Abstract
Three docetaxel (DX) lipid conjugates: 2′-lauroyl-docetaxel (C12-DX), 2′-stearoyl-docetaxel (C18-DX), and 2′-behenoyl-docetaxel (C22-DX) were synthesized to enhance drug loading, entrapment, and retention in liquid oil-filled lipid nanoparticles (NPs). The three conjugates showed ten-fold higher solubility in the liquid oil phase Miglyol 808 than DX. To further increase the drug entrapment efficiency in NPs, orthogonal design was performed. The optimized formulation was composed of Miglyol 808, Brij 78, and Vitamin E tocopheryl polyethylene glycol succinate (TPGS). The conjugates were successfully entrapped in the reduced-surfactant NPs with entrapment efficiencies of about 50%–60% as measured by gel permeation chromatography (GPC) at a final concentration of 0.5 mg/mL. All three conjugates showed 45% initial burst release in 100% mouse plasma. Whereas C12-DX showed another 40% release over the next 8 hours, C18-DX and C22-DX in NPs showed no additional release after the initial burst of drug. All conjugates showed significantly lower cytotoxicity than DX in human DU-145 prostate cancer cells. The half maximal inhibitory concentration values (IC50) of free conjugates and conjugate NPs were comparable except for C22-DX, which was nontoxic in the tested concentration range and showed only vehicle toxicity when entrapped in NPs. In vivo, the total area under the curve (AUC0–∞) values of all DX conjugate NPs were significantly greater than that of Taxotere, demonstrating prolonged retention of drug in the blood. The AUC0–∞ value of DX in Taxotere was 8.3-fold, 358.0-fold, and 454.5-fold lower than that of NP-formulated C12-DX, C18-DX, and C22-DX, respectively. The results of these studies strongly support the idea that the physical/chemical properties of DX conjugates may be fine-tuned to influence the affinity and retention of DX in oil-filled lipid NPs, which leads to very different pharmacokinetic profiles and blood exposure of an otherwise potent chemo-therapeutic agent. These studies and methodologies may allow for improved and more potent nanoparticle-based formulations.
Introduction
Docetaxel (DX) is a potent anticancer drug used to treat various cancers including metastatic androgen-independent prostate cancer, breast cancer, and advanced non-small cell lung cancer.Citation1–Citation3 DX inhibits cell growth by binding to microtubules, stabilizing them, and preventing their depolymerization.Citation4 Currently, Taxotere® (Sanofi–Aventis, Bridgewater NJ) is the only commercial formulation of DX on the market. The formulation contains a solvent system of polysorbate 80 and ethanol. Side-effects related to these excipients have been reported, including hypersensitivity and fluid retention.Citation5–Citation9 Great effort has been made to develop safer formulations to effectively deliver DX, including micelles, liposomes, nanoemulsions, solid lipid nanoparticles, nanocapsules, and polymeric nanoparticles.Citation10–Citation15 Given the hydrophobic property of DX, lipid-based NPs, especially liquid oil-filled NPs, serve as a viable alternative delivery system. Lipid-based NPs have the advantages of low toxicity, capability for controlled drug release, and the potential to penetrate the leaky vasculature of tumors.
The enhanced permeability and retention (EPR) effect is the major mechanism for passive targeting of anticancer drug formulations to accumulate in the tumor site. To ensure that the NPs take advantage of the EPR effect, the NPs need to maintain two aspects of stability in vivo: long circulation of delivery vehicles and long retention of anticancer agents in the NPs. The importance of long circulation of NPs has been widely recognized and extensively demonstrated for decades. Various polyethylene glycol (PEG)-coated nano-formulations have shown prolonged circulation time in vivo.Citation16–Citation19 On the other hand, the enhancement of drug retention in long circulating NPs increases drug uptake and accumulation in the tumor tissue. To study the retention of drugs in the NPs, many in vitro release studies have been conducted in aqueous buffers (eg, phosphate buffered saline [PBS]) and are expected to predict the in vivo retention behaviors of the nano-formulation. However, the correlation between the in vitro release in PBS and in vivo release behavior is often poor, especially when the entrapped drug has extremely low aqueous solubility and/or high protein binding affinity. Given that DX has poor water solubility and high protein binding,Citation20 we developed a more predictive “ex vivo” release method to better mimic the in vivo environment with the goal to achieve better correlation with the pharmacokinetic profiles.
Previously, we developed liquid-oil filled Brij 78, Vitamin E TPGS and Miglyol (BTM) 808 NPs to deliver DX. However, despite the desirable formulation properties (eg, monodisperse particle size, apparent drug entrapment efficiency, etc), DX was found to be rapidly released in mouse plasma in vitro and nearly superimposable pharmacokinetic profiles with Taxotere were observed (unpublished data). Despite the fact that DX is a poorly water-soluble drug, which favors the oil phase over the aqueous phase during preparation, DX has appreciable solubility in aqueous solutions and is highly protein bound. Moreover, the affinity of DX with the oil core is not high enough to prevent its rapid diffusion from the oil core to an aqueous phase comprised of 100% plasma. These factors led to the poor retention of DX in the BTM 808 NPs in plasma and in vivo.
The objective of this study was to improve the affinity and retention of DX in the NPs to thereby achieve prolonged in vivo blood exposure. To this end, we synthesized three lipid-DX prodrugs with different fatty acid chain lengths. The chain lengths (12, 18, and 22) were chosen to be compatible with the liquid oil core, Miglyol 808, which is composed of caprylic acid triglycerides. By utilizing the new release method to investigate the in vitro release of the conjugates from these NPs, a correlation between the in vitro release and in vivo pharmacokinetics was achieved. The superior pharmacokinetic profiles of the three conjugates in NPs compared to Taxotere makes them promising candidates for preclinical anticancer efficacy studies.
Materials and methods
Materials and animals
Docetaxel, paclitaxel (PX), lauroyl chloride (98%), stearoyl chloride (97%), behenoyl chloride (>99%) and 4-(dimethylamino) pyridine (DMAP) were purchased from Sigma-Aldrich (St Louis, MO). Miglyol 808 was obtained from Sasol (Witten, Germany). Polyoxyl 20-stearyl ether (Brij 78) was obtained from Uniqema (Wilmington, DE). D-alpha-tocopheryl polyethylene glycol succinate (Vitamin E TPGS) was purchased from Eastman Chemicals (Kingsport, TN). BALB/c mouse plasma was purchased from Innovative Research Inc (Novi, MI). Sepharose CL-4B was purchased from GE Healthcare (Uppsala, Sweden). Hybrid-SPE® cartridge was purchased from Sigma-Aldrich Supelco (St Louis, MO).
The human prostate cancer cell line, DU-145, was obtained from the American Type Collection (Manassas, VA) and was maintained in RPMI-1640 medium with 10% fetal bovine serum (FBS). Male athymic nude (nu/nu) mice, 4–5 weeks old, were obtained from the University of North Carolina, Division of Laboratory Animal Medicine (Chapel Hill, NC) and housed in a pathogen-free room. All experiments involving mice were conducted according to an approved animal protocol by the University of North Carolina Institutional Animal Care and Use Committee.
Synthesis
General procedure for the synthesis of 2′-lauroyl-docetaxel (1, C12-DX)Citation21
A flame-dried round-bottom flask was charged with DX (0.2 g, 2.48 × 10−4 mol, 1 equiv) and 4-(dimethylamino)pyridine (DMAP) (0.06 g, 4.95 × 10−4 mol, 2 equiv) in dry CH2Cl2 (8 mL) under argon. The solution was stirred for 10 minutes at 0°C. Lauroyl chloride (57.2 μl, 2.48 × 10−4 mol, 1 equiv) was added, and the reaction mixture was stirred for 6 hours at 0°C. The reaction was monitored by thin layer chromatography (TLC) (CH2Cl2: MeOH 9:1 v/v; Rf = 0.7) for completion. After completion, the solvent was removed by rotary evaporation in vacuo and the crude product was dissolved in diethyl ether (50 mL) and washed with 5% HCl (3 × 40 mL), and finally with brine (40 mL) to remove the salt byproducts. The organic phase was dried over anhydrous sodium sulfate, and the solvent was evaporated in vacuo. The product was purified by silica-packed column chromatography (9:1 CH2Cl2:MeOH) to give the desired DX derivative as a white solid (0.21 g, yield 85%). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.12 (t, 3H, −CH3(CH2)10), 1.23 (s, 6H, −H16,17), 1.34 (s, 9H, −H7′–9′), 1.72 (s, 3H, −H19), 1.75 (m, 14H, −(CH2)7CH2CH3), 1.81 (m, 2H, −CH2CH2C1″), 1.84 (s, 3H, −H18), 1.87 (m, 2H, −H14), 2.26 (d, 2H, −CH2C1″), 2.36 (s, 3H, −H22), 2.67 (m, 1H, −H3), 3.43 (s, 1H, −H7), 3.9 (d, 1H, −H4), 4.17 (d, 1H, −H6), 4.24 (m, 1H, −H5), 4.3 (d, 1H, −H20), 4.61 (s, 1H, −H10), 4.93 (d, 1H, −H13), 5.21 (d, 1H, −H10), 5.67 (d, 1H, −H2), 6.21 (t, 1H, −H2′), 7.31 (m, 1H, −H3′), 7.36–7.61 (m, 8H, −Ar-H26–28 and Ar-H30–35), 8.1 (d, 2H, −Ar-H25,29). 13C NMR (100 MHz, CD3OD): δ (ppm) = 9.9 (−C19), 14.4 (−CH3(CH2)10), 20.6 (−C18), 22.5 (−C22), 24.8 (−(CH2)9CH2CH3), 26.44 (−C16,17), 28.2 (−C7′–9′), 31.9 (−(CH2)8C1″), 34.4 (−C6,14), 43.1 (−C15), 46.4 (−C3), 56.4 (−C3′), 57.6 (−C8), 72 (−C13), 72.4 (−C7), 74.5 (−C2), 74.8 (−C10), 76.6 (−C20), 78.8 (−C6′), 80.2 (−C1), 81.1(−C4), 84.1 (−C5), 126. 7 (−C31,33,35), 128 (−C32,34), 128.7 (−C26,28), 130.2 (−C24,25,29), 133.7 (−C27), 135.9 (−C11), 138.5 (−C12), 155.3 (−C5′), 167.1, 167 (−C23), 172.7 (−C21), 174 (−C1), 177.8 (−C1″), 211.6 (−C9).
General procedure for the synthesis of 2′-stearoyl-docetaxel (2, C18-DX)
2′-stearoyl-docetaxel was synthesized following the same procedure outlined above for 2′-lauroyl-docetaxel using stearoyl chloride to give the final conjugate as a white solid (0.17 g, yield 65%). 1H NMR (400 MHz, CDCl3): δ (ppm) = 0.8 (t, 3H, −CH3(CH2)16), 1.05 (s, 6H, −H16,17), 1.14 (s, 9H, −H7′–9′), 1.16 (s, 3H, −H19), 1.26 (m, 14H, −(CH2)13CH2CH3), 1.45 (m, 2H, −CH2CH2C1″), 1.68 (s, 3H, −H18), 1.88 (m, 2H, −H14), 2.25 (d, 2H, −CH2C1″), 2.37 (s, 3H, −H22), 2.38 (m, 1H, −H3), 3.43 (s, 1H, −H7), 3.85 (d, 1H, −H4), 4.12 (d, 1H, −H6), 4.24 (m, 1H, −H5), 4.3 (d, 1H, −H20), 4.88 (d, 1H, −H13), 5.14 (s, 1H, −H10), 5.3 (d, 1H, −H10), 5.61 (d, 1H, −H2), 6.2 (t, 1H, −H2′), 7.2 (m, 1H, −H3′), 7.25–7.53 (m, 8H, −Ar-H26–28 and Ar-H30–35), 8.05 (d, 2H, −Ar-H25,29). 13C NMR (100 MHz, CD3OD): δ (ppm) = 8.9 (−C19), 13.2 (−CH3(CH2)16), 19.9 (−C18), 21.6 (−C22), 23.7 (−(CH2)15CH2CH3), 25.3 (−C16,17), 27.1 (−C7′–9′), 30.9 (−(CH2)14C1″), 32.7 (−C6,14), 42.1 (−C15), 45.4 (−C3), 56.4 (−C3′), 57.6 (−C8), 70.8 (−C13), 73.1 (−C7), 73.5 (−C2), 74 (−C10), 77.9 (−C20), 79.4 (−C6′), 79.9 (−C1), 83.2 (−C4), 84.1 (−C5), 125.3 (−C31,33,35), 127.1 (−C32,34), 127.8 (−C26,28), 129.2 (−C24,25,29), 132.6 (−C27), 134.5 (−C11), 138.2 (−C12), 154.1 (−C5′), 166.1 (−C23), 167.2 (−C21), 168.7 (−C1), 171.8 (−C1″), 210.6 (−C9).
General procedure for the synthesis of 2′-behenoyl-docetaxel (3, C22-DX)
2′-behenoyl-docetaxel was synthesized following the same procedure outlined above for 2′-lauroyl-docetaxel using behenoyl chloride to give the final conjugate as a white solid (0.26 g, yield 95%). 1H NMR (400 MHz, CDCl3): δ (ppm) = 0.81 (t, 3H, −CH3(CH2)20), 1.05 (s, 6H, −H16,17), 1.14 (s, 9H, −H7′–9′), 1.16 (s, 3H, −H19), 1.23 (m, 36H, −(CH2)18CH2CH3), 1.26 (m, 2H, −CH2CH2C1″), 1.45 (s, 3H, −H18), 1.62 (m, 2H, −H14), 1.67 (d, 2H, −CH2C1″), 1.9 (s, 3H, −H22), 2.26 (m, 1H, −H3), 2.4 (s, 1H, −H7), 3.86 (d, 1H, −H4), 4.12 (d, 1H, −H6), 4.23 (m, 1H, −H5), 4.26 (d, 1H, −H20), 4.61 (s, 1H, −H10), 4.9 (d, 1H, −H13), 5.14 (d, 1H, −H10), 5.62 (d, 1H, −H2), 6.2 (t, 1H, −H2′), 7.2 (m, 1H, −H3′), 7.22–7.53 (m, 8H, −Ar-H26–28 and Ar-H30–35), 8.05 (d, 2H, −Ar-H25,29). 13C NMR (100 MHz, CD3OD): δ (ppm) = 8.9 (−C19), 13.2 (−CH3(CH2)20), 19.9 (−C18), 21.6 (−C22), 23.7 (−(CH2)19CH2CH3), 25.3 (−C16,17), 27.1 (−C7′–9′), 30.9 (−(CH2)18C1″), 32.7 (−C6,14), 42.1 (−C15), 45.4 (−C3), 56.5(−C3′), 57.2 (−C8), 70.8 (−C13), 73.1 (−C7), 73.5 (−C2), 74.0 (−C10), 76.3 (−C20), 77.9 (−C6′), 79.9 (−C1), 83.2(−C4), 84.1 (−C5), 125.3 (−C31,33,35), 127.1 (−C32,34), 127.8 (−C26,28), 129.2 (−C24,25,29), 132.6 (−C27), 134.5 (−C11), 138.2 (−C12), 154.2 (−C5′), 166.1, 167.2 (−C23), 168.4 (−C21), 171.8 (−C1), 177.8 (−C1″), 210.6 (−C9).
Characterization of DX and DX conjugates
Mass spectrometry
Electrospray ionization (ESI) coupled with direct injection was employed to determine the m/z of the final synthetic conjugate products by Thermo Scientific TSQ Quantum Access (Waltham, MA) with positive ionization. The mass of the observed molecular ions were m/z = 1012.6, 1096.7, and 1152.8, which clearly corresponded to the Na+ adducts of C12-DX, C18-DX, and C22-DX respectively.
High performance liquid chromatography (HPLC)
The DX conjugate concentrations were quantified by HPLC using a Finnigan Surveyor HPLC system (Thermo Finnigan, San Jose, CA) with a Photodiode Array (PDA) plus detector, autosampler, and LC pump plus with a Inertsil® ODS-3 column (4 μm, 4.6 × 150 mm, [GL Sciences, Torrance, CA]) at 25°C. Chromatographic separation was achieved by gradient elution using mobile phase 2-propanol, acetonitrile (ACN), and water (5: 55: 40 v/v/v). The flow rate was 1.0 mL/minute and the total run time was 25 minutes for each 25 μL injection. The wavelength was 230 nm.
The DX concentration was quantified by LC/MS/ MS using a Finnigan Surveyor Autosampler Plus and Finnigan Surveyor MS Pump Plus (Thermo Finnigan). Chromatographic separations were achieved using a SunFire ™ C18 column (2.1 × 30 mm, 3.5 μm particle size, [Waters, Milford, MA]) at 25°C. The mobile phase consisted of 0.1% formic acid in water and methanol using gradient separation. The flow rate was 0.5 mL/minute and the total run time was 8 minutes for each 25 μL injection. Mass spectrometric analysis was performed using a Thermo Scientific TSQ Quantum Access with positive ionization. The capillary temperature was set up to 390°C, and the spray voltage was 4000V. For DX analysis, m/z 830.0 → 549.0 was monitored with PX (m/z 876.3 → 308.0) as an internal standard.
Evaluation of DX conjugate solubility in Miglyol 808 and mouse plasma
Approximately 5 mg of DX or 50 mg of each DX conjugate was added to individual vials containing 50 μL of Miglyol 808. For the evaluation of DX and DX conjugate solubility in mouse plasma, around 1 mg of each DX conjugate or DX was added to a vial containing 1 mL of BALB/c mouse plasma. The mixtures were stirred at room temperature for 24 hours. The samples were then centrifuged for 1 hour at 14,000 rpm to remove undissolved drugs. After centrifugation, the saturated supernatant was diluted with ACN and analyzed by HPLC.
Preparation and characterization of BTM NPs
Preparation of BTM NPs containing DX conjugates
DX conjugates containing NPs were prepared using a warm oil-in-water (o/w) microemulsion precursor method previously developed in our laboratory.Citation22 Briefly, defined amounts of Miglyol 808 and surfactants (Brij 78 and Vitamin E TPGS) were accurately weighed into glass vials and heated to 50°C–60°C. Drugs dissolved in ACN were added and the organic solvent was removed by nitrogen flow. One milliliter of pre-heated 10% lactose in water was added into the mixture of melted oil, surfactants and drugs. The mixture was stirred for 20 minutes at 50°C–60°C then cooled to room temperature. Orthogonal design was performed to optimize nanoparticles with desirable properties, including particle size and drug entrapment efficiency.
For in vivo studies, NPs were concentrated and PEGylated. The formulation was concentrated four-fold by adding four-fold less 10% lactose continuous phase while keeping the other components of the formulation unchanged. The NPs were PEGylated by adding 8% Brij 700 (w/w Brij 700/ Miglyol 808) during the preparation.
Characterization of BTM NPs containing DX conjugates
Particle size and zeta potential
Particle size and size distribution of NPs were determined using N5 Submicron Particle Size Analyzer (Beckman Coulter, Brea, CA). Five microliters of NPs was diluted with 1 mL of water to reach the intensity required by the instrument. Particle size was determined at 90° light scattering at 25°C. The zeta potential of NPs was determined using the Zetasizer Nano Z (Malvern Instruments, Southborough, MA).
Drug entrapment efficiency
Drug entrapment efficiency was determined by GPC. DX conjugate NPs were separated from the free drugs using a 15 cm Sepharose CL-4B GPC column (GE Healthcare, Pittsburg, PA). NPs were eluted by PBS in fraction 5–8 (1 mL/fraction, confirmed by dynamic light scattering intensity). Each fraction was evaporated to dryness in vacuo, resuspended in 1 mL ACN, and analyzed by HPLC to determine the concentration of DX conjugate in each fraction. The percentage of drug entrapment efficiency was defined as 100% × the ratio of the weight of drug detected in fraction 5–8 to the total drug weight detected.
Drug release in mouse plasma
In vitro release studies were performed in 100% plasma from BALB/c mice. Briefly, 100 μL of purified DX conjugate NPs were spiked into 2 mL of mouse plasma. The release mixture was incubated at 37°C in a water bath shaker. At designated time points from 0–8 hours, two aliquots of release mixture were removed. One aliquot (100 μL) was used to determine the total drug concentration by solid phase extraction (SPE) using Hybrid-SPE precipitate method. Briefly, one volume of release mixture was mixed with three volumes of 2% formic acid in ACN. Following vortex and centrifugation, the supernatant was applied to a Hybrid-SPE cartridge. The eluate was collected for HPLC analysis. Another aliquot (100 μL) was used to determine the drug remaining in the NPs using the method described previously. The percentage of DX released at any time point was calculated as 100% × [(total drug detected – drug remaining in the NPs)/total drug detected].
Evaluation of in vitro cytotoxicity
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was utilized to assess cytotoxicity of free DX conjugates and the DX conjugate NPs. Serial dilutions of free drugs or drug containing NPs were added to the DU-145 cells and incubated for 48 hours. The cells were then incubated with MTT solution and solubilized by dimethyl sulfoxide (DMSO). The absorbance was measured using the Synergy 2 Multi-Detection Microplate Reader (BioTek Instruments, Winooski, VT) at 570 nm, and the concentration of drug that inhibited cell survival by 50% (IC50) was determined from cell survival plots.
In vitro esterase digestion
The esterase digestion study was performed in fresh mouse plasma. The 5 mg/mL DX conjugate solutions in DMSO were spiked into the plasma to make a final concentration of 10 μg/mL. The mixture was incubated at 37°C in a water bath shaker. At designated time points, 100 μL of digestion mixture was removed. The concentration of DX conjugates was determined by Hybrid-SPE precipitate method as described above followed by HPLC analysis. The percentage of DX conjugate remaining at any time point was calculated as 100% × the ratio of remaining drug amount to the total drug spiked into this volume of plasma.
In vivo pharmacokinetic studies
Male athymic nude mice were randomly divided into four groups. The mice (n = 3/time point) were injected via tail vein with test samples (Taxotere, C12-DX NPs, C18-DX NPs, and C22-DX NPs), all at a DX dose of 10 mg/kg. At designated time points from 3 minutes to 24 hours, the mice were given an overdose of ketamine (100 mg/kg) and domitor (0.5 mg/kg) for deep anesthesia prior to cardiac puncture to collect blood and a cervical dislocation was then performed to euthanize the mice. For plasma separation, the blood collected in heparin-coated tubes was centrifuged at 12,300 rpm for 15 minutes. The obtained plasma was processed with Hybrid-SPE precipitate method as described above. The concentrations of DX conjugates in plasma were determined by HPLC, and the DX concentrations were quantified by LC-MS. Pharmacokinetic analysis and modeling was performed by Winnonlin (v 5.2.1; Pharsight Corp, Mountain View, CA).
Statistical analysis
Statistical comparisons were performed using one-way analysis of variance (ANOVA) (GraphPad Prism Software Inc, La Jolla, CA). Results were considered significant at the 95% confidence interval (P < 0.05). Orthogonal experimental design for formulation optimization was performed and statistically analyzed using Design Expert® (v 7.1 Trial; Stat-Ease, Minneapolis, MN).
Results
Synthesis and characterization of DX conjugates
The DX-lipid conjugates in these studies were prepared by a one-step esterification reaction using acid chloride derivatives of various chain length fatty acids (). Although there are multiple hydroxyl groups in DX molecule, the 2′-OH is the most reactive and accessible one, followed by 7-OH.Citation21 It has been previously reported that the conjugation of fatty acids to DX and PX occurs preferentially on 2′-OH.Citation21,Citation23,Citation24 By controlling the molar ratio of the fatty acid chloride to DX carefully, 2′-mono substituted DX conjugates were obtained with minimal formation of 2′,7-di substituted byproducts and unreacted DX. In the case of C22-DX, only the 2′-OH ester derivative was obtained after washing with 5% HCl and brine as determined by thin layer chromatography and nuclear magnetic resonance, without further chromatography required. The yield for this reaction was as high as 95%.
Figure 1 Synthesis of 2′-docetaxel conjugates.
Solubility of DX conjugates in Miglyol 808 and mouse plasma
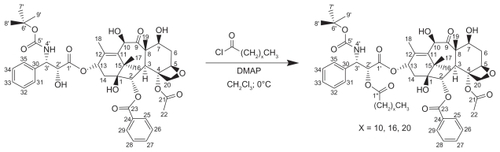
To enhance the drug loading capacity and retention of drug in the NPs, DX conjugates were synthesized and investigated for their solubility in Miglyol 808 (). The solubility of all three DX conjugates in Miglyol 808 was about 10-fold higher than the solubility of DX. The solubility showed no chain-length dependency. The chemical composition of Miglyol 808 is caprylic acid triglyceride, so DX conjugates with a 12–22 carbon chain are more compatible than DX with Miglyol 808 due to the similarity of the chemical structure.
Table 1 Solubility of DX and DX conjugates in Miglyol 808 (N = 3)
Since the in vitro release of DX conjugates from NPs was studied in mouse plasma, the solubility of DX conjugates in BALB/c mouse plasma was determined and compared as well (). In contrast to solubility in Miglyol 808, the solubility of DX conjugates in plasma showed significant chain-length dependency. With an increase in lipid chain, the solubility of the conjugate in plasma decreased. The solubility of C12-DX (377.0 ± 21.5 μg/mL) was about 10-fold higher than that of C22-DX (34.4 ± 0.6 μg/mL) and 6.5-fold higher than that of C18-DX (57.5 ± 2.6 μg/mL). Given the extremely low water solubility of the DX conjugates, the solubility of the conjugates in plasma was attributed almost entirely to their binding with plasma proteins.
Figure 2 Solubility of DX conjugates in mouse plasma.
Abbreviation: DX, docetaxel.
Optimization of DX conjugate-containing nanoparticles by orthogonal design
The orthogonal design was based on the NP previously developed to formulate DX, which was composed of Miglyol 808 (2.5 mg/mL), Brij 78 (3.7 mg/mL), TPGS (1.2 mg/mL), and DX (0.3 mg/mL). In the present study, particle size and drug entrapment efficiency were chosen as responses in the optimization process. A criterion in the orthogonal design strategy was to reduce the total amount of surfactant used in the formulation, as it had been previously established that increased levels of surfactants in the formulation decreased drug entrapment, especially for drugs that have amphipathic properties.
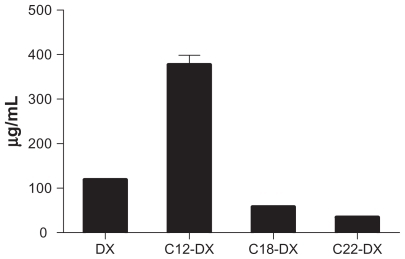
Based on the preliminary studies, a 3-level variable orthogonal experiment (L-9 33) was designed as shown in . The two responses selected were particle size and percent of entrapment efficiency. In this experiment, C12-DX was used as a representative DX conjugate. The resulting NP compositions based on the optimization using C12-DX were applied to other conjugates. Nine batches of C12-DX NPs were prepared and characterized. Results are also shown in . Statistical analysis showed that temperature as a variable was not significant to the model (P > 0.05). The particle size, as a defined model response, was not responsive to the variables (P > 0.05). It should be noted that the general placebo composition for this formulation was previously optimized.Citation22 Thus, it was anticipated that continued optimization with DX conjugates would lead to a relatively narrow response range. When the model focused on the effect of surfactant concentrations on the percent of entrapment efficiency, it was clear that decreasing the surfactant concentrations increased drug entrapment in the NPs (). Although the percent of entrapment efficiency of batch 2 was slightly higher than batch 5, batch 5 was more stable over long-term (1 month) storage at 4°C (data not shown). The final composition was selected as shown in . Due to the enhanced solubility of drugs in the oil core, the newly developed formulation was capable of loading more DX conjugate (0.5–1 mg/mL conjugates vs 0.3 mg/mL DX) without significantly changing the physical properties of the resulting NPs. The optimal NPs had a mean particle size of 200 nm with a zeta potential around 0 mV (). The entrapment efficiencies of the three DX conjugates were 55.2% ± 2.3%, 56.3% ± 7.6%, and 59.6% ± 1.6% for C12-DX, C18-DX, and C22-DX, respectively. The similar entrapment efficiencies of three DX conjugates was predicted by their comparable solubility in Miglyol 808 as shown in .
Table 2 Orthogonal design and responses
Table 3 Compositions and properties of BTM 808 NPs
Figure 3 3D surface plot for the modeling of the effect of Brij 78 and TPGS concentrations on percent of entrapment.
Abbreviations: DX, docetaxel; TPGS, tocopheryl polyethylene glycol succinate.
In vitro release of DX conjugates from NPs in mouse plasma
In this study, a novel in vitro release method was developed to study the release of DX conjugates from the NPs in 100% mouse plasma. The in vitro drug release of nano-formulations is usually studied in aqueous buffer using a dialysis method. However, the correlation between in vitro release and in vivo pharmacokinetic profile is often poor due to the more complex in vivo environment, such as the presence of large amounts of proteins and enzymes in the plasma. In our developed “ex vivo” release method, NPs were spiked directly into 100% mouse plasma. Drug-containing NPs were separated from protein-bound DX conjugates and free DX conjugates using a Sepharose CL-4B column (GE Healthcare). The 15 cm, gravity-packed Sepharose CL-4B column was able to achieve baseline separation of the NPs with plasma proteins and free drugs, validated by dynamic light scattering intensity, BSA assay, and HPLC analysis (data not shown).
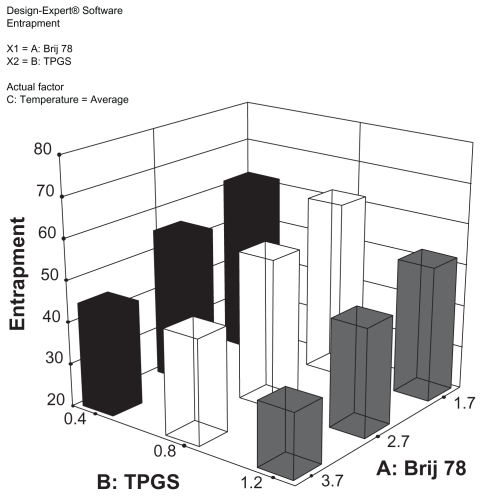
For the release studies, the DX conjugate-containing NPs were first purified by GPC and only NPs with size around 200 nm (fraction 5–8) were collected. For all three DX conjugate NPs, an initial 45% burst release was observed upon spiking into the mouse plasma (). After the initial burst release, C12-DX was slowly released to 86% in 8 hours, while no additional C18-DX and C22-DX were released from the NPs within 8 hours. A longer time point (96 hours) release study was carried out for the C18-DX NPs; however, no drug was released from the NPs after the burst (data not shown).
Figure 4 Release of DX conjugates from BTM NPs in mouse plasma.
Abbreviations: DX, docetaxel; BTM, Brij 78, Vitamin E TPGS and Miglyol 808.
In vitro cytotoxicity
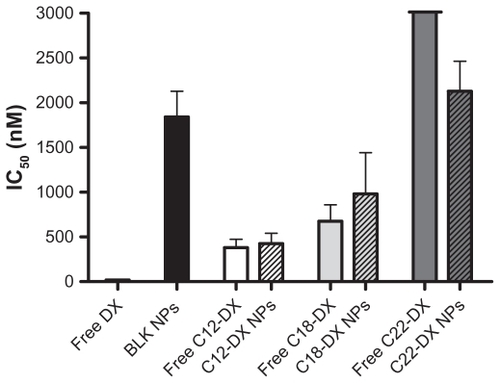
The cytotoxicity of DX conjugate NPs was studied in human prostate cancer DU-145 cells using the MTT assay (). Both free DX conjugates and DX conjugate NPs showed a dose-dependent cytotoxicity in DU-145 cells. In general, all three DX conjugates had significantly lower cytotoxicity than unmodified DX in DU-145 cells. The decrease in cytotoxicity was chain-length dependent. As shown in , free C12-DX was 20.6-fold less active than DX, and free C18-DX was 36.5-fold less active than DX. Free C22-DX was almost nontoxic to DU-145 cells. C12-DX and C18-DX NPs showed comparable IC50 values with their free forms, while C22-DX NPs showed similar toxicity to the blank NPs. The blank NPs IC50 was 1842 ± 287 nM in DX conjugate equivalent dose.
Figure 5 In vitro cytotoxicity of free DX and DX conjugates and their NPs in DU-145 cells.
Abbreviation: DX, docetaxel.
In vitro esterase digestion
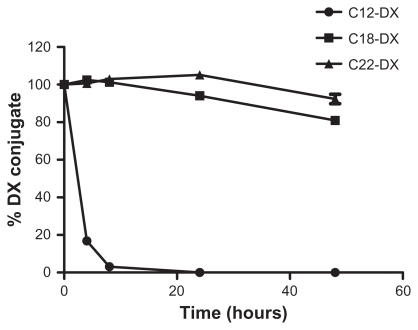
The digestion rate of DX conjugates in mouse plasma bearing murine esterase activity was chain-length dependent (). C12-DX with the shortest fatty acid chain length disappeared in 8 hours, while about 80% and 90% of C18-DX and C22-DX were detected after 48 hours incubation, respectively.
Figure 6 The digestion of free DX conjugates in fresh mouse plasma at 37°C.
Note: Data are shown as mean ± SD (n = 3).
Abbreviation: DX, docetaxel.
In vivo pharmacokinetics
The plasma concentration-time curves in mice receiving intravenous bolus injections of Taxotere, C12-DX NPs, C18-DX NPs, and C22-NPs at a dose of 10 mg DX/kg are shown in . Pharmacokinetic parameters obtained using a noncompartmental model of analysis are listed in . The AUC0–∞ values of all NP-formulated DX conjugates were significantly higher than that of Taxotere. The AUCs increased as the conjugate chain length increased. The AUC0–∞ values of DX were 8.3-fold, 358-fold, and 454.5- fold lower than that of NP-formulated C12-DX, C18-DX, and C22-DX, respectively. The terminal half-lives of NP-formulated C18-DX and C22-DX were 1.9-fold and 3.4-fold longer than that of DX, respectively. The terminal half-life of NP-formulated C12-DX was shorter than that of DX. The volume of distribution of DX conjugates after administration of C12-DX NPs, C18-DX NPs, and C22-DX NPs were comparable. Overall, the volume of distribution of DX conjugate NPs was 20-fold lower than that of Taxotere.
Table 4A Pharmacokinetic parameters of DX conjugates and Taxotere in mice after iv bolus administration
Figure 7 Plasma concentration-time curves for (A) DX, C12-DX, C18-DX, and C22-DX after administration of Taxotere, C12-DX NPs, C18-DX NPs, and C22-DX NPs, respectively, and (B) DX as an active metabolite from C12-DX NPs and C18-DX NPs using Taxotere as a reference. The plasma concentrations of DX from C22-DX NP were below the lower limit of quantification.
Note: Data are shown as mean ± SD (n = 3).
Abbreviation: DX, docetaxel.
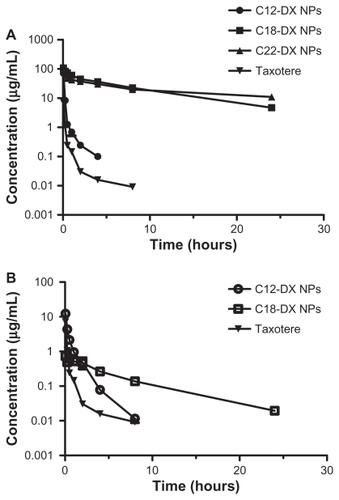
The plasma concentrations of DX as an active metabolite hydrolyzed from C12-DX, C18-DX, and C22-DX were determined and are shown in . DX concentrations of Taxotere are also shown as a reference for comparison. Pharmacokinetic parameters of the noncompartmental model are shown in . The plasma concentrations of DX from C22-DX NP were below the lower limit of quantification. C12-DX NPs and C18-DX NPs improved DX AUC about 3-fold compared to Taxotere. The AUC of DX from C12-DX NP was slightly higher than that of C18-DX NP and the Cmax of DX from C12-DX NPs was 16.7-fold higher than that of C18-DX NPs. However, the terminal half-life of DX from C18-DX NPs was 5-fold higher than that of C12-DX NPs. The DX from C12-DX NPs decreased promptly below the level of DX from C18-DX NPs 4 hours post-injection. Eight hours post-administration, the DX concentration from C12-DX NP decreased to the same level as Taxotere, whereas DX from C18-DX NP could be detected after 24 hours.
Table 4B Pharmacokinetic parameters of DX after iv bolus administration of DX conjugates and Taxotere in mice
Discussion
In the present study, three DX-lipid conjugates, C12-DX, C18-DX, and C22-DX were synthesized and characterized. The solubility of all three DX-lipid conjugates in Miglyol 808 was enhanced >10-fold over that of DX. Following optimization of the DX conjugate NP using orthogonal design, the final optimized NPs contained significantly reduced surfactant concentrations and increased drug entrapment. The improved solubility of the DX conjugates in the NP liquid oil-phase led to very different pharmacokinetic profiles and blood exposure of DX.
A novel liquid oil-filled NP was previously developed in our laboratory, which was composed of Miglyol 812, Brij 78, and Vitamin E TPGS.Citation25 During the initial development of NP to formulate DX, Miglyol 808 was selected over Miglyol 812 due to the significantly higher solubility of DX in Miglyol 808 (52.07 ± 0.84 mg/mL) compared to that in Miglyol 812 (36.11 ± 0.10 mg/mL, P < 0.01). The oil phase with higher drug solubility represents better compatibility and affinity of the drug with the inner liquid oil core of the delivery vehicles thereby leading to higher drug loading capacity and longer retention of drugs in the NPs. The high drug-loading capacity minimizes the amount of the delivery vehicle needed to achieve the necessary drug dosage, thereby reducing the potential toxicity of the delivery vehicle. The >10-fold increase in the solubility of DX conjugates in Miglyol 808 compared to DX allowed for a significant increase in drug loading and entrapment. The comparable entrapment efficiency of the three DX conjugates in BTM 808 NPs was consistent with solubility of the conjugates in the Miglyol 808 liquid oil core.
While the solubility of DX conjugates in Miglyol 808 strongly influenced the drug loading and entrapment efficiency in the NP formulation, the partition of DX conjugates in plasma was the driving force for their release in vivo. Results from the in vitro release studies showed that after an initial burst, an additional 40% of C12-DX having a relatively higher solubility in plasma was released from the NPs in 8 hours. In contrast, the C18-DX and C22-DX were extensively retained in the NPs after the initial burst release. It should be noted that all three DX-lipid conjugates showed an initial 45% burst release in mouse plasma using this “ex vivo” method, which suggests that the burst release was not related to the lipid chain length in this range. It is likely that the relatively more hydrophilic head group of the DX-lipid conjugates resided on the surface of NPs and promptly partitioned to plasma proteins upon mixing with plasma. The burst release may not be a desirable property of NPs; however, it almost certainly reflects the true release behavior in vivo.
In vivo, NP-formulated C12-DX, C18-DX, and C22-DX achieved much higher AUCs compared to Taxotere, which was expected due to their better anchoring in the long-circulating NPs. The low volume of distribution of DX conjugates was attributed to the size of NPs which limited their distribution to the normal tissues.Citation26 The elimination routes and the relative contribution of each route for each conjugate varied. In addition to the elimination of conjugate containing NPs by the reticuloendothelial system (RES), the elimination of the conjugates was also attributed to two other possible mechanisms including release of the conjugate from the NPs and hydrolysis of the DX conjugates. The DX plasma concentration-time curves indicated that C12-DX was more quickly hydrolyzed to DX in vivo than the other two conjugates. C12-DX, with a shorter acyl chain, likely has less steric hindrance and is more readily cleaved to release DX. C12-DX in NPs was either released from the NPs followed by hydrolysis to DX or was hydrolyzed to DX first, followed by quick release as DX. Regardless of the mechanism, C12-DX had the shortest terminal half-life in vivo, which was even shorter than Taxotere. However, the plasma exposure of DX was the highest after C12-DX NP injection. In contrast, the longer acyl chain of C18-DX makes it less susceptible to hydrolysis and better anchored in the NPs in vivo resulting in a significant increase of the plasma exposure of the conjugate. As a result, the AUC of NP-formulated C18-DX was 43-fold higher than that of C12-DX. Because of the slower hydrolysis, the DX AUC from C18-DX NPs was lower compared to C12-DX NPs. However, as shown in , the duration of DX exposure after C18-DX NP administration was much longer than C12-DX NPs. C18-DX NPs served as a drug reservoir and released DX in a sustained manner. It has been reported that prolonged time above a threshold concentration is ideal for cell cycle-specific drugs.Citation27 Although DX was not detected after C22-DX NPs were injected into the blood, it is possible that the hydrolysis kinetics of C22-DX was too slow and the released DX was too quickly eliminated to be detected. It is worth noting that in the tumor site the overall anti-tumor toxicity comes from three forms: DX taken up by the tumors from the systemic circulation, the accumulating DX conjugates on their own, and the DX hydrolyzed from the accumulating conjugates in the site. It is possible that C22-DX is hydrolyzed to DX slowly after accumulating in the tumors.
While conjugating fatty acid chains to DX decreased its cell growth inhibitory activity in vitro, it may enhance the in vivo efficacy. Many studies have reported the reduction of PX or DX activity by conjugating fatty acid chains to 2′-OH.Citation21,Citation23,Citation24 This study is consistent with previous reports that in general, all three DX-lipid conjugates were less potent than DX against DU-145 cells, and increasing the lipid chain length decreased the cell growth inhibitory activity in vitro. It has been previously demonstrated that esterification at 2′-OH or 7-OH abolished the microtubule binding affinity of the conjugates but not the total toxicity.Citation28 The DX conjugates as ester prodrugs are expected to be cleaved to release free DX and exert their antitumor toxicity after cleavage. However, in the present 48 hours in vitro cytotoxicity study, the hydrolysis rate may not be fast enough to release all the DX. In addition, it is possible that DX conjugates as intact parent drugs have alternative cytotoxic mechanisms other than microtubule binding. These additional mechanisms, if any, still remain to be investigated. The chain length-dependent cytotoxicity reduction may be explained by their different rate/extent of cellular uptake, different cellular compartmental sequester, and/or different rate/extent of hydrolysis/ degradation. However, many studies have reported that in vivo efficacy does not necessarily correlate with in vitro cytotoxicity. In a previous study,Citation23 a series of PX prodrugs with various linkers and lipid anchors were synthesized. Among the lipophilic prodrugs of PX, the most potent compound (compound 7) in vivo showed only moderate in vitro cytotoxicity. Another study showed that the 2′-(2-bromo)- hexadecanoyl DX with lowest in vitro cytotoxicity in its kind was most effective in vivo, showing 100% survival at day 300 for OVCAR-3-bearing SCID mice.Citation24 The DHA-PX on clinical trial III prepared by linking PX to docosahexaenoic acid (DHA) was less toxic than PX in vitro but cured 10/10 M109 tumor-bearing mice, whereas PX cured 0/10.Citation29 Our pharmacokinetic results provided the basis for enhanced in vivo efficacy.
In conclusion, the NPs developed in these studies have low toxicity, long circulation in the blood, and released DX-lipid conjugates in a slow and sustained manner in the plasma. Thus, the NPs have the potential to exert superior anti-tumor efficacy and less systemic toxicity in vivo. The results of these studies strongly support that the physical/chemical properties of DX conjugates may be fine-tuned to influence the affinity and retention of DX in oil-filled lipid NPs which therefore leads to very different pharmacokinetic profiles and blood exposure of DX. These studies demonstrate that the affinity of a drug for the core NP material may influence the retention and release rate of a drug from these NPs. In addition, a new “ex vivo” method was developed to better correlate and predict the release rate of a drug from a NP formulation to the resulting pharmacokinetic profile. These studies and methodologies may allow for improved and more potent nanoparticle-based formulations.
Acknowledgments
This research was supported by NIH-NCI R01 CA115197 and NIH-NCI U54 CA151652. The authors thank Mianmian Sun for providing technical support for HPLC and mass spectrometry. The authors are grateful to Charlene M Santos and her group for their assistance with animal applications.
Disclosure
The authors report no conflicts of interest in this work.
References
- GoldspielBRClinical overview of the taxanesPharmacotherapy1997175 Pt 2110S125S9322878
- van OosteromATSchrijversDSchrijversDDocetaxel (Taxotere), a review of preclinical and clinical experience. Part II: Clinical experienceAnticancer Drugs1995633563687670133
- CortesJEPazdurRDocetaxelJ Clin Oncol19951310264326557595719
- RingelIHorwitzSBStudies with RP 56976 (taxotere): a semisynthetic analogue of taxolJ Natl Cancer Inst19918342882911671606
- ExtraJMRousseauFBrunoRClavelMLe BailNMartyMPhase I and pharmacokinetic study of Taxotere (RP56976;NSC 628503) given as a short intravenous infusionCancer Res1993535103710428094996
- BissettDSetanoiansACassidyJPhase I and pharmacokinetic study of taxotere (RP 56976) administered as a 24-hour infusionCancer Res19935335235278093854
- BurrisHIrvinRKuhnJPhase-I clinical-trial of taxotere administered as either a 2-hour or 6-hour intravenous-infusionJ Clin Oncol19931159509588098059
- PazdurRNewmanRANewmanBMPhase I trial of Taxotere: five-day scheduleJ Natl Cancer Inst19928423178117881359154
- TomiakEPiccartMJKergerJPhase-I study of docetaxel administered as a 1-hour intravenous-infusion on a weekly basisJ Clin Oncol1994127145814677912725
- CuiZRYanasarnNSloatBRNanoparticles engineered from lecithin-in- water emulsions as a potential delivery system for docetaxelInt J Pharmaceut20093791174180
- KhalidMNSimardPHoarauDDragomirALerouxJCLong circulating poly(ethylene glycol)-decorated lipid nanocapsules deliver docetaxel to solid tumorsPharm Res200623475275816550475
- XuZChenLGuWThe performance of docetaxel-loaded solid lipid nanoparticles targeted to hepatocellular carcinomaBiomaterials200930222623218851881
- ElsabahyMPerronMEBertrandNYuGELerouxJCSolubilization of docetaxel in poly(ethylene oxide)-block-poly(butylene/styrene oxide) micellesBiomacromolecules2007872250225717579476
- ImmordinoMLBrusaPArpiccoSStellaBDosioFCattelLPreparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing docetaxelJ Control Release200391341742912932719
- StraubingerRMBalasubramanianSVPreparation and characterization of taxane-containing liposomesMethods Enzymol20053919711715721376
- IsacchiBArrigucciSMarcaGLConventional and long-circulating liposomes of artemisinin: preparation, characterization, and pharmacokinetic profile in miceJ Liposome Res201121323724421158702
- ColeAJDavidAEWangJGalbanCJHillHLYangVCPolyethylene glycol modified, cross-linked starch-coated iron oxide nanoparticles for enhanced magnetic tumor targetingBiomaterials20113282183219321176955
- KimCKKimJYKimJKParkJSByunYThe use of PEGylated liposomes to prolong circulation lifetimes of tissue plasminogen activatorBiomaterials200930295751575619656559
- ImmordinoMLDosioFCattelLStealth liposomes: review of the basic science, rationale, and clinical applications, existing and potentialInt J Nanomedicine20061329731517717971
- De ValeriolaDBrassinneCGaillardCStudy of excretion balance, metabolism and protein binding of 14C radiolabelled Taxotere (RP56976, NSC628503) in cancer patientsProc Am Cancer Res199334373
- HuynhLLerouxJCAllenCEnhancement of docetaxel solubility via conjugation of formulation-compatible moietiesOrg Biomol Chem20097173437344619675898
- DongXWMattinglyCATsengMChoMAdamsVRMumperRJDevelopment of new lipid-based paclitaxel nanoparticles using sequential simplex optimizationEur J Pharm Biopharm200972191719111929
- AnsellSMJohnstoneSATardiPGModulating the therapeutic activity of nanoparticle delivered paclitaxel by manipulating the hydrophobicity of prodrug conjugatesJ Med Chem200851113288329618465845
- AliSAhmadIPetersAHydrolyzable hydrophobic taxanes: synthesis and anti-cancer activitiesAnticancer Drugs200112211712811261884
- MumperRJDongXWMattinglyCADoxorubicin and paclitaxel-loaded lipid-based nanoparticles overcome multidrug resistance by inhibiting P-glycoprotein and depleting ATPCancer Res20096993918392619383919
- AllenTMCullisPRDrug delivery systems: entering the mainstreamScience200430356651818182215031496
- GardnerSNA mechanistic, predictive model of dose-response curves for cell cycle phase-specific and -nonspecific drugsCancer Res20006051417142510728708
- DeutschHMGlinskiJAHernandezMSynthesis of congeners and prodrugs. 3. Water-soluble prodrugs of taxol with potent antitumor activityJ Med Chem19893247887922564894
- BradleyMOSwindellCSAnthonyFHTumor targeting by conjugation of DHA to paclitaxelJ Control Release2001741–323323611489499