugates of colchicineChemistry200511133941395015861378
- CrielaardBJvan der WalSLammersTLiposomes as carriers for colchicine-derived prodrugs: vascular disrupting nanomedicines with tailorable drug release kineticsEur J Pharm Sci Epub 2011 Sep 1
- ParveenSSahooSKNanomedicine: clinical applications of polyethylene glycol conjugated proteins and drugsClin Pharmacokinet2006451096598816984211
- GreenwaldRBPendriABolikalDGilbertCWHighly water soluble taxol derivatives: 2′-polyethyleneglycol esters as potential prodrugsBioorg Med Chem Lett199442024652470
- GreenwaldRBChoeYHMcGuireJConoverCDEffective drug delivery by PEGylated drug conjugatesAdv Drug Deliv Rev200355221725012564978
- DennyWATumor-activated prodrugs – a new approach to cancer therapyCancer Invest200422460461915565818
- ScudieroDAShoemakerRHPaullKDEvaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell linesCancer Res19884817482748333409223
- Wet op de dierproeven. stb. 1977, 6. BWBR0003081; Article 9.
- European Treaty Series. 18 III 1986. ETS No. 123
- BrossiASharmaPNAtwellLBiological effects of modified colchicines. 2. Evaluation of catecholic colchicines, colchifolines, colchicide, and novel N-acyl- and N-aroyldeacetylcolchicinesJ Med Chem19832610136513696620299
- NguyenTLMcGrathCHermoneARA common pharmacophore for a diverse set of colchicine site inhibitors using a structure-based approachJ Med Chem200548196107611616162011
- FensMHHillKJIssaJLiposomal encapsulation enhances the antitumour efficacy of the vascular disrupting agent ZD6126 in murine B16.F10 melanomaBr J Cancer20089981256126418797467
- LammersTHenninkWEStormGTumour-targeted nanomedicines: principles and practiceBr J Cancer200899339239718648371
- DanquahMKZhangXAMahatoRIExtravasation of polymeric nanomedicines across tumor vasculatureAdv Drug Deliv Rev201163862363921144874
- YamadaTHosokawaMSatohTImmunohistochemistry with an antibody to human liver carboxylesterase in human brain tissuesBrain Res19946581–21631677834338
- XieMYangDLiuLXueBYanBHuman and rodent carboxylesterases: Immunorelatedness, overlapping substrate specificity, differential sensitivity to serine enzyme inhibitors, and tumor-related expressionDrug Metab Dispos200230554154711950785
- RedinboMRPotterPMMammalian carboxylesterases: from drug targets to protein therapeuticsDrug Discov Today200510531332515749280
- SatohTHosokawaMThe mammalian carboxylesterases: from molecules to functionsAnnu Rev Pharmacol Toxicol1998382572889597156
- CrowJAHerringKLXieSBorazjaniAPotterPMRossMKInhibition of carboxylesterase activity of THP1 monocytes/macrophages and recombinant human carboxylesterase 1 by oxysterols and fatty acidsBiochim Biophys Acta201018011314119761868