 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Background
Inhaled corticosteroids provide unique systems for local treatment of asthma or chronic obstructive pulmonary disease. However, the use of poorly soluble drugs for nebulization has been inadequate, and many patients rely on large doses to achieve optimal control of their disease. Theoretically, nanotechnology with a sustained-release formulation may provide a favorable therapeutic index. The aim of this study was to determine the feasibility of using sterically stabilized phospholipid nanomicelles of budesonide for pulmonary delivery via nebulization.
Methods
PEG5000-DSPE polymeric micelles containing budesonide (BUD-SSMs) were prepared by the coprecipitation and reconstitution method, and the physicochemical and pharmacodynamic characteristics of BUD-SSMs were investigated.
Results
The optimal concentration of solubilized budesonide at 5 mM PEG5000-DSPE was 605.71 ± 6.38 μg/mL, with a single-sized peak population determined by photon correlation spectroscopy and a particle size distribution of 21.51 ± 1.5 nm. The zeta potential of BUD-SSMs was −28.43 ± 1.98 mV. The percent entrapment efficiency, percent yield, and percent drug loading of the lyophilized formulations were 100.13% ± 1.09%, 97.98% ± 1.95%, and 2.01% ± 0.02%, respectively. Budesonide was found to be amorphous by differential scanning calorimetry, and had no chemical interaction with PEGylated polymer according to Fourier transform infrared spectroscopy. Transmission electron microscopic images of BUD-SSMs revealed spherical nanoparticles. BUD-SSMs exhibited prolonged dissolution behavior compared with Pulmicort Respules® (P < 0.05). Aerodynamic characteristics indicated significantly higher deposition in the lungs compared with Pulmicort Respules®. The mass median aerodynamic, geometric standard deviation, percent emitted dose, and the fine particle fraction were 2.83 ± 0.08 μm, 2.33 ± 0.04 μm, 59.13% ± 0.19%, and 52.31% ± 0.25%, respectively. Intratracheal administration of BUD-SSMs 23 hours before challenge (1 mg/kg) in an asthmatic/chronic obstructive pulmonary disease rat model led to a significant reduction in inflammatory cell counts (76.94 ± 5.11) in bronchoalveolar lavage fluid compared with administration of Pulmicort Respules® (25.06 ± 6.91).
Conclusion
The BUD-SSMs system might be advantageous for asthma or chronic obstructive pulmonary disease and other inflammatory airway diseases.
Introduction
Asthma, chronic obstructive pulmonary disease, and other pulmonary diseases can be efficiently treated via the pulmonary route if high and prolonged drug concentrations are maintained in the lungs.Citation1,Citation2 Local treatment of lung disorders via pulmonary drug delivery offers many advantages over oral or intravenous routes of administration, because direct deposition of drug at the diseased site could increase local drug concentrations, improve the pulmonary receptor occupancy, and reduce the overall dose required and the side effects that result from high doses of drug.Citation3 In addition, sustained-release formulations for pulmonary delivery may result in a favorable therapeutic index by prolonging drug action at the target site, reducing its side effects, and enhancing patient compliance.Citation4
Microparticle and nanoparticle drug carrier systems have been extensively studied.Citation3,Citation5–Citation8 However, the majority of these formulations have not successfully controlled the inhaled therapeutics in the pulmonary system for more than a few hours due to efficient clearance of the therapeutics from the deep lung either through phagocytosis or via the rapid absorption of the delivered therapeutics by the alveoli, making them more suitable for enhancing systemic bioavailability.Citation4,Citation9 Controlling the drug in the respiratory tract may be achievable by employing suitable carrier systems with appropriate drug-release characteristics. Among these drug carrier systems, polymeric micelles are particularly promising and are receiving increasing attention.Citation10,Citation11 So-called polymeric micelle colloidal dispersions are self-assembled core-shell nanostructures formed in an aqueous solution consisting of hydrophobic fragments of amphiphilic molecules forming the core of a micelle, which is segregated from the environment by hydrophilic parts of the molecules that form the micelle corona.Citation12,Citation13 The cargo space (core) formed from the hydrophobic segment solubilizes a variety of poorly soluble therapeutic and diagnostic agents. This solubilization increases the bioavailability and circulation time after parenteral administration, as well as modifying the pharmacokinetics and biodistribution of the therapeutic agents.Citation14,Citation15 The small size of the micelles permits their extravasation and accumulation in a variety of pathological sites such as tumors.Citation16 Additionally, polymeric micelles are easily prepared on a large scale.Citation14
A variety of block copolymers may be used to form polymeric micelles, including conjugates of polyethylene glycol and phosphatidylethanolamine (PEG-DSPE), to form so-called sterically stabilized phospholipid nanomicelles (SSMs).Citation17 Polymeric micelles have the ability to prolong the circulation time (in vivo experiments) and the ability to accumulate in target organs.Citation18 In addition, the fact that humans secrete phospholipase A2, which is able to degrade PEG-DSPE, makes them biodegradable.Citation19
However, to date, no in vitro or in vivo study of the efficiency of sterically stabilized phospholipid nanomicelles containing a poorly water-soluble corticosteroid drug (budesonide) as a pulmonary delivery system has been reported. Therefore, this study focused on the formulation, characterization, and in vitro and in vivo evaluation of sterically stabilized phospholipid nanomicelles containing budesonide as a pulmonary delivery system via nebulization.
Materials and methods
Materials
1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-Nmethoxy- poly(ethylene glycol 5000) (PEG5000-DSPE) was purchased from NOF Corporation (Tokyo, Japan) and budesonide (molecular weight 430.50 g/mol) was purchased from Symbiotica Specialty Ingredients Sdn Bhd (Kedah, Malaysia). High-pressure liquid chromatography (HPLC)-grade methanol and chloroform were purchased from Fisher Scientific (Franklin Lakes, NJ, and Leicestershire, UK, respectively). Phosphate-buffered saline tablets were purchased from Sigma-Aldrich Chemie Gmbh (Steinheim, Germany).
Preparation of BUD-SSMs
Aqueous dispersions of BUD-SSMs were prepared by the coprecipitation and reconstitution method using some modifications, as previously reported.Citation20 Known amounts of budesonide and PEG5000-DSPE were mixed at different molar ratios. The mixtures were subsequently sonicated for 2 minutes using a sonicator processor (Bransonic Ultrasonic 8510, Danbury, CT). The content was transferred to a 50 mL round-bottomed flask, mixed, and vortexed for one minute. The organic solvents were evaporated under vacuum at 40°C using a rotary evaporator (Eyela N-1001S-W, Tokyo, Japan). Any traces of remaining solvent in the film obtained were removed under vacuum overnight.Citation21 The dried films were rehydrated with phosphate-buffered saline, and the SSMs were formed by shaking at 40°C for 10 minutes. Drug that was not encapsulated was separated by filtration of the micellar solutions using a 0.2 μm Minisart microfilter (Sartorius, Germany). The maximum solubility of budesonide in the SSMs was determined by keeping the PEG5000-DSPE concentration fixed (5 mM) and changing the drug concentration (budesonide to PEG5000-DSPE molar ratios ranged from 0.20 to 0.34) until a homogeneous system was confirmed by photon correlation spectroscopy as a singlesize peak population using a Zetasizer (Malvern 1000HSA, Worcestershire, UK). The amount of solubilized budesonide in the SSMs was determined spectrophotometrically using an ultraviolet spectrophotometer (Hitachi, model U-2000, Japan) at 244 nm after diluting the clear aqueous dispersion with methanol (drug-SSMs:methanol, 1:3 [v/v]). BUD-SSMs with the maximum solubility of budesonide were lyophilized (Labconco 7753501, Labconco Corporation, Kansas City, MO) to form a lyophilized cake ready for reconstitution.
Physicochemical characterization of BUD-SSMs
The following parameters of the BUD-SSM formulations were characterized: particle size, using a Zetasizer (Malvern 1000HSA); zeta potential, using a Zeecom zeta potential analyzer (Symphotic TII Corporation, Camarillo, CA); and entrapment efficiency (%EE), yield percentage (%Y), and drug loading percentage (%DL) using the following equations according to a previously published method:Citation22
where (a) is the concentration of drug loaded in SSMs (μg/mL) and (b) is the amount of drug used in SSMs preparation (μg/mL):
We also performed Fourier transform infrared spectroscopy using an infrared spectrophotometer (Thermo Nicolet Corporation, Nexus model, Madison, WI), equipped with OMNIC 6.1 version software, differential scanning calorimetry (Perkin Elmer, Pyris 6, Boston, MA), and morphological examinations of the rehydrated BUD-SSMs using a transmission electron microscope (Phillips CM12, Eindhoven, The Netherlands) with Docu version 3.2 image analysis.
Solubility and dissolution study
To determine the sink condition, the solubility of budesonide in phosphate-buffered saline was measured by placing an excess amount of budesonide (300 mg) into 5 mL of phosphate- buffered saline (pH 7.4) in a dialysis bag (Spectra/pro, molecular weight cutoff 10,000 kD, Spectrum, Torrance, CA) placed in a 100 mL phosphate-buffered saline Pyrex bottle and shaken horizontally at 100 rpm in a thermostatic water bath (Memmrete WB22, Germany) at 37°C ± 0.5°C. Samples of the drug solution were withdrawn from the bottle (200 μL, after 30 hours) and analyzed by a validated HPLC method, as previously described.Citation23 The resulting equilibrium drug concentration was used to calculate the saturated solubility drug concentration.Citation24
For the dissolution study, rehydrated BUD-SSMs or Pulmicort Respules® (containing the equivalent amount of budesonide [400 μg] required for the sink condition) were diluted in 5 mL of phosphate-buffered saline (pH 7.4) and transferred into a dialysis bag. The two ends of the dialysis bag were fastened securely with special clips spaced 5 cm apart. The dialysis bag was placed into a Pyrex screw-capped bottle containing 100 mL of phosphate-buffered saline (pH 7.4) as the dissolution medium and 0.02% of sodium azide (Sigma-Aldrich Chemie Gmbh) to prevent microbial growth. The bottle was shaken horizontally at 100 rpm in a thermostatic water bath (Memmrete WB22, Germany) at 37°C ± 0.5°C. At predetermined time intervals, 200 μL samples were withdrawn from the bottle and analyzed by a validated HPLC method, as described previously.Citation23 Briefly, the system consisted of a Shimadzu LC-20AD delivery pump (Shimadzu, Tokyo, Japan) equipped with a SIL-20A HT prominence autosampler (Shimadzu) fitted with a 100 μL sample loop, an ultraviolet-visible detector (SPD-20A, Shimadzu), a DGU-20A3 prominence degasser (Shimadzu), and a chromato-integrator (CBM-20A prominence Communications Bus Model, Shimadzu). The chromatographic separation of the analyte was performed at 40°C (CTO-10AS VP, Shimadzu column oven) using a Zorbax Eclipse Plus (250 × 4.6 mm, 5 mm) analytical column connected to a SecurityGuard™ cartridge system (Zorbax, Agilent Technologies, Colorado Springs, CO). The mobile phase consisted of 10 mM ammonium acetate (pH 5, adjusted with orthophosphoric acid) to acetonitrile (37:63). The mobile phase was filtered through a 0.45 μm nylon membrane filter (Whatman, Maidstone, UK) under vacuum and degassed prior to use. The analysis was conducted at a flow rate of 1.0 mL/ minute at an ultraviolet detection wavelength of 254 nm. The injection volume was 50 μL.
The kinetics of drug release were examined using the previously described add-in DDSolver program.Citation25 Different mathematical kinetic models (zero-order, first order, Higuchi, Hixson-Crowell, and Baker-Lonsdale) were used to determine the drug release kinetics. The criteria for selecting the most appropriate model were based on the goodness of fit (adjusted coefficient of determination, R2 adjusted) and the Akaike information criterion.Citation26
Aerodynamic characterization
In vitro deposition was investigated using a next generation impactor (Model 170) connected to a vacuum pump (HCP4) and Pari LC nebulizer (Pari LC plus nebulizer connected to an airjet compressor Pari Master type 84G73, Munich, Germany). A flow meter (DFM2) (Copley Scientific, UK) was used to calibrate the airflow through the next generation impactor. Two mL of an aqueous preparation containing 0.5 mg of rehydrated BUD-SSMs was nebulized for 15 minutes at a room temperature of 28°C and a relative humidity of 65%. The effective cutoff diameters for the impactor at a flow rate of 60 L/min were: 8.06 μm (stage 1), 4.46 μm (stage 2), 2.82 μm (stage 3), 1.66 μm (stage 4), 0.94 μm (stage 5), 0.55 μm (stage 6), and 0.34 μm (stage 7). The flow rate of 60 L/minute simulated the mean peak inspiratory flow rate of adult asthmatic patients.Citation27 Each stage of the next generation impactor, the induction port, and the inhaler device were rinsed with 10 mL of the respective HPLC mobile phase and collected for quantitative analysis by HPLC.Citation23
The mass median aerodynamic diameter and geometric standard deviation were calculated after plotting the cumulative amount of drug under size deposited in each stage of the cascade impactor versus their corresponding aerodynamic diameter as specified by the cascade impactor using logprobability paper.Citation28 On this graph, the mass median aerodynamic diameter of the aerosolized particles is the particle size at which the line crosses the 50% mark (mass median aerodynamic diameter [μm] = D50%).
From a log-normal distribution that is normal with respect to aerodynamic diameter,Citation28 the geometric standard deviation (GSD) becomes:
The emitted dose (ED) was determined as the percentage of total amount of drug available for nebulization:Citation23,Citation27
The fine particle fraction was calculated as the total amount of drug deposited on Stage 2 or 3 to filter:Citation23,Citation27,Citation29
Inhibitory duration of inflammatory cell infiltration in airways after BUD-SSMs and Pulmicort Respules®
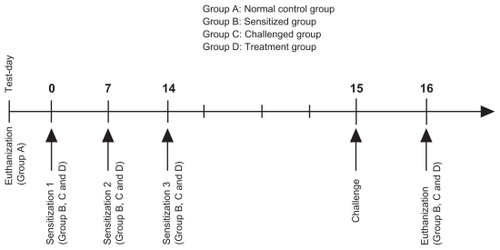
The experimental protocol (see ) was designed to study the duration of budesonide inhibition with respect to total cell counts and differential cell counts in the bronchoalveolar lavage fluid of ovalbumin-sensitized and ovalbumin-challenged male Sprague-Dawley rats. The protocol for ovalbumin sensitization and challenge () was modified from a previously published method.Citation30 The experimental protocol was approved by the Animal Ethics Committee, Health Campus, Universiti Sains Malaysia. Pathogen-free male Sprague-Dawley rats (8–11 weeks of age) weighing 200 ± 50 g, were housed six per cage in the laboratory with free access to food and water, and maintained on a 12-hour dark/light cycle. Briefly, following acclimatization, the rats were randomly divided into four groups: Group A, untreated rats, required to illustrate and define the physiological norm of inflammatory cell infiltration (controls); Group B, rats sensitized by intraperitoneal injection of 100 μg ovalbumin precipitated in 5 mg aluminum hydroxide (ovalbumin precipitate) per rat on days 0, 7, and 14 (negative control) and used to assess the effect of sensitization on inflammatory cell infiltration; Group C rats, sensitized by intraperitoneal injection of 100 μg ovalbumin precipitate on days 0, 7, and 14, followed by anesthesia and intratracheal instillation of ovalbumin challenge (100 μg/rat) 24 hours after the last ovalbumin sensitization, ie, on day 14 (positive control), and used to assess the effect of ovalbumin challenge on inflammatory cell infiltration; and Group D, which comprised the treatment group, and was divided into nine subgroups as follows:
Pulmicort Respules® group, in which treatment was administered via intratracheal instillation 23 hours (DP1), 12 hours (DP2), or one hour (DP3) before ovalbumin challenge.
BUD:PEG5000-DSPE SSMs group (F43, A, intervention group, n = 18) in which treatment was administered via intratracheal instillation 23 hours (DIA1), 12 hours (DIA2), or one hour (DIA3) before ovalbumin challenge.
Rehydrated PEG5000-DSPE SSMs group (F39), in which treatment was administered via intratracheal instillation one hour (D1) before ovalbumin challenge to examine the pharmacodynamic effect of the placebo treatment on the ovalbumin model rats.
Rats were sensitized by intraperitoneal injection of 100 μg/rat ovalbumin with 5 mg aluminum hydroxide on days 0, 7, and 14. They were anesthetized by intraperitoneal injection of ketamine + xylazine (80 mg/kg + 8 mg/ kg intraperitoneally)Citation31 and received intratracheal instillation of ovalbumin (100 μg/rat) 24 hours after the last ovalbumin sensitization using a Penn-Century Liquid MicroSprayer Aerosolizer® (Model IA-1B, Penn-Century Inc, Wyndmoor, PA) with the aid of a small animal laryngoscope (Model LS-2, Penn-Century Inc). At one hour, 12 hours, or 23 hours before ovalbumin exposure, 1 mg/kg of BUD-SSMs or Pulmicort Respules® was administered via intratracheal instillation. At 24 hours after ovalbumin challenge, the rats were euthanized in a carbon dioxide chamber, and the lungs with trachea were excised and lavaged.
Bronchoalveolar lavage was performed by flushing the airways with 5 mL of Dulbecco’s phosphate-buffered saline (SAFC Biosciences, Kansas City, MO) through the tracheal cannula. The total leukocyte cell counts and differential cell counts of the bronchoalveolar lavage fluid were recorded by a single blinded expert in a private laboratory (Pro-Lab, Penang, Malaysia). The total leukocyte cell counts in the bronchoalveolar lavage fluid were determined using a hematocytometer. Cell smears (8 × 104 cells/mL) were prepared by centrifugation in a bronchoalveolar lavage fluid centrifuge at 4000 rpm for five minutes at 20ºC, after which the supernatants were removed and the cells were resuspended in Dulbecco’s phosphate-buffered saline. Differential cell counts of 200 leukocytes (eosinophils, lymphocytes, neutrophils, and macrophages) were conducted for each animal under light microscopy (400× magnification). Briefly, a thin film of resuspended cells was obtained by spreading a drop of the cells evenly across a clean grease-free slide. A few drops of Leishman’s stain (Laboratory Labchem Chemical, Selangor Darul Ehsan, Malaysia) were poured onto the slide, and after 3 minutes, 20 drops of buffered water were added. After a further 7 minutes, the stain was washed off with distilled water for 2–3 minutes, following which the slides were rinsed with tap water and stood in a rack to drain and dry in air. These methods are commonly employed and are fully described elsewhere.Citation32,Citation33 Percent inhibition of inflammatory cell infiltration into the airways was calculated according to the following equation:
where EP.control, EN.control, and EGroup are inflammatory cell counts in bronchoalveolar lavage fluid in the positive control (Group C), negative control (Group B), and treatment group (Group D), respectively.
Statistical analysis
For statistical comparisons, analysis of variance with pairwise comparison and dependent and independent t-test procedures were used where applicable. A P value of less than 0.05 was considered significant for all analyses.
Results and discussion
Drug loading and maximum solubility
Budesonide was successfully incorporated into SSMs of PEG5000-DSPE using the coprecipitation and reconstitution method. The SSMs’ potential to solubilize budesonide was assessed with eight different budesonide concentrations ranging from 430.5 to 731.85 μg/mL in 5 mM PEG5000-DSPE. Surprisingly, all samples at or below 605.71 ± 6.38 μg/mL of budesonide (drug-to-PEGylated polymer molar ratio of 0.28) had a single uniform size distribution determined by photon correlation spectroscopy; a second signal was observed above this concentration (). For a given PEGylated polymer, the poorly soluble drug only solubilized up to a certain concentration of drug, and when the maximum threshold was achieved, populations of other species, namely sterically stabilized particles, were observed by photon correlation spectroscopy ().Citation34 The sterically stabilized particles were probably stabilized by the PEG5000-DSPE on their surfaces, which helped the steroids form aggregates and thus to maximize the hydrophobic interactions and hydrogen bonding network with water.Citation20,Citation21 Formulations containing sterically stabilized particles were not considered the optimum formulation due to difficulties reproducing the formulations, as previously reported.Citation20
Figure 1 Effect of the budesonide: PEGylated polymer molar ratio on solubilization of budesonide; PEGylated polymer molar concentration kept at 5 mM.
Note: Gray bars indicate second population signal in photon correlation spectroscopy.
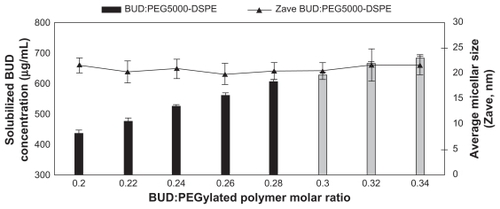
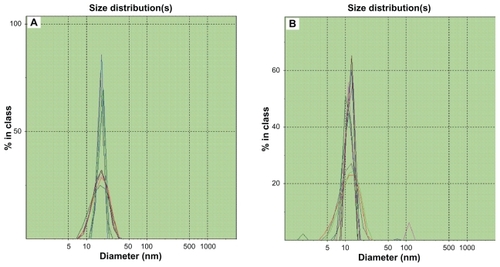
Figure 2 Photon correlation spectroscopic size distribution of BUD-SSMs. (A) BUD-SSMs at optimum drug concentration (0.28 drug to polymer molar ratio); (B) BUD-SSMs at excessive drug concentration (0.30 drug to polymer molar ratio).
Abbreviation: BUD-SSMs, PEG5000-DSPE polymeric micelles containing budesonide.
Hydrodynamic particle size and zeta potential of SSMs
The particle size of the final rehydrated BUD-SSMs (drug to PEGylated polymer molar ratio of 0.28) was 21.51 ± 1.5 nm, with a narrow polydispersity index (0.22 ± 0.02) that did not differ significantly (P > 0.05) from that observed in the prelyophilization process (20.45 ± 1.65), indicating formation of SSMs with a typical particle size of polymeric micelles between 10 and 100 nm.Citation35 Due to their small size, the likelihood that BUD-SSMs would undergo phagocytosis in the alveoli is much lower than that for micron-sized particles, because it has been reported that particles of less than 260 nm can escape phagocytosis by macrophages.Citation36
The zeta potential measurement gives an indication of the stability of the colloidal system. Dispersions with a large negative or positive zeta potential tend to have better stability against aggregation.Citation37 The zeta potential of BUD-SSMs was −28.43 ± 1.98 mV, confirming the stability potential. The negative charge of the BUD-SSM formulation also gives it a potential advantage as a drug carrier because it has been reported that negatively charged particles are retained in the lungs more efficiently than positively charged molecules.Citation38
Drug loading, yield, and entrapment efficiency of BUD-SSMs
According to the spectrophotometric and particle size analysis, all budesonide molecules were incorporated into the SSMs after lyophilization. This was supported by the lack of a significant difference (P > 0.05) in percentage entrapment efficiency before (100.49 ± 1.06) and after lyophilization (100.13 ± 1.09). In addition, the BUD-SSM formulations had a high percentage yield (97.98 ± 1.95) and considerable percentage drug loading (2.01% ± 0.02%). The percentage drug loading value was consistent with findings for other steroid formulations.Citation39,Citation40 In addition, the BUD-SSMs formed from PEG5000-DSPE were robust to the lyophilization conditions, with the lyophilized cakes having a fluffy and elegant appearance and dissolving easily upon reconstitution with distilled water to form clear and colorless solutions. This was due to the hydrophilic PEG polymer on the outer surface (corona), which can act as both a cryoprotectant and a lyoprotectant.Citation20 A 5 mM concentration of PEG5000-DSPE was appropriate for lyophilization because high-density PEG blocks can serve as a steric barrier between hydrophobic cores and prevent contact between and agglomeration of the core materials of the micelles, as previously reported when using 10 mM PEG2000-DSPE.Citation17,Citation21
Differential scanning calorimetry study
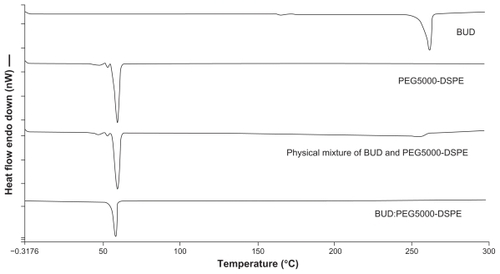
Differential scanning calorimetry analysis was performed to determine the nature of the drug inside the polymer matrix (molecular dispersion or in crystallized form).Citation41 The differential scanning calorimetry thermograms for the various budesonide samples are shown in . Budesonide melted at approximately 261.58°C ± 0.32°C. The melting peak for PEG5000-DSPE was at 59.91°C ± 0.04°C. The peaks of the physical mixtures of budesonide and PEG5000-DSPE were approximately 256°C ± 2.27°C and 59.82°C ± 0.09°C, respectively, with a sharp decrease in the peak intensity for budesonide. Samples of the lyophilized BUD-SSMs gave only one peak at approximately 58.08°C ± 0.17°C for PEG5000-DSPE, with no endothermic budesonide peak. These results indicated that the physical mixing of budesonide and PEGylated polymer did not affect the structure of the block copolymer. Conversely, BUD-SSMs caused a slight but significant (P < 0.05) downward shift in the endothermic peak of the PEGylated polymer, which indicated that there were physical interactions between budesonide and the PEGylated polymer. The interaction suggested that budesonide was molecularly dispersed in the PEGylated polymer matrix. The disappearance of the endothermic peak of budesonide at low drug concentrations in the BUD-SSMs was due to its complete miscibility in the molten PEGylated polymer. However, at higher drug ratios (physical mixture), only partial miscibility was achieved, and the drug only began to melt at 249°C ± 2.00°C. Similar results were reported in a study of the interaction between PEG6000 and oxazepam.Citation42 Zhang et al showed that the glass transition temperature of paclitaxel-loaded polymeric nanoparticles from differential scanning calorimetry analysis was lower than that of the polymeric blanks, which suggested that the descent peak shift might be an indication that the drug is either molecularly dispersed in the block copolymer or distributed in the block copolymer in an amorphous state.Citation43
Figure 3 Differential scanning calorimetric thermograms of budesonide, PEG5000-DSPE, physical mixture and BUD-SSMs.
Abbreviations: BUD-SSMs, PEG5000-DSPE polymeric micelles containing budesonide; PEG5000-DSPE, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-methoxypoly(ethylene glycol 5000).
Fourier transform infrared spectroscopy
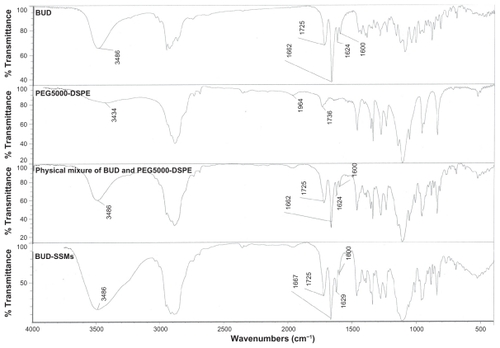
The Fourier transform infrared spectrum of budesonide showed carbonyl stretching bands at 1725 and 1662 cm−1 (). Vibrations in the 1600–1900 cm−1 regions indicated a C=O stretch. The budesonide molecule has two C=O groups, ie, a dihydrobenzoquinone C=O group and an acetyl C=O group. Typically, acetyl C=O and dihydrobenzoquinone C=O groups exhibit stretching bands in the regions of 1700–1900 cm−1 and 1600–1750 cm−1, respectively. Thus, the bands at 1725 cm−1 and 1662 cm−1 in the budesonide spectrum corresponded to the nonconjugated acetyl C=O stretch and conjugated dihydrobenzoquinone C=O groups, respectively.Citation44 In addition, there was a (OH) vibrational band at 3486 cm−1. The physical mixture of budesonide and the PEGylated polymers (PEG5000-DSPE) showed similar spectra. However, the lyophilized BUD-SSMs exhibited a small shift in the conjugated C=O stretching band from 1662 to 1667 cm−1. This shift was caused by an altered environment around the interatomic C=O bonds due to the solvation process in the hydrophobic portion of the PEGylated polymer. Formation of intermolecular hydrogen bonds between the OH group of budesonide and the C=O groups of the PEGylated polymer may explain this shift. Conversely, there was no shift in the nonconjugated C=O of budesonide. In addition, the vibration band of (OH) for both budesonide and the PEGylated polymer changed to a broad peak. Changes in the Fourier transform infrared spectrum of budesonide have also been reported by other investigators. Tajber et al showed that spray-dried budesonide exhibited a shift in the conjugated C=O stretching band when converted from the crystalline to the amorphous form.Citation45 In addition, they found that the OH bands appeared as a broad peak located in the 3300–3700 cm−1 regions. They suggested a possible change in the environment of the conjugated C=O group of budesonide. As a result, no pronounced interactions were identified between budesonide and the PEGylated polymer because the entire relevant spectral band (conjugated and nonconjugated C=O band) had not disappeared, which would indicate that the budesonide molecules were in an amorphous state. This result was confirmed by differential scanning calorimetry analysis.
Figure 4 Fourier transform infrared spectra of budesonide, PEG5000-DSPE, physical mixture, and BUD-SSMs.
Abbreviations: BUD-SSMs, PEG5000-DSPE polymeric micelles containing budesonide; PEG5000-DSPE, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-methoxypoly(ethylene glycol 5000).
Morphology of BUD-SSMs
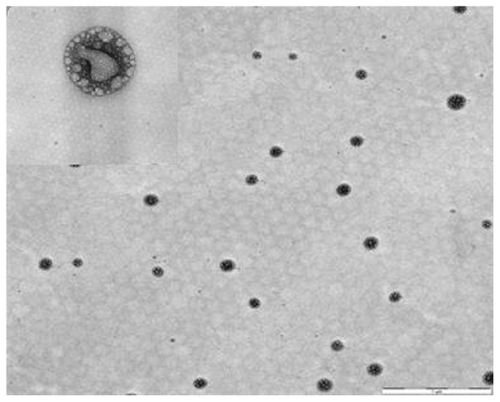
Transmission electron microscopy is commonly used to examine nanoscopic colloids such as SSMs. The mean diameter of these SSMs was measured directly from randomly selected formulations in transmission electron microscopic images. The mean particle size of BUD-SSMs was 104.08 ± 27.30 nm (). These measurements differed significantly from those obtained by photon correlation spectroscopy. This was due to the tendency for particles to aggregate as a result of the drying process during sample preparation for transmission electron microscopy, as also reported by other researchers.Citation46 Closer observation of the BUD-SSMs revealed spherical nanoparticles with bright and dark areas and a core-shell with a multivesicular appearance.
Figure 5 Morphological examinations of BUD-SSMs by transmission electron microscopy.
Note: Scale bar = 1 μm and 50 nm.
Abbreviation: BUD-SSMs, PEG5000-DSPE polymeric micelles containing budesonide.
Solubility and budesonide release from SSMs
The equilibrium dialysis method was used to determine the solubility of poorly water-soluble corticosteroids because it enabled drug equilibration to be achieved. Furthermore, it overcame the inherent wetting problem of these highly hydrophobic powders and minimized errors associated with sample processing.Citation24 The aim of the solubility study was to calculate the concentration that was required for the drugrelease studies under the sink condition.
Budesonide has low solubility in phosphate-buffered saline (21.95 ± 1.15 μg/mL) and this result was consistent with previously reported results (23 μg/mL).Citation24 The sink condition is achieved if the final concentration of the drug in the dissolution medium is less than 20% of the saturation solubility of the drug.Citation47 The final concentration of budesonide after complete release in phosphate-buffered saline was less than 4.39 μg/mL and was consistent with the sink condition.
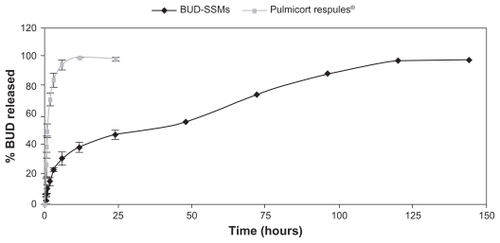
Comparison of the dissolution release profiles for BUD-SSMs and Pulmicort Respules® at the end of the first 6 hours revealed that more than 90% of budesonide was released from Pulmicort Respules®, while less than 35% of budesonide was released from BUD-SSMs, with completion of release within 6 days (). The drug release kinetics () of the various preparations were evaluated by fitting the release data to the zero-order, first-order, and Higuchi equations using the DDSolver software. The most appropriate drug-release model was selected based on its highest adjusted coefficient of determination (R2 adjusted) value and lowest Akaike information criterion. BUD-SSMs best fitted the Higuchi release kinetics (P < 0.05). In contrast, Pulmicort Respules® followed first-order release kinetics (P < 0.05).
Table 1 Kinetic drug release values of different budesonide formulations
Figure 6 Drug-release profiles of BUD-SSMs and Pulmicort Respules®.
Abbreviation: BUD-SSMs, PEG5000-DSPE polymeric micelles containing budesonide.
According to the Higuchi model, two possible mechanisms may be involved in drug release from BUD-SSMs, ie, diffusion of the drug from the micelles and micelle erosion resulting from the degradation of the PEGylated polymer. In order to determine the best possible mechanism, release data for the BUD-SSMs were fitted to the Baker-Lonsdale and Hixson-Crowell models. The Baker-Lonsdale model best describes the release of drug incorporated in a spherical matrix following the diffusion mechanism, while the Hixson-Crowell model best describes the release of drug from the delivery system according to the change in surface area and diameter of the particles over time.Citation48–Citation50 The results showed that the BUD-SSMs most closely followed the Baker-Lonsdale model (P < 0.05) and further supported the postulation that the release of budesonide from SSMs is more consistent with a diffusion mechanism than a matrix erosion mechanism. Derakhshandeh et al reported a similar result with 9-nitrocamptothecin-PLGA-PEG nanoparticles, where drug release followed the diffusion mechanism using the Baker-Lonsdale model.Citation50 Abdulla et al found that the release profile of rifampicin-SSMs followed both the first-order and Higuchi kinetics models.Citation51 However, they only used the correlation coefficient to evaluate the goodness of fit. In our study, there were insignificant differences among the R2 adjusted values following the first-order and Higuchi equation (P > 0.05), while significant (P < 0.05) differences were observed among the Akaike information criterion values for the SSM formulations following the first-order (94.47 ± 1.67) and Higuchi equations (77.82 ± 3.90). Our results indicated that using only a single criterion to evaluate the goodness of fit is not sufficient. When comparing several models, the model with the smallest Akaike information criterion value is a further indication for the best goodness of fit. In addition, from a statistical point of view, the R2 adjusted value is regarded as a more powerful measure than the unadjusted coefficient of R2 determination.Citation52
Many methods can be used to compare in vitro drug-release profiles. For the analysis of variance (ANOVA)-based method, the most suitable approach is to use repeated measures ANOVA because the release data comprise repeated measurements collected over time during the same experiment (ie, time is the repeated factor and percent drug released is the dependent variable). Repeated measures ANOVA is regarded as a more precise method than independent or paired t-tests.Citation53 The one-way ANOVA method compares pairs of values at each time point but ignores the correlation between the dissolution time points (this method treats each time point as independent of the other, which is definitely not the case).Citation54 Repeated measures ANOVA has been used in many studies.Citation53,Citation55–Citation57
The model-dependent method evaluates the dissolution profiles using different model parameters (eg, t50%, release rate constant).Citation53 This method cannot be used to compare the release profiles of formulations having different release mechanisms.Citation58 The estimated model parameters can be compared statistically using the t-test.Citation26,Citation58 The third method for comparison of in vitro release is the model-independent method (using similarity and difference factors), but this was not used in this study because the coefficients of variation (release profile within batch) during the first 15 minutes were higher than 15%.Citation48,Citation54,Citation58 When the results of our study were analyzed, the repeated measures ANOVA and one-way ANOVA showed that there were significant differences between the percent of drug released from Pulmicort Respules® and BUD-SSMs. These results indicated that the corticosteroid-loaded SSMs successfully prolonged the in vitro drug release in comparison with the reference product.
Aerodynamic characterization
To demonstrate the ability to nebulize BUD-SSM formulations, aerodynamic parameters were studied using a next generation impactor connected to a Pari LC nebulizer and comparisons were made with a commercially available product (Pulmicort Respules®). The aerodynamic distribution and characteristics of Pulmicort Respules® using the Pari LC nebulizer have previously been reported.Citation23 The aerodynamic characteristics are shown in . The mass median aerodynamic diameter of BUD-SSMs was significantly smaller than that of the Pulmicort Respules®. These results were due to the formulation effect. Solutions (our formulations) and microsuspensions (Pulmicort Respules®) behave differently during nebulization. The particles in Pulmicort Respules® are 2–3 μm in size, as reported by other researchers, while the particle size of the BUD-SSMs is 20.45 ± 1.64 nm.Citation59 The nebulized suspension will therefore generate larger aerosol droplets (ie, larger mass median aerodynamic diameter values) containing a smaller number of suspended particles (2–3 μm), whereas the aerosolized SSM droplets are smaller (with lower mass median aerodynamic diameter values) and contain many thousands of nanoparticles.Citation60
Table 2 Aerodynamic characteristics of different budesonide formulations
This postulation can be proved by using simple theoretical calculations, as suggested elsewhere.Citation60 The average droplet size produced by Pari LC nebulizers is approximately 4 to 5 μm according to the manufacturer. As such, the maximum particle load for an average 4.5 μm diameter droplet generated by the nebulizers can be estimated using the following calculations:
where d is the diameter of the droplet in μm and π = 3.14.
The volume of an average 4.5-μm droplet produced by the nebulizer will be:
The volume of a 2 μm Pulmicort Respules® particle (the lowest value) will be:
The approximate volume of a 20 nm (0.02 μm) SSM particle will be:
Number of particles per 4.5-μm droplet
The number of 2 μm Pulmicort Respules® particles per 4.5-μm droplet will be:
The number of 20 nm SSM particles per 4.5-μm droplet will be:
Thus, based on the above calculations, it is possible in theory to pack as many as 7.99 × 106 SSM nanoparticles of size 20 nm into a 4.5 μm diameter droplet as opposed to only 7.99 Pulmicort Respules® particles of 4 to 5 μm diameter into a similar droplet. This concept is based on the fact that there is a 100-fold difference in the particle diameter and that the volumetric manipulation is a cubic relationship. Of course, this situation also assumes that the particles are perfectly spherical in shape and does not demonstrate the effect of drug concentration in the formulation. All values for mass median aerodynamic diameter, emitted dose, fine particle fraction, percent of drug remaining in the nebulizer, and percent of drug deposited in the induction port of the BUD-SSMs were significantly superior compared with Pulmicort Respules® due to the particle size and presence of the hydrophilic polymer (PEG) in the outer shell of the SSMs, which induced a repulsive steric interaction between the particles that could effectively decrease the overall adhesive forces, thereby allowing more efficient aerosolization and stabilizing the colloidal suspension in air.Citation29,Citation61,Citation62 The geometric standard deviation values of BUD-SSMs were significantly (P < 0.05) higher than those of the Pulmicort Respules® due to the effect of operating the cascade impactor at ambient temperature, which affected smaller droplets more than the larger droplets generated by the nebulizer, and produced a wider spread of geometric standard deviation values, as previously reported.Citation63 The improved aerodynamic performance of the BUD-SSMs leads to a decrease in the therapeutic dose and a reduction in systemic side effects.
Inhibitory duration of inflammatory cell infiltration in airways following BUD-SSMs and Pulmicort Respules®
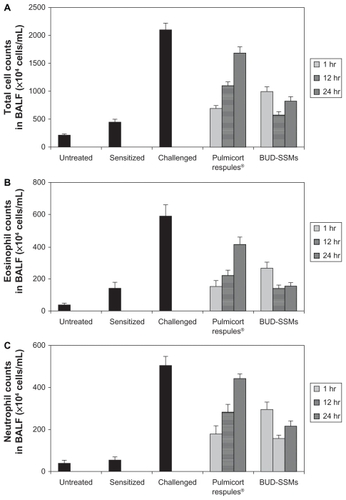
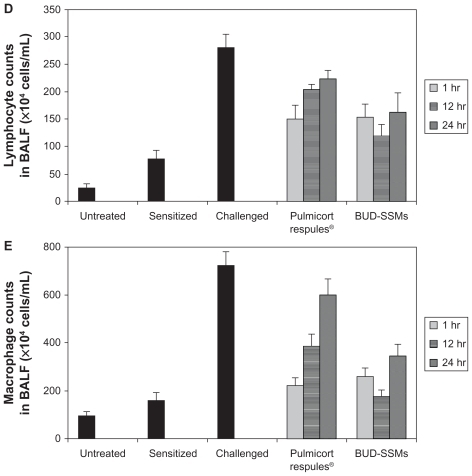
Ovalbumin-sensitized animals are commonly used as an asthma/chronic obstructive pulmonary disease model, because recurrent exposure of ovalbumin in actively immunized rats induces production of ovalbumin-specific IgE, airway hyperresponsiveness, peribronchial inflammation, and an increase in eosinophilia, neutrophilia, lymphocytes, and macrophages in bronchioalveolar lavage fluid.Citation64 Challenge with ovalbumin led to a significant inflammatory response (P < 0.05) in the airways of sensitized Sprague-Dawley rats according to changes in total and differential cell counts of inflammatory cells in the bronchioalveolar lavage fluid in comparison with the untreated and sensitized groups (). Treatment with Pulmicort Respules® or with BUD-SSMs one hour before challenge (1 mg/kg) reduced inflammatory cell infiltration (P < 0.05) in comparison with the challenged group (). However, this inhibitory effect was significantly attenuated (P < 0.05) when the same dose of Pulmicort Respules® was administered 12 or 24 hours before the final challenge in comparison with the BUD-SSM formulations. Although the inhibitory effect was reduced, the results differed significantly from those of the positive group (P < 0.05). This is because the inhaled budesonide formed its fatty acid ester in the lung (budesonide-21 oleate), hence prolonging its half-life in the lung to 18–20 hours and consequently showing a prolonged duration of action, compared with other inhaled steroids.Citation30,Citation65 Treatment with BUD-SSMs led to an extreme and prolonged (P < 0.05) inhibitory effect compared with Pulmicort Respules® at 12 and 24 hours before challenge (). It should be noted that the antigen-induced cell infiltration in the airways was not affected by the placebo formulation (PEG5000-DSPE), which was similarly prepared by coprecipitation and reconstitution, and administered as a rehydrated powder ready for aerosolization, demonstrating that the polymer had no inhibitory effect in this animal model (data not shown).
Table 3 Inhibitory duration of inflammatory cell infiltration after intratracheal instillation of different budesonide formulations
Figure 7 Total cell counts (A), eosinophil counts (B), neutrophil counts (C), lymphocyte counts (D), and macrophage counts (E) in bronchoalveolar lavage fluid in each experimental group of animals.
Notes: Error bar represents the standard error of the mean. for n = 6. Untreated, no sensitization or challenge; sensitized (negative control), multiple antigen sensitization, no challenge; challenged (positive control), multiple antigen sensitization with challenge.
Scheme 1 Sensitization, challenge and treatment protocol.
Because budesonide undergoes extensive hepatic inactivation (first-pass metabolism) by virtue of its molecular design, with a half-life of about 2.8 hours, it is likely that this inhibitory effect () resulted from the local effect of budesonide in the lung.Citation66 In addition, pulmonary absorption differs considerably due to the physicochemical properties of the drug, such as solubility.Citation67,Citation68 After particles deposit on the surface of the airways, they are wetted and dissolve in the airway lining fluid. Inhaled drug particles with low solubility (such as budesonide in Pulmicort Respules®) take a substantial period of time for solubilization and partitioning between the phases of the airway lining, and are preferentially cleared from the airways by mucociliary transport and phagocytosis. Inhaled drug particles with high solubility (such as budesonide in SSMs) enter into and dissolve in the airway lining fluid more rapidly, and are therefore less susceptible to mucociliary clearance.Citation69–Citation71 Therefore, the microsuspension (Pulmicort Respules®) is transported out of the lungs via cilia, whereas nanoparticles can adhere onto the mucosal surface for a longer period and in that way increase the residence time of the drug in the lung.Citation72 Existing pharmacokinetic-pharmacodynamic studies have demonstrated that the pulmonary residence time of inhaled glucocorticoids defines their anti-inflammatory action in the lung, providing the opportunity for sustained-release formulations to be beneficial.Citation73 A recent pharmacokinetic study of paclitaxel + PEG5000-DSPE found a significantly higher area under the curve for paclitaxel in rat lung with a low systemic concentration (plasma, liver, and spleen) compared with its area under the curve following intravenous administration due to the localization of chemotherapy to the lungs, which in turn avoids unwanted side effects. The same study found that paclitaxel + PEG5000-DSPE produced similar drug levels to that of paclitaxel in the first hour after intratracheal inhalation, but that higher paclitaxel concentrations were found in the lungs at 12 hours compared with paclitaxel at the same time.Citation37 These results confirmed the impact of SSMs as sustained-release formulations, from which budesonide is slowly released over time. As a result, BUD-SSMs inhibited airway inflammation in the asthma/chronic obstructive pulmonary disease model more effectively than the commercial dosage form. The SSM system for budesonide would help to prevent the side effects associated with systemic exposure to budesonide by decreasing the dose due to the prolonged duration of action. The new BUD-SSM system might thus be beneficial as a treatment for asthma, chronic obstructive pulmonary disease, and other inflammatory airway diseases.
Conclusion
We successfully incorporated budesonide into SSMs of the PEGylated polymer, PEG5000-DSPE, with appropriate physicochemical properties. The in vitro release studies indicated the sustained-release potential of BUD-SSMs. Furthermore, the BUD-SSMs showed superior aerodynamic characteristics compared with Pulmicort Respules®, being fine enough to be inhaled and reach into the deep lung tissue. The pharmacodynamic study showed a significantly longer duration of inhibition of inflammatory cell infiltration in the airways of antigen-induced asthmatic rats which was prolonged for up to 24 hours, compared with Pulmicort Respules®. In general, the SSM system has the ability to keep budesonide at or near the desired pharmacological site of action and to provide selective and prolonged activity in the lung, thereby reducing systemic toxicity. Overall, the in vitro and in vivo studies of BUD-SSMs resulted in a good understanding of the potential use of SSMs as a pulmonary delivery system.
Acknowledgments
This work was supported by a grant from the Universiti Sains Malaysia (RU Grant 815001). MNS and SAM gratefully acknowledge the Universiti Sains Malaysia for granting them a postgraduate student fellowship to help fund this research.
Disclosure
The authors report no conflicts of interest in this work.
References
- ZaruMMourtasSKlepetsanisPFaddaAMAntimisiarisSGLiposomes for drug delivery to the lungs by nebulizationEur J Pharm Biopharm200767365566617540552
- JaspartSBertholetPPielGDogneJMDelattreLEvrardBSolid lipid microparticles as a sustained release system for pulmonary drug deliveryEur J Pharm Biopharm2007651475616962749
- BaileyMMBerklandCJNanoparticle formulations in pulmonary drug deliveryMed Res Rev200929119621218958847
- CookROPannuRKKellawayIWNovel sustained release microspheres for pulmonary drug deliveryJ Control Release20051041799015866336
- FuJFiegelJKraulandEHanesJNew polymeric carriers for controlled drug delivery following inhalation or injectionBiomaterials200223224425443312219833
- GrenhaAGraingerCIDaileyLAChitosan nanoparticles are compatible with respiratory epithelial cells in vitroEur J Pharm Sci2007312738417408932
- SmolaMVandammeTSokolowskiANanocarriers as pulmonary drug delivery systems to treat and to diagnose respiratory and non respiratory diseasesInt J Nanomedicine20083111918488412
- El-SherbinyIMSmythHDCBiodegradable nano-micro carrier systems for sustained pulmonary drug delivery: (I) self-assembled nanoparticles encapsulated in respirable/swellable semi-IPN microspheresInt J Pharm20103951–213214120580794
- GrenhaASeijoBRemunan-LopezCMicroencapsulated chitosan nanoparticles for lung protein deliveryEur J Pharm Sci2005254–542743715893461
- GaucherGDufresneMHSantVPKangNMaysingerDLerouxJCBlock copolymer micelles: preparation, characterization and application in drug deliveryJ Control Release20051091–316918816289422
- MikhailASAllenCBlock copolymer micelles for delivery of cancer therapy: transport at the whole body, tissue and cellular levelsJ Control Release2009138321422319376167
- RiessGMicellization of block copolymersProg Polym Sci200328711071170
- GaoZLukyanovANSinghalATorchilinVPDiacyllipid-Polymer Micelles as Nanocarriers for Poorly Soluble Anticancer DrugsNano Lett200229979982
- TorchilinVPStructure and design of polymeric surfactant-based drug delivery systemsJ Control Release2001732–313717211516494
- JonesMCLerouxJCPolymeric micelles – a new generation of colloidal drug carriersEur J Pharm Biopharm199948210111110469928
- SezginZYükselNBaykaraTPreparation and characterization of polymeric micelles for solubilization of poorly soluble anticancer drugsEur J Pharm Biopharm200664326126816884896
- LimSBRubinsteinIÖnyükselHFreeze drying of peptide drugs self-associated with long-circulating, biocompatible and biodegradable sterically stabilized phospholipid nanomicellesInt J Pharm20083561–234535018289811
- SmolaMVandammeTSokolowskiANanocarriers as pulmonary drug delivery systems to treat and to diagnose respiratory and non respiratory diseasesInt J Nanomedicine20083111918488412
- VermehrenCJørgensenKSchiffelersRFrokjaerSActivity of mammalian secreted phospholipase A(2) from inflammatory peritoneal fluid towards PEG-liposomes. Early indicationsInt J Pharm20012141–2939811282244
- KooOMRubinsteinIOnyukselHCamptothecin in sterically stabilized phospholipid micelles: a novel nanomedicineNanomedicine200511778417292061
- CesurHRubinsteinIPaiAÖnyükselHSelf-associated indisulam in phospholipid-based nanomicelles: a potential nanomedicine for cancerNanomedicine20095217818319071064
- ZhangLHuYJiangXYangCLuWYangYHCamptothecin derivative- loaded poly(caprolactone-co-lactide)-b-PEG-b-poly(caprolactone-co-lactide) nanoparticles and their biodistribution in miceJ Control Release200496113514815063036
- SahibMNDarwisYKhiangPKTanYTFAerodynamic characterization of marketed inhaler dosage forms: High performance liquid chromatography assay method for the determination of budesonideAfr J Pharm Pharmacol2010412878884
- DaviesNMFeddahMRA novel method for assessing dissolution of aerosol inhaler productsInt J Pharm20032551–217518712672613
- ZhangYHuoMZhouJDDSolver: an add-in program for modeling and comparison of drug dissolution profilesAAPS J201012326327120373062
- CostaFOSousaJJPaisAAFormosinhoSJComparison of dissolution profiles of ibuprofen pelletsJ Control Release200389219921212711444
- SahibMNDarwisYKhiangPKFung TanYTAerodynamic characterization of beclomethasone dipropionate from beclate-50 inhaler® by HPLC-UVJ Liq Chromatogr Relat Technol2011348613621
- WigginsNAThe Development of a Mathematical Approximation Technique to Determine the Mass Median Aerodynamic Diameter (MMAD) and Geometric Standard Deviation (GSD) of Drug Particles in an Inhalation Aerosol SpratDrug Dev Ind Pharm1991171419711986
- YamamotoHKunoYSugimotoSTakeuchiHKawashimaYSurface-modified PLGA nanosphere with chitosan improved pulmonary delivery of calcitonin by mucoadhesion and opening of the intercellular tight junctionsJ Control Release2005102237338115653158
- OnoueSSatoHKawabataYMizumotoTHashimotoNYamadaSIn vitro and in vivo characterization on amorphous solid dispersion of cyclosporine A for inhalation therapyJ Control Release20091381162319376169
- GreenCKnightJPreciousSSimpkinSKetamine alone and combined with diazepam or xylazine in laboratory animals: a 10 year experienceLab Anim19811521631707278122
- AnderssonSEZackrissonCBehrensKEffect of allergen provocation on inflammatory cell profile and endothelin-like immunoreactivity in guinea-pig airwaysAllergy19955043493587573819
- SakagamiMKinoshitaWSakonKSatoJMakinoYMucoadhesive beclomethasone microspheres for powder inhalation: their pharmacokinetics and pharmacodynamics evaluationJ Control Release2002801–320721811943399
- ZhangJXHansenCBAllenTMBoeyABochRLipid-derivatized poly(ethylene glycol) micellar formulations of benzoporphyrin derivativesJ Control Release2003862–332333812526828
- RijckenCJSogaOHenninkWENostrumCFTriggered destabilisation of polymeric micelles and vesicles by changing polymers polarity: an attractive tool for drug deliveryJ Control Release2007120313114817582642
- YangWPetersJIWilliamsROIIIInhaled nanoparticles – a current reviewInt J Pharm20083561–223924718358652
- GillKKNazzalSKaddoumiAPaclitaxel loaded PEG5000-DSPE micelles as pulmonary delivery platform: Formulation characterization, tissue distribution, plasma pharmacokinetics, and toxicological evaluationEur J Pharm Biopharm57201179227628421575719
- FidlerIJRazAFoglerWEKirshRBugelskiPPosteGDesign of liposomes to improve delivery of macrophage-augmenting agents to alveolar macrophagesCancer Res19804012446044667002293
- ZhangJXLiXJQiuLYIndomethacin-loaded polymeric nanocarriers based on amphiphilic polyphosphazenes with poly (N-isopropylacrylamide) and ethyl tryptophan as side groups: Preparation, in vitro and in vivo evaluationJ Control Release2006116332232917109985
- AliabadiHMElhasiSMahmudAGulamhuseinRMahdipoorPLavasanifarAEncapsulation of hydrophobic drugs in polymeric micelles through co-solvent evaporation: The effect of solvent composition on micellar properties and drug loadingInt J Pharm20073291–215816517008034
- MuLFengSSFabrication, characterization and in vitro release of paclitaxel (Taxol) loaded poly (lactic-co-glycolic acid) microspheres prepared by spray drying technique with lipid/cholesterol emulsifiersJ Control Release200176323925411578739
- AriasMJMoyanoJRGinésJMStudy by DSC and HSM of the oxazepam-PEG 6000 and oxazepam-D-mannitol systems: Application to the preparation of solid dispersionsThermochim Acta19983211–23341
- ZhangXJacksonJKBurtHMDevelopment of amphiphilic diblock copolymers as micellar carriers of taxolInt J Pharm19961321–2195206
- AliHREdwardsHGKendrickJMunshiTScowenIJVibrational spectroscopic study of budesonideJ Raman Spectrosc2007387903908
- TajberLCorriganDOCorriganOIHealyAMSpray drying of budesonide, formoterol fumarate and their composites – I. Physicochemical characterisationInt J Pharm20093671–2798518929631
- FalamarzianALavasanifarAOptimization of the hydrophobic domain in poly(ethylene oxide)-poly([var epsilon]-caprolactone) based nano-carriers for the solubilization and delivery of Amphotericin BColloids Surf B Biointerfaces201081131332020674292
- MuthuMSinghSPoly (D, L-Lactide) nanosuspensions of risperidone for parenteral delivery: formulation and in-vitro evaluationCurr Drug Deliv200961626819418957
- CostaPSousa LoboJMModeling and comparison of dissolution profilesEur J Pharm Sci200113212313311297896
- DashSMurthyPNNathLChowdhuryPKinetic modeling on drug release from controlled drug delivery systemsActa Pol Pharm201067321722320524422
- DerakhshandehKSoheiliMDadashzadehSSaghiriRPreparation and in vitro characterization of 9-nitrocamptothecin-loaded long circulating nanoparticles for delivery in cancer patientsInt J Nanomedicine2010546347120957168
- AbdullaJMTanYTDarwisYRehydrated lyophilized rifampicin-loaded mPEG-DSPE formulations for nebulizationAAPS Pharm Sci Tech2010112663671
- FieldAPDiscovering statistics using SPSS: (and sex and drugs and rock ‘n’ roll)3London, UKSAGE publications Ltd2009
- YukselNKanikAEBaykaraTComparison of in vitro dissolution profiles by ANOVA-based, model-dependent and -independent methodsInt J Pharm20002091–2576711084246
- ChowSCKiFYStatistical comparison between dissolution profiles of drug productsJ Biopharm Stat1997722412589136067
- PolliJERekhiGSAugsburgerLLShahVPMethods to compare dissolution profiles and a rationale for wide dissolution specifications for metoprolol tartrate tabletsJ Pharm Sci19978666907009188051
- GonjariIDKarmarkarABHosmaniAEvaluation of in vitro dissolution profile comparison methods of sustained release tramadol hydrochloride liquisolid compact formulations with marketed sustained release tabletsDigest J Nanomaterials Biostructures20094651661
- AdamsECoomansDSmeyers-VerbekeJMassartDLApplication of linear mixed effects models to the evaluation of dissolution profilesInt J Pharm20012261–210712511532575
- O’HaraTDunneAButlerJDevaneJA review of methods used to compare dissolution profile dataPharm Sci and Technolo Today199815214223
- VaghiABergELiljedahlSSvenssonJOIn vitro comparison of nebulised budesonide (Pulmicort Respules®) and beclomethasone dipropionate (Clenil® per Aerosol)Pulm Pharmacol Ther200518215115315649857
- OstranderKDBoschHWBondanzaDMAn in-vitro assessment of a NanoCrystal beclomethasone dipropionate colloidal dispersion via ultrasonic nebulizationEur J Pharm Biopharm199948320721510612031
- DaileyLASchmehlTGesslerTNebulization of biodegradable nanoparticles: impact of nebulizer technology and nanoparticle characteristics on aerosol featuresJ Control Release200386113114412490379
- CoombesAGTaskerSLindbladMBiodegradable polymeric microparticles for drug delivery and vaccine formulation: the surface attachment of hydrophilic species using the concept of poly(ethylene glycol) anchoring segmentsBiomaterials19971817115311619259512
- MitchellJNewmanSChanHKIn vitro and in vivo aspects of cascade impactor tests and inhaler performance: a reviewAAPS Pharm Sci Tech200784E110
- DawkinsPAStockleyRAAnimal models of chronic obstructive pulmonary diseaseThorax2001561297297711713362
- LeungSYWilliamsASNathPDose-dependent inhibition of allergic inflammation and bronchial hyperresponsiveness by budesonide in ovalbumin-sensitised Brown-Norway ratsPulm Pharmacol Ther20082119810417331766
- Miller-LarssonAJanssonPRunstromABrattsandRProlonged airway activity and improved selectivity of budesonide possibly due to esterificationAm J Respir Crit Care Med20001624 Pt 11455146111029361
- TrondeANordénBMarchnerHWendelAKLennernäsHBengtssonUHPulmonary absorption rate and bioavailability of drugs in vivo in rats: structure-absorption relationships and physicochemical profiling of inhaled drugsJ Pharm Sci20039261216123312761811
- TrondeANordénBJeppssonABDrug absorption from the isolated perfused rat lung-correlations with drug physicochemical properties and epithelial permeabilityJ Drug Target2003111617412852442
- GeiserMGerberPMayeIIm HofVGehrPRetention of Teflon particles in hamster lungs: a stereological studyJ Aerosol Med2000131435510947323
- JohnJWollmerPDahlbackMLutsAJonsonBTidal volume and alveolar clearance of insoluble particlesJ Appl Physiol19947625845888175567
- LayJCStangMRFisherPEYankaskasJRBennettWDAirway retention of materials of different solubility following local intrabronchial deposition in dogsJ Aerosol Med200316215316612823909
- JacobsCMüllerRHProduction and characterization of a budesonide nanosuspension for pulmonary administrationPharm Res200219218919411883646
- CerasoliFJrDeveloping the ideal inhaled corticosteroidChest20061301 Suppl54S64S16840368