Abstract
Objectives
In situ formation of nanocrystals and dissolution profiles of fenofibrate (FFB) from a self-microemulsifying drug delivery system (SMEDDS) were characterized.
Methods
SMEDDS formulated with Myritol® and surfactant mixture (Smix) of D-α-Tocopheryl polyethylene glycol 1000 succinate (TPGS) and either Tween® 20 (A, C, E, G, M, S, N, T, O) or Tween® 80 (B, D, F, H, P, U, Q, V, R) at various oil/Smix ratios (Group I: A and B of 0.42, C and D of 0.25, E and F of 0.11; Group II: G and H of 1.38, M and P of 1.11, S and U of 0.9, N and Q of 0.73, T and V of 0.58, and O and R of 0.46) and water contents (1: 9.5%, 2: 5.0%, 3: 0.0%, G–V: 4.5%). Their dissolutions were conducted at different rotation speeds. Two optimal SMEDDSs containing Tween 80(B2) or a higher oil/Smix ratio(Q) and B2(solution) were selected for pharmacokinetic study.
Results
FFB particles formed within the nanosize range from Group I gradually increased with time but decreased with increasing stirring rates. However, the mean size of FFB formed by B series was as low as 200 nm, which was smaller than that of A series at three stirring rates. The release rate from both groups obviously increased with increasing stirring rate. However, incomplete release was observed for S and N in Tween 20 series, whereas a faster release rate and complete release were observed for Tween 80 series with an insignificant difference among them. Results of pharmacokinetic study demonstrated that the highest-ranked area under the curve and Cmax values were for Q(SMEDDS) and B2(solution), respectively. The relative bioavailability of Q(SMEDDS) with respect to Tricor® was enhanced by about 1.14–1.22-fold.
Conclusion
SMEDDS, consisting of Myritol 318 and TPGS combined with Tween 80 at 4:1, was able to enhance the oral bioavailability of FFB.
Introduction
Since microemulsions were first introduced some 60 years ago by Hoar and Schuaman,Citation1 many applications have been found, especially for improving the oral bioavailability of poorly soluble drugs and nutraceuticals.Citation2 Currently, water/oil (w/o) types of microemulsions,Citation3 or oil/water (o/w) microemulsions separated by a two-phase region,Citation4,Citation5 self-microemulsifying drug delivery system (SMEDDS),Citation6–Citation8 and U-type phase diagrams capable of forming microemulsions at any given water dilution level (U-type preconcentrate) have been utilized to solubilize and increase the bioavailability of drugs and nutraceuticals.Citation9–Citation13 W/o or o/w microemulsions are excellent and effective carriers that increase the solubility and improve the bioavailability of poorly water-soluble drugs.Citation14 Optimally, SMEDDSs and U-type preconcentrate are clear and transparent isotropic systems, which have the ability to produce o/w microemulsion systems when in contact with aqueous media (ie, the gastrointestinal fluid) under gentle agitation (ie, digestive motility of the stomach and intestines), which would be advantageous in resolving the problem of the deterioration of soft capsules due to the water content of either w/o or o/w type microemulsions.Citation15,Citation16
A SMEDDS is used to form preconcentrates of surfactants, oil, cosolvents, so that the drug can be diluted with water or pH 1.2 simulated gastric acid when orally administered to form o/w microemulsions with droplet sizes within the nanosize range. Nanosized droplets formed this way have very high surface-to-volume ratios and are expected to solubilize the drug, speed up drug release, and, subsequently, improve the bioavailabilityCitation17 as well as release the drug in a more reproducible manner, which is less dependent on the gastrointestinal physiology and feeding/fasting state of the patient.Citation18 SMEDDSs have been reported to improve the in vivo dissolution and therefore enhance the bioavailability of lipophilic drugs.Citation6,Citation19–Citation25 Commercially available drugs that make use of SMEDDSs include cyclosporin A as well as preparations of ritonavir and saquinavir (HIV protease inhibitors).Citation26,Citation27 However, although the SMEDDS preconcentrates containing 10% celecoxib revealed by Subramanian et al were not fully dilutable and only formed fine o/w droplets upon dilution in an aqueous phase, the results of drug absorption in humans were very encouraging, with a 1.32-fold increase in the relative bioavailability compared with a conventional dosage form.Citation28 Moreover, unlike drug-free SMEDDS preconcentrates, when flurbiprofen was loaded, the dilution mixtures generated by the SMEDDS preconcentrates containing either Tween® 20 (ICI Americas Inc, Wilmington, DE) or Cremophor® EL (BASF Corp, Ludwigshafen, Germany) with Capmul® PG8 (ABITEC Corp, Columbus, OH) produced emulsions with observed cloudiness, the particle size was greatly increased compared with that of drug-free microemulsions (~11–13 nm).Citation29 This effect became more apparent as the drug loading was increased.
Fenofibrate (FFB), used to treat high-cholesterol and high-triglyceride levels, is a neutral lipophilic compound (log P = 5.24) with very low solubility (<0.5 mg/L).Citation30 A low dissolution rate in aqueous media (including gastrointestinal fluids) is expected, which will result in incomplete and irregular bioavailability after oral ingestion. Reduction in the particle size of FFB by a micronization process can improve its solubility and the bioavailability is subsequently increased.Citation30 A new dosage form of FFB, called a suprabioavailable tablet, has been developed, which combines micronization technology and microcoating processes. In this way, the increase in the amount of drug dissolved in the aqueous medium of the gastrointestinal tract also improves the extent of absorption.Citation31–Citation33 Another type of formulation developed for FFB is a hard gelatin capsule with a semisolid content into which FFB is homogenously dispersed within a lipid excipient mixture supplemented with hydroxypropyl methyl cellulose. The resulting formulation has increased drug solubility and dissolution rates as well as improved oral bioavailability equivalent to micronized FFB formulations.Citation34
A SMEDDS composed of LabrafacTM (Gattefossé, Lyon, France) CM10 (31.5%), Tween® 80 (ICI Americas Inc) (47.3%), and polyethylene glycol 400 (12.7%) was formulated for FFB and produced significant reductions in serum lipid levels in Phases I and II of the Triton test compared with plain FFB.Citation35 Based on the previous study,Citation36 the authors of the present study were intrigued that microemulsifying SMEDDSs in an aqueous medium or gastrointestinal tract fluid could decrease the drug loading, causing the solubilized FFB in the SMEDDS preconcentrate to precipitate. The authors wished to characterize how formulation factors of SMEDDSs affected the resultant drug particles for dissolution after microemulsification with aqueous medium or gastrointestinal tract fluid, which influenced the in vivo absorption. Thus, the main objective of this study was to characterize the in situ formation of nanocrystals and dissolution profiles of FFB from SMEDDSs containing an oil of medium-chain triglyceride (MCT), Myritol® 318, and nonionic surfactant mixture, D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) combined with Tweens for a lipophilic model drug, FFB, in terms of the formulation factors and dissolution conditions to optimally correlate them with in vivo oral absorption.
Experimental
Materials
Myritol 318 (C8/C10 triglycerides) (Cognis Ltd, Tokyo, Japan) was used as the oil phase. TPGS was purchased from Eastman Chemical Company (Kingsport, TN, USA). FFB and fenofibric acid (FBA) were supplied from Sigma Aldrich (St Louis, MO). Tween 20 and 80 were purchased from E Merck (Darmstadt, Germany). Hard gelatin capsules were supplied by Shing Lih Fang Enterprise (Taichung, Taiwan). Tricor® 54 mg tablets (lot 028362E21; exp date 2005/03/01) were supplied by Abbott Laboratories (North Chicago, IL; manufactured by Laboratories Fournier, Chenôve, France). All materials were either pharmaceutical or reagent grade.
Preparation and viscosity measurement of SMEDDS
The pseudoternary phase diagrams of the SMEDDSs constructed previouslyCitation14 comprised Myritol 318 and surfactant mixtures (Smix) of TPGS/polysorbates (Tween 20 or 80), incorporating 10% FFB. Formulations for the SMEDDSs, either within or outside the microemulsion region, were selected; their preliminary physical characteristics are listed in and , respectively. The formulation components were accurately weighed into screw-capped glass tubes with water-tight closures and heated to approximately 80°C in a thermostat-controlled water bath. When all formulation components had been melted/liquefied, the mixtures were thoroughly vortex mixed until a homogeneous solution was obtained, which was then allowed to stand at room temperature. The viscosity of the SMEDDSs was measured with a viscometer (model DV-II+ viscometer, Brookfield Engineering Labs, Middleboro, MA) using an LV spindle set of sp16 at 37°C. The mixture of the SMEDDS was manually filled into size 1 transparent hard gelatin capsules to a weight of about 540 mg. The capsules were kept at ambient temperature.
Table 1 Formulation compositions and physical characteristics (appearance, mean particle size, and polydispersity index) of fenofibrate (FFB) self-microemulsifying drug delivery system (SMEDDS) composed of D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) as the surfactant and Tween® 20 (A, C, and E) or Tween® 80 (B, D, and F) as the cosurfactant at various oil/Smix ratios (A and B at 0.42, C and D at 0.25, and E and F at 0.11) and water contents (−1, −2, and −3)
Table 2 Formulation compositions and physical characteristics (appearance, mean particle size, and polydispersity index [PI]) of fenofibrate (FFB) self-microemulsifying drug delivery system (SMEDDS) composed of D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) as the surfactant and Tween® 20 (G, M, O, S, and T) or Tween® 80 (H, P, R, U, and V) as the cosurfactant at various oil/Smix ratios (G and H at 1.38, M and P at 1.11, S and U at 0.9, T and V at 0.58, and O and R at 0.46) and the same water content (4.5%)
Dissolution studies and particle characterization
The dissolution profiles of the FFB SMEDDS capsules were determined using the USP 25 dissolution apparatus II (VK7020S; Varian Inc, Palo Alto, CA) method. The dissolution media consisted of 500 mL of simulated gastric fluid (pH 1.2). The temperature of the medium was maintained at 37.0°C ± 0.5°C, and the rotation speed was set to three different speeds of 10, 25, and 50 rpm, since rotation speeds of 75 and 100 rpm were found to result in an insignificant difference in the release rate. Samples (5 mL) were automatically withdrawn at 0, 5, 10, 20, 30, 60, 90, 120, 180, and 270 minutes and fresh medium replaced. The sample was then diluted with a threefold volume of absolute alcohol to achieve an appropriate linear concentration range and prevent the drug from precipitating. Samples were then measured spectrophotometrically at 287 nm (using a Jasco V-550 UV/VIS spectrophotometer; JASCO International Co, Ltd, Tokyo, Japan) to determine the amount of drug released. The average percentage drug dissolved at each sampling time (n = 3) was calculated after correcting for the cumulative amount removed in previous samples. The UV method for quantification of FFB was validated to ensure acceptable precision and accuracy for intra- and interday measurements (data not shown).
The mean particle size and polydispersity index of FFB particles in the portion of each sample were determined using a nanoparticle size analyzer (BIC 90 PLUS; Brookhaven Instruments, Holtsville, NY). Before the measurement, samples were diluted with double-distilled water to a suitable scattering intensity. Processing the fluctuating signal with a digital autocorrelator yielded the particle’s diffusion coefficient, from which the equivalent spherical particle size was calculated using the Stokes–Einstein equation.
Pharmacokinetic studies
Healthy volunteers were recruited after signing an informed consent agreement approved by the Ethics Committee of Taipei Medical University Hospital. A parallel design was conducted with four subjects for each treatment of three formulations (SMEDDS B2 and Q, and a B2 solution) at the same strength (54 mg). After dosing, blood samples (10 mL) were collected in BD Vacutainers™ (Becton, Dickinson and Company, Franklin Lakes, NJ) containing K3EDTA by means of an indwelling venous cannula in the cubital vein according to a predetermined time schedule, which included a blank sample, just before dosing and then samples at 0.5, 1, 2, 3, 4, 5, 6, 7, 8, 10, 12, 24, 36, and 60 hours after dosing. Plasma was immediately separated by centrifugation at 1690 g for 10 minutes, then transferred to suitably labeled tubes, and stored at −80°C until analysis. The plasma drug concentration was assayed with the validated high-performance liquid chromatographic (HPLC) method described below.
The HPLC system consisted of an Intelligent HPLC pump (PU-980; JASCO), an autosampler (AS-1555–10; JASCO), an UV/VIS detector (UV-975; JASCO), and a column oven (40°C). A column of Gemini® C18 110A (Phenomenex, Inc, Torrance, CA) (5 μm, 150 × 4.6 mm) was employed with a mobile phase consisting of acetonitrile:H2O:acetic acid of 55:45:0.2 (v/v, %) at a flow rate of 1 mL/minute. The eluent was monitored with a UV detector at a wavelength of 287 nm. Ketoprofen (40 μg/mL) was used as the internal standard. FBA, a major in vivo metabolite of FFB and used as a standard, was prepared with the solvent mixture of acetonitrile:H2O of 4:1 in the concentration range of 0.1, 0.2, 0.5, 1.0, 2.0, and 5.0 μg/mL, and three quality control (QC) samples of 0.3, 2.5, and 4.0 μg/mL were dispensed in the same solvent mixture. Standard curves were constructed by adding 50 μL each of freshly prepared standard solutions and the internal standard stock solution to 0.5 mL of plasma in a 10 mL screw-top test tube. Plasma samples were then deproteinated by adding 0.1 mL of acetonitrile:70% perchloric acid at 1:1 and briefly vortexed for 1 minute. The sample mixtures were centrifuged at 1690 g for 10 minutes at 4°C. The supernatant aqueous layer was pipetted out, and 400 μL was injected into a column for the HPLC analysis. Based on plasma profiles, all parameters of the pharmacokinetic study as defined were calculated using noncompartmental models for the period of 0–60 hours. The maximum concentrations (Cmax) and the time to reach Cmax (Tmax) were determined from the respective observed plasma concentrations versus time data. The elimination rate constant (ke) was obtained by linear regression analysis of at least three sampling points of the terminal log-linear declining phase to the last measurable concentration. The elimination half-life (T1/2) was calculated as ln2/ke. The area under the curve (AUC) to the last measurable concentration (AUC0–last) was calculated by the linear trapezoidal rule. The apparent oral clearance (Cl) was calculated as Dose/AUC0–inf and the mean residence time was calculated as AUMC0–inf/AUC0–inf. The relative bioavailability (F%) was calculated as AUC0–60,sample/AUC0–60,Tricor × 100. Results of inter- and intraday validations indicated that the correlation coefficients were all >0.999, and the variabilities of the slopes and intercepts were all <5% for all calibration curves constructed from the inter- and intraday assays. The relative standard error (RSE%) and coefficient of variation (CV%) were better than 20% at the lower limit of quantification (0.1 μg/mL) and better than 15% at the remaining concentrations in both the intra- and interday analyses.
Statistical analysis
All results are presented as the mean ± standard deviation (SD). A one-way analysis of variance (ANOVA) was used to determine statistical significance of particle size, time when 50% drug released, and pharmacokinetic parameters between groups (PASW Statistics 18.0; IBM Corp, Armonk, NY), and P < 0.05 was considered to be statistically significant.
Results and discussion
Physical characterizations of the SMEDDS formulations
The solubility of FFB is known to vary with those components employed to formulate SMEDDSs examined in this study. It has been reported that 1 g of Myritol 318 could dissolve 94.14 ± 3.15 mg/g of FFB. The solubilities of FFB in 10% TPGS, 10% Tween 20, and 10% Tween 80 solutions were 1.56 ± 0.08, 0.52 ± 0.01, and 0.80 ± 0.02 mg/g, respectively.Citation36 Thus, the physical characteristics, including the appearance and solution status, of the SMEDDS formulations composed of Myritol 318 as the oil phase and TPGS/Tween 20 or 80 as the surfactant/cosurfactant at the same level of 10% FFB based on the pseudoternary phase diagrams as reported previouslyCitation14 were examined to select those SMEDDSs with optimal solubility for further evaluation. The detailed compositions of the two groups (Group I with various lower levels of oil/Smix ratios: A and B of 0.42, C and D of 0.25, and E and F of 0.11 and water contents: 0.0%, 5.0%, and 9.5%; Group II with various higher levels of oil/Smix ratios: G and H of 1.38, M and P of 1.11, S and U of 0.9, T and V of 0.58, and O and R of 0.46 and the same water content of 4.5%) of SMEDDS formulations containing either TPGS/Tween 20 or 80 are listed in and , respectively. For Group I (formulations A–F), it was found that only the A and B series of SMEDDS formulations maintained a dissolved status with no precipitation of FFB at 12 hours after preparation. Whereas, 1 week after preparation, FFB was observed to have precipitated out to a small extent in both A and B series of SMEDDS formulations but at a higher extent from the other formulations (C–F) in this group. For Group II with a higher oil/Smix ratio (formulations G–O containing Tween 20 and formulations H–R containing Tween 80), FFB was completely dissolved in SMEDDS formulations using either Tween 20 or 80 as the cosurfactant at all oil/Smix ratios 12 hours after preparation, but phase separation was only observed for those SMEDDS formulations containing the Tween 20 at the higher oil/Smix ratio (formulations G, M, and S). At 1 week after preparation, FFB was found to have precipitated from all SMEDDS formulations containing either Tween 20 or 80, with the former showing a greater level of precipitation than the latter, except the SMEDDS formulations of T (containing Tween 20) and V (containing Tween 80) at an oil/Smix ratio of 0.58, which maintained complete dissolution with no phase separation.
In vitro dissolution characterizations
Those SMEDDS formulations selected above were further subjected to dissolution studies excluding formulations C, D, E, and F in Group I, for which severe FFB precipitation was observed 1 week after preparation, and formulations G, M, H, and P in Group II. For the first two, Tween 20 as the cosurfactant was insufficient for a higher level of oil content to be microemulsified into a single phase, and Tween 80 was the cosurfactant at the same oil/Smix ratio as formulations G and M in the latter two.
Particle size characterizations
The results of mean particle size versus time profiles at three stirring rates are illustrated in and , respectively, for Group I (series A and B) and II (Tween 20 series: N, O, S, and T; Tween 80 series: Q, R, U, and V) SMEDDS formulations. The corresponding mean size and their size distributions expressed by the polydispersity index for the last time point of the dissolution study are listed in . Comparing series A and B SMEDDS formulations at three stirring rates () showed that FFB drug particles formed within the nanosize range gradually increased with time but decreased with an increase in the stirring rate at the same time point for both series. A minor difference was shown in the size of the FFB drug particles formed by these SMEDDS formulations containing various water contents within the same series. However, the mean particle size of FFB formed by the series B was as small as 200 nm, which was smaller than the corresponding SMEDDS formulations of series A at all three stirring rates. Similar results were observed for the particle characteristics determined for the final sampling time point, as shown in . It was concluded that for those formulations within the SMEDDS region, Tween 80 was better than Tween 20 for precipitating FFB in SMEDDS formulations to form drug particles in a smaller size range. This effect was dependent on the stirring rate but independent of the water content. Because the hydrophilic–lipophilic balance value of Tween 80 (15) was lower than that of Tween 20 (16.7), it was expected that Tween 80 would have a higher capacity to solubilize FFB than Tween 20 resulting in the precipitation of FFB in a smaller nanosize range than those SMEDDSs containing Tween 80 as the cosurfactant.
Figure 1 Dissolution profiles of fenofibrate (FFB) from self-microemulsifying drug delivery system (SMEDDS) formulations composed of D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) as the surfactant and Tween® 20 (A1, A2, and A3) or Tween® 80 (B1, B2, and B3) as the cosurfactant at various stirring rates: (A) 10 rpm, (B) 25 rpm, (C) 50 rpm.
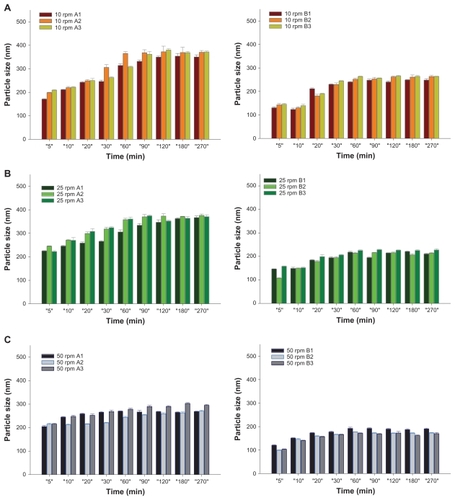
Figure 2 Dissolution profiles of fenofibrate (FFB) from self-microemulsifying drug delivery system (SMEDDS) formulations composed of D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) as the surfactant and Tween® 20 (N, O, S, and T) or Tween® 80 (Q, R, U, and V) as the cosurfactant at various oil/Smix ratios (S and U at 0.9, N and Q at 0.73, T and V at 0.58, and O and R at 0.46) with the same water content (4.5%) at various stirring rates: (A) 10 rpm, (B) 25 rpm, (C) 50 rpm.
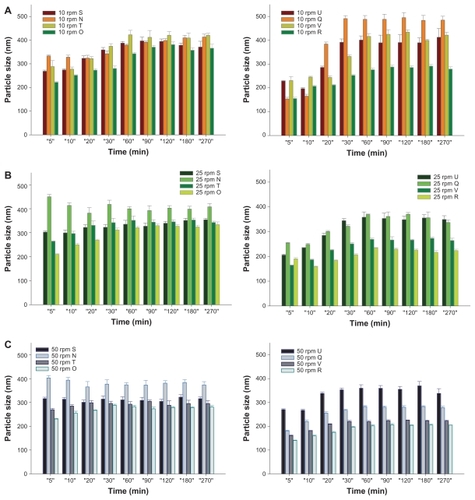
Comparing the Tween 20 (N, O, S, and T) and Tween 80 series (Q, R, U, and V) of SMEDDS formulations in Group II at three stirring rates () also demonstrated that FFB drug particles formed within a similar nano-size range gradually increased with time except for the N formulation at the two higher stirring rates, but decreased with increasing stirring rates at the same time point for both series. A decrease was shown in the mean particle size of FFB particles formed from those SMEDDS formulations with a decreasing oil/Smix ratio within the same series except for formulation S in the Tween 20 series at all three stirring rates, and formulation U in the Tween 80 series at the lowest stirring rate of 10 rpm. However, the mean particle size of FFB formed by the Tween 80 series reached the 200 ~ 300 nm range at the highest stirring rate of 50 rpm, which was smaller than the corresponding SMEDDS formulations of the Tween 20 series at the same stirring rate except for formulation Q in the Tween 80 series at the lowest stirring rate of 10 rpm. Similar results were observed for the particle characteristics determined for the final sampling time point as demonstrated in . Because formulation S of the Tween 20 series was unable to form a SMEDDS leading to phase separation as demonstrated in , the portion of FFB which dissolved in the oil phase separated from the SMEDDS during dissolution, causing a smaller amount of FFB to be precipitated and form drug particles with a smaller mean size than for the other formulations of the Tween 20 series. On the other hand, a larger mean size of FFB particles formed from formulation Q at the lowest stirring rate of 10 rpm could be attributed to the higher viscosity of Tween 80 at a higher oil/Smix ratio of 0.73, which increased the droplet size of the microemulsified internal phase. Rationally, this viscosity effect on the formation of droplet size of the internal phase was only significant at a lower stirring rate (P < 0.001).
Dissolution profile characterizations
The drug released (%) from those SMEDDS formulations in the dissolution study was measured after dilution by the addition of absolute alcohol to dissolve those FFB nanoparticles in the dissolution medium to an appropriate concentration range. and , respectively, illustrate the release profiles for Groups I (series A and B) and II (Tween 20 series: N, O, S, and T; and Tween 80 series: Q, R, U, and V) at three stirring rates. As shown in , the release rates from both series A and B SMEDDSs obviously increased with an increasing stirring rate (P < 0.001 for series A and P = 0.003 for series B). At the lower stirring rate of 10 rpm (), the release rate from series A seemed to decrease with an increasing water content (A1 [9.5%] < A2 [5.0%] < A3 [0.0%]), whereas that from series B followed an opposite trend (B1 [9.5%] ≅ B2 [5.0%] > B3 [0.0%]). Further, only the release rate from series B containing 0% water content (B3) was slower than that from the corresponding series A containing the same water content. With an increase in the stirring rate to 25 rpm (), no difference was evident among the release rates from series B (P = 0.896), and all were faster than those from the corresponding series A formulations with the same water content. The release rate from A3 was a little faster than those from A1 and A2. With a further increase in the stirring rate to 50 rpm (), the release rates from series A were in the order of A1 ≅ A2 > A3, whereas those from series B were in the order of B3 > B2 ≅ B1, all of which were slower than those from the corresponding series A containing the same water content.
Figure 3 Mean particle size profiles of fenofibrate (FFB) from self-microemulsifying drug delivery system (SMEDDS) formulations composed of D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) as the surfactant and Tween® 20 (A1, A2, and A3) or Tween® 80 (B1, B2, and B3) as the cosurfactant at various stirring rates: (A) 10 rpm, (B) 25 rpm, (C) 50 rpm.
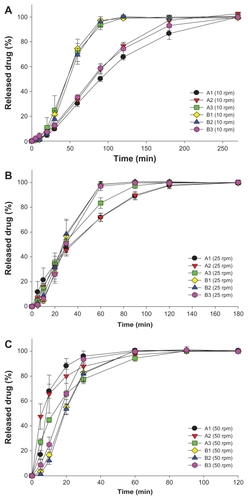
Figure 4 Mean particle size profiles of fenofibrate (FFB) from self-microemulsifying drug delivery system (SMEDDS) formulations composed of D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) as the surfactant and Tween® 20 (N, O, S, and T) or Tween® 80 (Q, R, U, and V) as the cosurfactant at various oil/Smix ratios (S and U of 0.9, N and Q of 0.73, T and V of 0.58, and O and R of 0.46) with the same water content (4.5%) at various stirring rates: (A) 10 rpm, (B) 25 rpm, (C) 50 rpm.
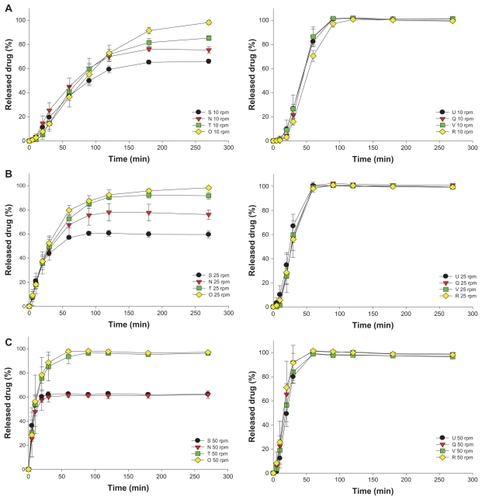
Similarly, as shown in , the release rates from both Tween 20 series (N, O, S, and T) and Tween 80 series (Q, R, U, and V) of SMEDDSs increased with an increasing stirring rate. Among the Tween 20 series, incomplete release was observed only for the S and N SMEDDSs at all three stirring rates. As described above, this was attributed to a failure of the S and N formulations to microemulsify all of the oil phase in which most of the FFB was dissolved. However, a faster release rate and complete release were observed for the Tween 80 series of SMEDDSs compared with the Tween 20 series, but no significant difference was shown among the release rates for the Tween 80 series examined (P = 0.758). Before the drug concentration was measured, the sampled dissolution medium was diluted with absolute alcohol, which possesses good solubility for FFB; thus, all drug particles existing in the dissolution medium would have been completely dissolved, meaning no differences in the amount released, even though different mean particle sizes were observed for the Tween 80 series in the respective dissolution medium. This result also implies that, at the same stirring rate, the microemulsification of those SMEDDSs of the Tween 80 series had to have reached the same extent to achieve the same amount of drug existing in the dissolution medium and for the same amount released to be measured.
Pharmacokinetic studies
Two optimal SMEDDSs were selected for the pharmacokinetic study conducted with a parallel design: one contained Tween 80 (B2[SMEDDS]) or a higher oil/Smix ratio (Q[SMEDDS]) at a dosing strength of 54 mg FFB in capsule form, and the B2(solution) was prepared by dissolving B2 SMEDDS in the same volume of water (250 mL) as that usually taken with oral administration. The resultant plasma profiles for the three treatments (B2[SMEDDS], Q[SMEDDS], and B2[solution]) and that for Q(SMEDDS) compared with that for A2(SMEDDS) and Tricor conducted in a previous studyCitation36 are illustrated in , respectively, and the corresponding calculated pharmacokinetic parameters are listed in . Results demonstrated that the AUC and Cmax values were ranked in the order of Q(SMEDDS) > B2(solution) > B2(SMEDDS) and B2(solution) > Q(SMEDDS) > B2(SM EDDS), respectively, for this study, whereas overall both the AUC and Cmax values were ranked, respectively, in the order of Q(SMEDDS) > Tricor ≅ B2(solution) > B2(SMEDDS) ≅ A2(SMEDDS) and B2(solution) > Q(SMEDDS) > Tricor ≅ B2(SMEDDS) > A2(SMEDDS). These results indicate that the overall relative bioavailability based on either AUC0–last or AUC0–inf with respect to Tricor for Q(SMEDDS) was enhanced approximately 1.14–1.22-fold. The relative bioavailabilities of A2(SMEDDS) and B2(SMEDDS) were similar but less than that for B2(solution), which was close to that for Tricor. Furthermore, as indicated by the large Cmax value and the short Tmax, the in vivo absorption rate for B2(solution) was the fastest of all, followed in order by Q(SMEDDS) > Tricor ≅ B2(SMEDDS) > A2(SMEDDS).
Table 3 Mean pharmacokinetic parameters for B2 and Q self-microemulsifying drug delivery system (SMEDDS) and B2 solution compared with A2 SMEDDS and Tricor® tablets conducted in a previous study36
Figure 5 Plasma fenofibric acid (FBA) concentration profiles of self-microemulsifying drug delivery system (SMEDDS) formulations (all containing 54 mg fenofibrate [FFB]). (A) B2(solution), B2(SMEDDS), and Q(SMEDDS); (B) A2(SMEDDS), Q(SMEDDS), and Tricor® tablets.
![Figure 5 Plasma fenofibric acid (FBA) concentration profiles of self-microemulsifying drug delivery system (SMEDDS) formulations (all containing 54 mg fenofibrate [FFB]). (A) B2(solution), B2(SMEDDS), and Q(SMEDDS); (B) A2(SMEDDS), Q(SMEDDS), and Tricor® tablets.](/cms/asset/d9717185-7f6f-41bd-827d-479c3da37228/dijn_a_25339_f0005_c.jpg)
As indicated by the particle sizes at the three stirring rates shown in , the fastest in vivo absorption rate for B2(solution) can potentially be attributed to the formation of a particle size in the smallest range for the B2 solution when the B2 SMEDDS was dissolved in water, making it readily available for absorption. After oral administration, the three SMEDDSs (A2, B2, and Q) would take some time for the microemulsification to dissolve before the FFB was available for absorption. Further, the mean particle size after microemulsification at the three different stirring rates for B2(SMEDDS) were all smaller than that for A2(SMEDDS). This can be explained by the fact that the in vivo absorption rate for B2(SMEDDS) was faster than that for A2(SMEDDS). Nevertheless, this particle size effect could not be explained by the in vivo absorption rate for Q(SMEDDS) being faster than that for B2(SMEDDS). Perhaps the oil content in the higher oil/Smix ratio of Q(SMEDDS) played a role as a carrier providing an additional oral absorption route, such as via the lymph system, to increase the in vivo absorption rate.Citation16 But the time required for microemulsification of Q(SMEDDS) in the gastrointestinal tract might cause some reduction in its in vivo absorption rate compared with the B2(solution), which was completely microemulsified before oral administration.
The oral bioavailability of B2(solution) was slightly lower than that for Tricor, but both were lower than that for Q(SMEDDS), and greater than those for B2(SMEDDS) and A2(SMEDDS), which did not significantly differ (P = 0.058). This result seems to indicate that the expected difference in the mean particle size after oral administration between B2(SMEDDS) and A2(SMEDDS) as deduced from the in vitro dissolution results did affect the in vivo absorption rate but not the oral bioavailability. However, the oral bioavailability of the B2(solution) formulated by completely microemulsifying B2(SMEDDS) in the same volume of water as that usually taken with oral administration was greater than that for B2(SMEDDS) and only slightly lower than that for Tricor. Therefore, this result might be attributed to the incomplete microemulsification of B2(SMEDDS) and A2(SMEDDS) in the gastrointestinal tract leading to a lower bioavailability compared with B2(solution) that was completely microemulsified before oral administration. As discussed above, the oil content in a higher oil/Smix ratio of Q(SMEDDS) probably acted as a carrier to provide a favorable additional oral absorption route, such as the lymphatic system, which enhanced both the in vivo absorption rate and the oral bioavailability. Therefore, Q(SMEDDS) with an appropriate ratio of oil/Smix presented the highest oral bioavailability of all SMEDDSs examined and even had an approximately 1.14–1.22-fold higher enhancement over Tricor.
Conclusion
An optimal SMEDDS preconcentrate consisting of Myritol 318 (a medium-chain triglyceride) as the oil phase and TPGS combined with Tween 80 at a ratio of 4:1 as the surfactant/cosurfactant, was able to enhance the oral bioavailability of FFB. Tween 80 was better than Tween 20 in forming smaller particles, which enhanced the in vivo absorption rate by increasing the surface area available for drug dissolution but had little influence on the oral bioavailability due to the lower extent of microemulsification. With a higher oil/Smix ratio exceeding 0.46, the microemulsification effectiveness (rate) was enhanced making drug particles readily available for absorption at any in vivo agitation rate; the oral bioavailability was improved with the favorable role Myritol 318 might play as a carrier by providing an additional oral route of the lymphatic system for FFB absorption.
Acknowledgment
Financial support by the National Council of Science of the ROC is highly appreciated (NSC-96-2320-B-038-001).
Disclosure
The authors report no conflicts of interest, other than the funding mentioned in the Acknowledgment, in this work.
References
- HoarTPSchulmanJHTransparent water in oil dispersions: oleopathic hydromicelleNature1943152102105
- SpernathAAserinAMicroemulsions as carriers for drugs and nutraceuticalsAdv Colloid Interface Sci2006128–1304764
- LyonsKCCharmanWNMillerRPorterCJFactors limiting the oral bioavailability of N-acetylglucosaminyl-N-acetylmuramyl dipeptide (GMDP) and enhancement of absorption in rats by delivery in a water-in- oil microemulsionInt J Pharm2000199172810794923
- HuZPTawaRKonishiTShibataNTakadaKA novel emulsifier, Labrasol, enhances gastrointestinal absorption of gentamicinLife Sci2001692899291011720093
- KawakamiKYoshikawaTHayashiTNishiharaYMasudaKMicroemulsion formulation for enhanced absorption of poorly soluble drugs. II. In vivo studyJ Control Release200281758211992680
- KangBKLeeJSChonSKDevelopment of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogsInt J Pharm2004274657315072783
- ShaXYYanGJWuYJLiJCFangXLEffect of self-microemulsifying drug delivery systems containing Labrasol on tight junctions in Caco-2 cellsEur J Pharm Sci20052447748615784337
- KommuruTRGurleyBKhanMAReddyIKSelf-emulsifying drug delivery systems (SEDDS) of coenzyme Q10: formulation development and bioavailability assessmentInt J Pharm200121223324611165081
- SpernathAYaghmurAAserinAHoffmanREGartiNSelf-diffusion nuclear magnetic resonance, microstructure transitions, and solubilization capacity of phytosterols and cholesterol in Winsor IV food-grade microemulsionsJ Agric Food Chem2003512359236412670181
- SpernathAYaghmurAAserinAHoffmanREGartiNFood-grade microemulsions based on nonionic emulsifiers: media to enhance lycopene solubilizationJ Agric Food Chem2002506917692212405797
- AmarIAserinAGartiNSolubilization patterns of lutein and lutein esters in food grade nonionic microemulsionsJ Agric Food Chem2003514775478114705912
- GartiNAmarIYaghmurASpernathAAserinAInterfacial modification and structural transitions induced by guest molecules solubilized in U-type nonionic microemulsionsJ Disper Sci Technol200324397410
- GartiNAvrahamiMAserinAImproved solubilization of celecoxib in U-type nonionic microemulsions and their structural transitions with progressive aqueous dilutionJ Colloid Interface Sci200629935236516529763
- KeWTLinSYHoHOSheuMTPhysical characterizations of microemulsion systems using tocopheryl polyethylene glycol 1000 succinate (TPGS) as a surfactant for the oral delivery of protein drugsJ Control Release200510248950715653166
- PoutonCWFormulation of self-emulsifying drug delivery systemsAdv Drug Deliv Rev1997254758
- HolmRPorterCJEdwardsGAMüllertzAKristensenHGCharmanWNExamination of oral absorption and lymphatic transport of halofantrine in a triple-cannulated canine model after administration in self-microemulsifying drug delivery systems (SMEDDS) containing structured triglyceridesEur J Pharm Sci200320919713678797
- KhooSMHumberstoneAJPorterCJEdwardsGACharmanWNFormulation design and bioavailability assessment of lipidic self-emulsifying formulations of halofantrineInt J Pharm1998167155164
- FrimanSBackmanLA new microemulsion formulation of cyclosporine: pharmacokinetic and clinical featuresClin Pharmacokinet1996301811938882300
- KimJYKuYSEnhanced absorption of indomethacin after oral or rectal administration of a self-emulsifying system containing indomethacin in ratsInt J Pharm2000194818910601687
- ShenHRZhongMKPreparation and evaluation of self-emulsifying drug delivery systems (SMEDDS) containing atorvastatinJ Pharm Pharmacol2006581183119116945176
- GroveMMüllertzANielsenJLPedersenGPBioavailability of seocalcitol II: development and characterization of self-microemulsifying drug delivery systems (SMEDDS) for oral administration containing medium and long chain triglyceridesEur J Pharm Sci20062823324216650738
- ChenYLiGWuXGSelf-microemulsifying drug delivery system (SMEDDS) of vinpocetine: formulation development and in vivo assessmentBiol Pharm Bull20083111812518175953
- WooJSSongYKHongJYLimSJKimCKReduced food-effect and enhanced bioavailability of a self-microemulsifying formulation of itraconazole in healthy volunteersEur J Pharm Sci20083315916518178070
- FatourosDGNielsenFSDouroumisDHadjileontiadisLJMullertzAIn vitro-in vivo correlations of self-emulsifying drug delivery systems combining the dynamic lipolysis model and neuro-fuzzy networksEur J Pharm Biopharm20086988789818367386
- ZhangPLiuYFengNPXuJPreparation and evaluation of self-emulsifying drug delivery system for oridoninInt J Pharm200835526927618242895
- CooneyGFJeevanandamVChoudhurySFeutrenGMullerEAEisenHJComparative bioavailability of Neoral and Sandimmune in cardiac transplant recipients over 1 yearTransplant19983018921894
- PorterCJCharmanWNIn vitro assessment of oral lipid based formulationsAdv Drug Deliv Rev200150Suppl 1S12714711576699
- SubramanianNRaySGhosalSKBhadraRMoulikSPFormulation design of self-emulsifying drug delivery systems for improved oral bioavailability of CelecoxibBiol Pharm Bull2004271993199915577219
- ParkKMKimCKPreparation and evaluation of flurbiprofen-loaded microemulsion for parenteral deliveryInt J Pharm199918117317910370213
- MunozAGuichardJPReginaultPMicronisedFFBAtherosclerosis1994110SupplS45S487857384
- StammASethPFenofibrate pharmaceutical composition having high bioavailability and method for preparing itUnited States patent US 6589552782003
- GuichardJPBlouquinPQingYA new formulation of FFB: suprabioavailable tabletsCurr Med Res Opin20001613413810893657
- NajibJFFB in the treatment of dyslipidemia: a review of the data as they relate to the new suprabioavailable tablet formulationClin Ther2002242022205012581543
- SonetBVanderbistFStreelBHouinGRandomized crossover studies of the bioequivalence of two FFB formulations after administration of a single oral dose in healthy volunteersArzneim Forsch20025220020411963648
- PatelARVaviaPRPreparation and in vivo evaluation of SMEDDS (self-emulsifying drug delivery system) containing fenofibrateAAPS J200793E34435218170981
- WangGMChenCHKeWTHoHOSheuMTCharacterizations of fenofibrate dissolution delivered by a self-microemulsifying drug delivery systemJ Pharm Pharmacol2010621685169621054394