Abstract
Mithramycin (MTM), a natural product of soil bacteria from the Streptomyces genus, displays potent anticancer activity but has been limited clinically by severe side effects and toxicities. Engineering of the MTM biosynthetic pathway has produced the 3-side-chain-modified analogs MTM SK (SK) and MTM SDK (SDK), which have exhibited increased anticancer activity and improved therapeutic index. However, these analogs still suffer from low bioavailability, short plasma retention time, and low tumor accumulation. In an effort to aid with these shortcomings, two nanoparticulate formulations, poly(ethylene glycol)-poly(aspartate hydrazide) self-assembled and cross-linked micelles, were investigated with regard to the ability to load and pH dependently release the drugs. Micelles were successfully formed with both nanoparticulate formulations of each drug analog, with an average size of 8.36 ± 3.21 and 12.19 ± 2.77 nm for the SK and SDK micelles and 29.56 ± 4.67 nm and 30.48 ± 7.00 nm for the SK and SDK cross-linked micelles respectively. All of the drug-loaded formulations showed a pH-dependent release of the drugs, which was accelerated as pH decreased from 7.4 to 5.0. The micelles retained biological activity of SK and SDK entrapped in the micelles, suppressing human A549 lung cancer cells effectively.
Introduction
Mithramycin (MTM), an aureolic acid-type polyketide antibiotic produced naturally by soil bacteria from the Streptomyces genus, exhibits anticancer activity by cross-linking GC-rich DNA and shutting down proto-oncogenes, particularly those triggered by Sp (specificity protein) transcription factors, eg, Sp1 and Sp3.Citation1–Citation5 Overexpression of Sp1 has been observed in several cancers and has been linked to the control of cell growth, survival, and differentiation, all of which are important in cancer progression.Citation6–Citation8 MTM has been used clinically in the past to treat testicular cancer, Paget’s bone disease, and hypercalcemia but has been severely limited by its poor bioavailability and toxic side effects such as hepatic, gastrointestinal, bone marrow, and renal toxicities.Citation9–Citation13 In order to address these issues, extensive combinatorial biosynthesis has been performed on the MTM pathway and has resulted in several novel analogs.Citation14–Citation16 The present authors have shown that the inactivation of the mtmW gene, which codes for the last enzyme in the MTM biosynthetic pathway, yields the most favorable of the new analogs, MTM SK (SK) and MTMSDK (SDK).Citation15,Citation16 Both SK and SDK exhibited higher anticancer activity than the native MTM,Citation15,Citation16 yet their short plasma retention time and low accumulation in tumors remained to be improved.
A drug-delivery system is a technique that ensures drug molecules are delivered to disease lesions at the right amount and timing by using drug carriers. Nanoparticles are widely used as drug carriers because large molecules (>50 kDa) can avoid renal clearance and circulate in the body for prolonged time in comparison to small molecules.Citation17 Tumors are characterized with leaky blood vessels and immature lymphatic drainage, which allow nanoparticles (20–200 nm) to accumulate in tumor tissues.Citation18 Nanoparticles take advantage of both prolonged plasma retention time and preferential tumor accumulation to deliver drug payloads to tumors preferentially.Citation19 Liposomes, dendrimers, and polymer micelles are nanoparticulate formulations used widely for tumor-preferential delivery of therapeutic agents. Among these formulations, polymer micelles offer a versatile platform that can be prepared through self-assembling of block copolymers in aqueous solutions.Citation20 Self-assembled polymer micelles possess a core compartment enveloped by a hydrophilic shell. The core-shell structure protects drug payloads from precipitation, protein adsorption, and enzymatic degradation in the body. The polymer micelles can dissociate as the core becomes unstable under conditions accompanied by dilution, drug release, or polymer degradation.Citation21 Polymer micelles can be stabilized by cross-linking in the core or shell to avoid premature micelle dissociation during tumor-preferential drug delivery.Citation22,Citation23
In addition to tumor-preferential drug delivery, controlled drug release is another important factor for nanoparticles to maximize therapeutic efficacy of drug payloads. Drug binding linkers are normally designed to degrade in response to pH, enzymatic activity, light, or heat.Citation24 The hydrazone bond, involved in many biomolecular events, is stable at physiological Ph (7.4), yet is cleaved in acidic conditions (pH < 7.0) in a pH-dependent manner. Such a unique degradation pattern has been employed to design pH-controlled drug-delivery systems that can trigger drug release in intracellular lysosomal compartments (pH 5.0), following the cellular uptake of drug carriers.Citation25 Tumor tissues are also acidic (pH 6.5–7.0) due to the inefficient glucose consumption that massively produces lactic acids.Citation26 Low pH in tumors is also attributed to high flux at the glyceraldehydes- 3-phosphate dehydrogenase step of glycolysis.Citation27 These two factors contribute to tumor acidosis in combination. The authors of this paper previously demonstrated that polymer micelles to which drug molecules were conjugated through acid-sensitive hydrazone linkers could accelerate drug release in cancer cells and reduce systemic toxicity in animal tumor models.Citation28 The hydrazone is a chemical bond between ketone and hydrazide groups, and thus, drug molecules containing ketone groups are advantageous to design pH-sensitive drug-delivery systems.Citation29–Citation31
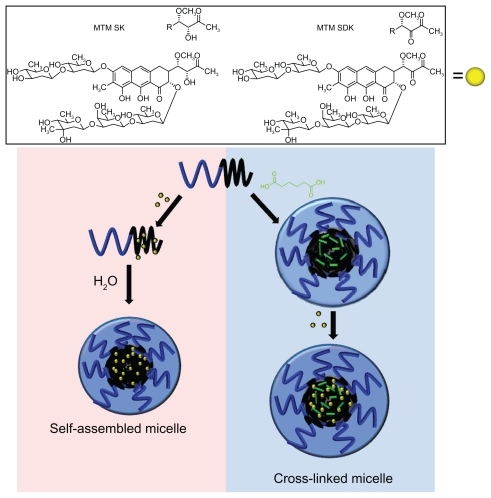
shows the likely chemical structures of MTM analogs (SK and SDK) and two nanoparticulate formulations (self-assembled and cross-linked polymer micelles) used in this study. SK and SDK possess one and two ketone groups, respectively, in their 3-side chains, plus an additional keto group in the tricyclic ring system. At least one of the ketone groups is subsequently bound to hydrazide groups of poly(ethylene glycol)-poly(aspartate hydrazide) [PEG-p(Asp- Hyd)] block copolymers to prepare drug-conjugated polymer micelles. The acid-labile hydrazone bonds between the block copolymers and MTM analogs are used to accelerate drug release in acidic conditions corresponding to tumor tissues in vivo. Self-assembled and cross-linked micelles are compared to investigate the influence of micelle stability on drug-release patterns. A549 cell lines (human nonsmall cell lung cancer) are used to test biological activity of nanoparticulate MTM formulations. This article, therefore, provides an insight and understanding of the design, development, and properties of MTM nanoparticulate formulations for effective cancer therapy.
Figure 1 Self-assembled and cross-linked polymer micelles as nanoparticulate formulations for MTM analogs.
Abbreviation: MTM, mithramycin.
Materials and methods
Materials
L-aspartic acid β-benzyl ester (BLA), triphosgene, anhydrous hydrazine, N,N′-diisopropylcarbodiimide (DIC), N-hydroxysuccinimide (NHS), anhydrous tetrahydrofuran (THF), anhydrous hexane, anhydrous ethyl ether, anhydrous dimethyl sulfoxide (DMSO), DMSO (molecular biology grade, ≥99.9%), DMSO-d6, resazurin sodium salt, acetate buffer solution, phosphate buffer solution, adipic acid, and sodium hydroxide (NaOH) were purchased from Sigma- Aldrich (St Louis, MO). α-Methoxy-ω-amino PEG (5 kDa) was purchased from NOF Corporation (Tokyo, Japan). Regenerated cellulose dialysis bags with molecular weight cutoff (MWCO) 6–8 kDa and Slide-A-Lyzer G2 dialysis cassettes with MWCO 10 kDa, methanol (MeOH), acetonitrile (ACN), celite, C18 RP silica gel, tryptic soy broth (TSB), LB broth, Difco™ agar, sucrose, potassium sulfate, magnesium chloride, glucose, casamino acids, yeast extract, MOPS (3-morpholinopropane-1-sulfonic acid), and trace elements were purchased from Fisher Scientific (Ottawa, ON). Streptomyces argillaceus ATCC 12956 was purchased from ATCC (Manassas, VA). Spectra/Por 6 dialysis tubing with MWCO 50 kDa was purchased from Spectrum Labs (Fort Lauderdale, FL).
Biosynthesis of MTM analogs
SK and SDK were produced by an adapted procedure reported previously.Citation15 Cells of S. argillaceus M7W1 were plated on R5A agar and grown for 4 days. After 4 days, the spores were used to inoculate 100 mL of TSB for 24 hours in an orbital shaker at 28°C, 250 rpm. After 24 hours, 4 mL of the TSB culture was used to inoculate 40, 100 mL flasks of R5A media. The cultures were grown for 3 days at 28°C, 250 rpm. After 3 days the cells were harvested and 50 g/L celite was added to the culture. The cells were removed by filtration, re-dissolved in MeOH and sonicated for 1 hour to lyse the cells. After sonication the cells were filtered again and the MeOH was removed from the filtrate, reconstituted in water, and loaded onto a 5 cm × 12 cm C18 RP column equilibrated with 10 column volumes of water. The column was washed with 10% ACN in water, followed by 20% ACN in water, and subsequently eluted with 100% ACN. The filtrate from the initial removal of the cells was also loaded onto the C18 column, followed by the same fractionation procedure. The samples were dried and re-dissolved in 80% MeOH and water. The compounds were further isolated by semi-preparative high-performance liquid chromatography (HPLC).
Block copolymer synthesis
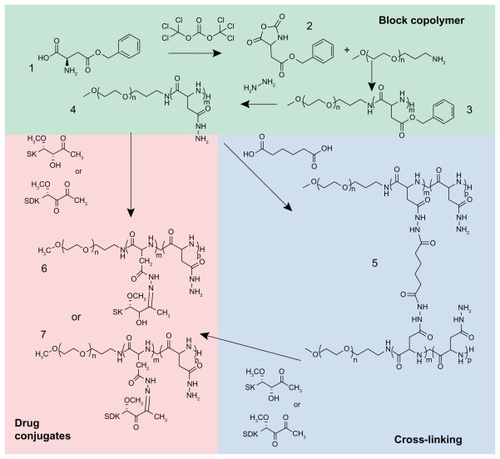
Block copolymers were synthesized as reported previously ().Citation22 In brief, BLA-N-carboxy anhydride (BLA-NCA) was prepared as a monomer by adding 2.88 g of triphosgene to 5.0 g β-benzyl-L-aspartate in 100 mL of dry THF at 45°C until the solution became clear. Anhydrous hexane was added to the solution slowly to form BLA-NCA crystal, which was recrystallized at −20°C. PEG was used as a macroinitiator to conduct the ring-opening polymerization of BLA-NCA, which produced PEG-(β-benzyl L-aspartate) [PEG-PBLA]. BLA-NCA and PEG were reacted in anhydrous DMSO at 50 mg/mL under N2 for 2 days at 45°C. The PEG-PBLA block copolymers were isolated from the reaction mixture by precipitation with anhydrous ethyl ether and subsequently freeze drying. PEG-PBLA (100 mg/mL) was reacted with 20-fold equivalent hydrazine in DMSO (40°C) for 1 hour to prepare PEG-p(Asp-Hyd) block copolymers. The materials were collected by ether precipitation and freeze drying. The block copolymer compositions were determined by proton nuclear magnetic resonance spectroscopy (1H-NMR), which is described as X-Y denoting the molecular weight of PEG × 10−3 (X) and the number of aspartate repeating units (Y), respectively.
Figure 2 Synthesis of block copolymers, cross-linked block copolymers, and drug conjugates.
Synthesis of self-assembled polymer micelles
MTM analogs were conjugated to the PEG-p(Asp-Hyd) block copolymers by adding both SK and SDK to the block copolymer at 50 weight % in DMSO. The reaction was allowed to proceed at room temperature for 72 hours, shaking at 500 rpm. The materials were collected and purified by repeating ether precipitation. The drug-polymer conjugates were then re-dissolved in deionized water to form self-assembled micelles. The drug-loaded micelles were collected by freeze drying.
Synthesis of cross-linked polymer micelles
Cross-linked polymer micelles were created by cross-linking PEG-p(Asp-Hyd) 5–40 with adipic acid. PEG-p(Asp-Hyd) 5–40 was dissolved in DMSO to a final concentration of 100 mg/mL. Adipic acid, NHS, and DIC were combined in DMSO to a mole ratio of 0.5:4:4 times the hydrazide groups of the block copolymer, respectively, and the reaction mixture was added to the block copolymer. The cross-linking reaction was conducted for 72 hours, stirring at room temperature. The reaction solution was then dialyzed against DMSO, followed by 50% DMSO, and finally deionized water. The solution was freeze dried to collect the purified materials. The cross-linked micelles were characterized by gel permeation chromatography (GPC) and 1H-NMR. MTM analogs were conjugated to cross-linked micelles at a 50 weight % in DMSO, followed by ether precipitation and freeze drying.
Micelle characterization
Drug loading efficiency in polymer micelles was determined by quantifying the absorbance of MTM analogs at 410 nm. Particle size of the micelles was determined by dynamic light scattering (DLS) on a Zetasizer Nano-ZS (Malvern Instruments Ltd, Worcestershire, UK) equipped with a He-Ne laser light source and 173° angle scattered light collection configuration.
Drug-release experiments
Drug-release patterns of the self-assembled or cross-linked polymer micelles were evaluated by using dialysis cassettes (Slide-A-Lyzer G2, MWCO 10 kDa). Dialysis cassettes containing 2 mL samples were placed in 4 L of phosphate buffer solution (10 mM, pH 7.4 or 5.0). The temperature was maintained at 37°C. An aliquot of 150 μL was taken out of each dialysis cassette at the time intervals of 0.0, 0.5, 1.0, 3.0, 6.0, 24.0, 48.0, and 72.0 hours. The aliquots taken at each time point were analyzed by ultraviolet-visible (UV-Vis) absorbance at 410 nm. All experiments were performed in triplicate.
Cell toxicity assays
The cytotoxic effects of four MTM nanoparticulate formulations were investigated in comparison to free drugs. A549 cells were cultured as specified from ATCC at 37°C, 5% CO2. The cells were seeded on a 96-well plate (5000 cells/well) and allowed to adhere for 24 hours. After 24 hours, growth media were replaced with fresh media containing samples at differing concentrations. 1% DMSO (biotechnological grade) was used as a control vehicle to prepare free drug formulations for SK and SDK. Self-assembled and cross-linked micelles were used as nanoparticulate formulations for SK and SDK. The cells were exposed to samples for 72 hours total (n = 8). Cell viability was assessed using a resazurin assay that indicates mitochondrial metabolic activity in live cells. A volume of 10 μL of a 1 mM resazurin solution in phosphate buffered saline was added to the vehicle- and drug-treated cells at the end of the treatment period. Cell viability was determined 3 hours later by reading the fluorescence at 560 nm (excitation wavelength)/590 nm (emission wavelength). The fluorescence signals were quantified using a Spectramax M5 plate reader (Molecular Devices, Sunnyvale, CA) equipped with SoftMax® Pro (Molecular Devices) software. Cytotoxicity was determined by calculating the half maximal inhibitory concentration (IC50) of each sample.
Results
Biosynthesis of SK and SDK
SK and SDK were successfully produced by the S. argillaceus M7W1, and isolated through an adapted procedure developed previously.Citation15 S. argillaceus cells of the M7W1 mutant strain were plated on R5A agar and allowed to grow until spores were formed. After 2 days, colonies began to appear on the plates; after 3 days, spores began to form; and after 4 days, the majority of the cells were present as spores. Spores from the plate were used to inoculate an initial culture of TSB media. The cells grew quickly in TSB media and were ready to transfer to R5A media after 24 hours. Once transferred to the R5A growth media, the cells were able to produce the drug analogs. Initially the media was adjusted to pH 6.85; however, after the observation of the production, the pH was adjusted to 7.0. The culture was monitored for the emergence of the MTM analogs using HPLC. After 24 hours, the presence of MTM analogs or precursors was not readily detected. After 48 hours, the MTM analogs began to appear; however, a large population of pre-MTM compounds was still observed. After 72 hours, the MTM compounds were the dominant compounds in the culture so the production was stopped. Much of the population of the MTM analogs was excreted in the culture media, thus the initial step of the isolation was to separate out the cells from the media by the addition of a celite cell-binding resin and subsequent filtration. Once the cells had been removed, the filtrate was passed over a C18 RP silica gel column to collect the compounds of interest. The initial washes of 10% and 20% ACN allowed many of the byproducts to be removed from the sample. Further fractionation of the culture on the C18 RP column did not aid in the purification of the individual MTM analogs, so it was decided to elute all the MTM compounds together to minimize sample loss and excessive dilution. The cells removed in the initial filtration step did still contain a substantial amount of SK and SDK, thus to take full advantage of the production of the analogs the cultured cells were covered in MeOH and sonicated to lyse the cells and release the drugs. Cellular debris was removed by again filtering the solution which was then able to be dried, reconstituted in water, and loaded on the C18 RP column. After the same wash and elution steps were performed, a similar solution to the initial cell filtrate was obtained; however, in a smaller quantity. The samples could then be loaded and the individual MTM analogs separated by using semi-preparative HPLC. The individual peaks were collected and analyzed for purity using HPLC-mass spectrometry. Both the SK and SDK products were collected at >95% purity through this procedure and were stored at −20°C for future use.
Synthesis of polymer micelles
1H-NMR showed that PEG-p(Asp-Hyd) was successfully synthesized as reported previouslyCitation22 and used to prepare self-assembled micelles. For cross-linked micelles, the cross-linking of PEG-p(Asp-Hyd) was confirmed by GPC. The cross-linked micelles had an average molecular weight of ~147 kDa, corresponding to an average of roughly 14 block copolymers per cross-linked micelle. NMR was also used to determine the cross-linking percentage. The carboxyl peak of adipic acid at around 12 ppm disappeared, indicating that no unreacted or partially reacted adipic acid was present in the sample. The cross-linking percentage was 12.5%, which was determined by integrating the newly appeared peaks at 2.1 ppm and 1.5 ppm (α and β CH2 groups of adipic acid) with respect to the PEG peak at 3.5 ppm.
Characterization of MTM-loaded micelles
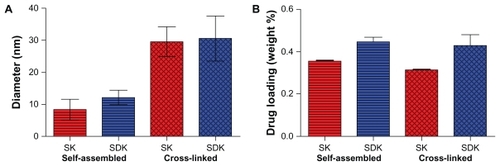
Following the drug conjugation reaction, ether precipitation was used to collect the polymers. Initial precipitations possessed a yellow supernatant corresponding to free drug that was not entrapped in the micelle structures. Ether precipitations were continued until the supernatant remained clear for several precipitations. The polymers maintained a yellowish color, after free drugs were removed, indicating the presence of the MTM analogs. UV-Vis absorption spectrometry at 410 nm confirmed that both SK and SDK were successfully entrapped in both the self-assembled micelles and cross-linked micelles. Drug loading in the selfassembled micelles was 35.4 ± 0.3 and 44.6 ± 2.2 weight % (wt %) for SK and SDK, respectively. Cross-linked micelles had an average drug loading of 31.3 ± 0.3 and 42.7 ± 5.3 wt % for SK and SDK, respectively. DLS showed that the particle size of self-assembled micelles entrapping SK and SDK was 8.36 ± 3.21 and 12.19 ± 2.77 nm. The particle size of cross-linked micelles was 29.56 ± 4.67 nm for SK and 30.48 ± 7.00 nm for SDK ().
Figure 3 (A) Particle size and (B) drug-entrapment yields.
Abbreviations: SDK, mithramycin SDK; SK, mithramycin SK.
Drug-release experiments
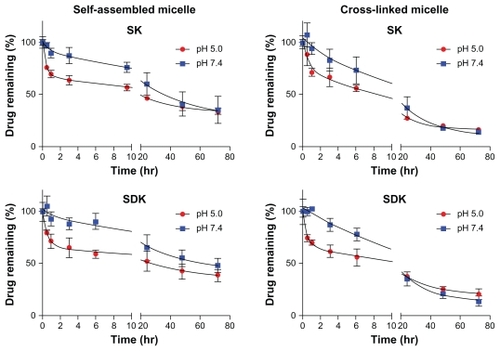
A pH sensitive hydrazone linkage was used to conjugate MTM analogs to the block copolymers to take advantage of the acidic tumor environment. To test pH-responsive drug-release patterns, MTM nanoparticulate formulations were incubated at pH 7.4 and 5.0. shows that all MTM nanoparticulate formulations released drugs effectively. At pH 7.4, self-assembled micelles entrapped more than 50% of MTM analogs for 24 hours, which will be stable enough to deliver drugs to tumor tissues following intravenous injections. Drug release from the self-assembled micelles was accelerated at pH 5.0, suggesting that SK and SDK will be released effectively in acidic tumor tissues. Cross-linked micelles appeared to release drugs more quickly at pH 7.4 than self-assembled micelles, yet retained 40% of drugs for 24 hours. Both SK and SDK were released more rapidly from cross-linked micelles at pH 5.0.
Figure 4 Drug-release patterns from self-assembled and cross-linked micelles at pH 7.4 and 5.0 for 72 hours.
Notes: All data points are the average of three measurements ± standard deviation; error bars not visible are obstructed by the point.
Abbreviations: SDK, mithramycin SDK; SK, mithramycin SK.
Cell toxicity assays
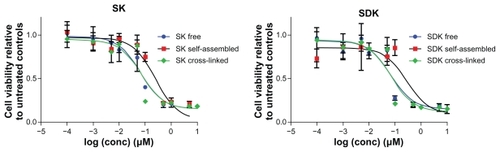
Cytotoxicity of MTM nanoparticulate formulations was tested with A549 cells (). Free SK and SDK showed IC50 values of 0.163 ± 0.092 μM and 0.093 ± 0.035 μM, respectively. IC50 values of self-assembled micelles entrapping MTM analogs were 0.262 ± 0.087 μM and 0.282 ± 0.131 μM for SK and SDK, which were 1.6- and 3.0-fold higher than those of free drugs. Interestingly, cross-linked micelles showed IC50 values of 0.073 ± 0.015 μM and 0.075 ± 0.009 μM for SK and SDK, which were 2.2- and 1.2-fold lower than free drugs (). No toxicity was observed with block copolymers, self-assembled micelles and cross-linked micelles alone up to 100 μM (data not shown).
Table 1 A summary of the characteristics and activity of the drug-loaded polymer nanoparticles and that of the free drugs for comparison
Figure 5 The cytotoxicity of free drug and nanoparticulate formulations for SK and SDK against A549 cell lines.
Notes: All points are the average of four measurements ± one standard deviation; error bars not visible are obstructed by the point.
Abbreviations: SDK, mithramycin SDK; SK, mithramycin SK.
Discussion
As mentioned above, MTM has been limited clinically by the severe negative side effects associated with the use of the drug. However, MTM has shown promise with respect to treating neurological diseases, glioblastomas, and other tumors in addition to showing the ability to inhibit the multidrug resistance gene, MDR1, in which smaller, less toxic doses are required.Citation32–Citation37 Recently, MTM was identified as the lead compound against the Friend leukemia virus integration 1 (EWS-FLI1) transcription factor.Citation38 EWS-FLI1 is believed to be responsible for malignant transformation and progression of Ewing sarcoma family tumors and was widely regarded as undruggable because it is a transcription factor.Citation38 A high-throughput screening of over 50,000 compounds identified MTM as the top candidate from the screen and greatly reduced and inhibited tumor volume in Ewing sarcoma family tumors in mouse xenograft models, and it is planned to reinstitute clinical trials for this application of MTM.Citation38 Additionally, a combinational approach of MTM with betulinic acid to treat pancreatic cancer showed the ability to synergistically treat the cancer with a nontoxic dose of the drugs to produce less discernible side effects.Citation35 As new treatments and uses for MTM continue to be uncovered, the development and optimization of a compatible drug-delivery system will be necessary.
The potential of MTM has been enhanced by the discovery of two new analogs produced through engineering of the MTM biosynthetic pathway. These analogs, SK and SDK, are a result of the inactivation of the mtmW gene and have shown improved activity compared with that of the native MTM. The biosynthesis and isolation of the SK and SDK analogs are pivotal as larger quantities of the drug will be required for testing and eventually clinical use. SK and SDK are produced alongside two other compounds, a demycarosyl-MTM SK and MTM SA (SA). The SA analog shows decreased anticancer activity and the demycarosyl- MTM SK shows activity similar to that of the native MTM, thus neither are the preferred products of the mutant strain. Initially, a culture pH of 6.85 was used for the production of the drugs. These conditions allowed for good production of the SK, but a large amount of SA and very little SDK were also being observed. The formation of SK, SDK, and SA are a result of the spontaneous rearrangement of the immediate product MTM DK, resulting from the MtmOIV reaction of the biosynthetic pathway.Citation15,Citation16 It has been shown by in-vitro work that the major product of the conversion of premithramycin B by MtmOIV can be shifted to SDK at a pH of 8.25.Citation39 In an effort to improve the production of SDK from the culture, the pH of the media was adjusted to 7.0. It was hoped that the adjustment of the pH, although small, would still allow optimal growth of the M7W1 mutant strain and production of the MTM analogs, but that the production yield of SDK would be improved. Increasing the pH of the growth media to 7.0 confirmed the hypothesis and increased the production yield of SDK. Increasing the pH also did not appear to adversely affect the production of SK. This will be critical as the production of the analogs is scaled up to meet the clinical demands.
As the drive for more specific and efficient chemotherapeutic options continues, delivery systems will undoubtedly play a pivotal role in their development and advancement. To that end, these novel MTM formulations still need to resolve issues such as low tumor accumulation, poor bioavailability, and short plasma retention time to be further explored clinically. Drug-delivery systems are expected to address these concerns by delivering MTM preferentially to tumors. Liposomal formulations have been previously tested, but the release of MTM analogs from liposomes has proved to be difficult.Citation40 Studies suggest that polymer micelles will offer an alternative platform for the delivery of MTM, not only delivering the drug to tumors but also controlling the drug-release patterns efficiently. It has been demonstrated that block copolymers offer a versatile platform in this direction. In this study, MTM analogs (SK and SDK) were entrapped in polymer micelles through an acid-labile hydrazone conjugation between drug molecules and micelle-forming block copolymers. Prepared MTM nanoparticulate formulations appeared to combine the favorable characteristics of drugdelivery systems with the ability to entrap drugs at high yields and to control the drug release at the area of interest.
SK and SDK have one and two ketone groups on the side chain. These ketone groups offer a prime location for the conjugation of the drugs to the PEG-p(Asp-Hyd) block copolymers through the hydrazone bond. Self-assembled micelles showed higher drug loading of SDK (44.6 wt %) than SK (35.4 wt %), suggesting that the second ketone group of the SDK side chain could facilitate drug loading rather than saturating the drug-binding sites. The particle size of self-assembled micelles was 44% larger with SDK (12.19 nm) than SK (8.36 nm), which is consistent with the increased SDK loading in micelles. The average particle size of cross-linked micelles was 26.79 nm in the absence of drugs. Cross-linked micelles were 29.56 and 30.48 nm after entrapping SK and SDK, respectively. Even though the particle size was retained, cross-linked micelles entrapped 31.3 wt % SK and 42.7 wt % SDK, which were comparable to the amount of drugs that self-assembled micelles entrapped.
Again, an acid labile hydrazone bond was used to conjugate the drugs to the block copolymers so the release of the drugs from the nanoformulations was investigated at both the physiological pH of 7.4 and an intracellular endosome or lysosome pH of 5.0. Effective drug release was observed in all MTM nanoparticulate formulations tested improving on previous attempts of MTM delivery with liposomes. MTM analogs were released slowly from polymer micelles at pH 7.4 regardless of formulations. The drug release was accelerated at pH 5.0 (), which is advantageous to release drugs in acidic tumor tissues. After 72 hours, however, the amount of drugs released was similar at both pH values. MTM nanoparticulate formulations are expected to minimize drug leakage in the blood and maximize therapeutic efficacy by releasing drugs in tumor tissues rapidly.
MTM nanoparticulate formulations were shown to be effective to suppress cancer cell growth, indicating that MTM analogs were delivered in a biologically active form. Self-assembled micelles entrapping MTM analogs showed lower cytotoxic effects than free drugs as expected, but interestingly, cross-linked micelles enhanced drug efficacy up to 2.2-fold (). Although further investigation is necessary to elucidate the mechanism behind this intriguing phenomenon, the results of this study suggest that drug release in the early stage would not play a crucial role in determining cellular response to MTM analogs as all nanoparticulate formulations released a similar amount of drugs between 1 and 3 hours. Drug release at pH 7.4 rather than 5.0 appeared to be the major factor that determines cytotoxicity. MTM analogs were effective to suppress cancer cell growth in order of cross-linked SK micelles, cross-linked SDK micelles, self-assembled SK micelles, and self-assembled SDK micelles, which is consistent with the drug-release patterns at pH 7.4, whereas all nanoparticulate formulations released drugs similarly at pH 5.0, which does not explain differential cytotoxicity observed in A549. Therefore, it is more reasonable to assume that drugs released from micelles at pH 7.4 comprise the major drug population that involved in cell killing, while intracellular drug release (pH 5.0) has less effect on overall cytotoxicity of MTM nanoparticulate formulations. However, detailed intracellular mechanisms on how cells respond to MTM nanoparticulate formulations (eg, particle size and intracellular drug distribution) should be investigated further in the future.
Cross-linked micelles had a 12.5% cross-linking yield, which was high enough to tether block copolymer chains before and after entrapping drugs without causing agglomeration between particles. In spite of the high cross-linking yield, cross-linked micelles entrapped and released MTM analogs effectively. Importantly cross-linked micelles released drugs more quickly than self-assembled micelles, implicating that MTM analogs could be entrapped in the vicinity rather than at the center of the micelle core. The rapid drug release from cross-linked micelles can be also attributed to the rigid environment in the cross-linked core of the micelles. Cross-linking the core could allow for a more porous and less tightly packed core in the micelles so that drug molecules can maneuver more easily out of the core. In contrast, self-assembled micelles have a flexible core that can continue to pull block copolymers to keep the particle stable as drugs are released. This may explain why drug release from self-assembled micelles was slower than cross-linked micelles.
Cytotoxic effects of MTM nanoparticulate formulations revealed an interesting phenomenon that can be used to design potent drug-delivery systems. Unlike self-assembled micelles that decreased the toxicity of MTM analogs, cross-linked micelles showed a higher potency against A549 cancer cells than free drugs. The exact mechanism and reason for the improved efficacy is yet to be confirmed, although the more stable cross-linked micelles could aid in the drug’s persistence in the cells, allowing more drugs to interact with the therapeutic targets in the cell. In addition to the known mechanism of the MTM family of compounds in which they bind the GC rich DNA present in Sp family of promoters, the expression of the gene encoding for a multidrug resistance efflux pump, MDR1, has also been shown to be inhibited by MTM and some of it derivatives.Citation36,Citation37 This would make the MTM nanoparticulate formulations excellent candidates for combinational therapy attacking hard-to-treat, multidrug-resistant cancer cell lines. Future studies will explore whether these results translate in vivo and dig deeper into the reasoning behind the improved activity.
Conclusion
Nanoparticulate formulations for controlled delivery of MTM analogs, SK and SDK, were prepared by using self-assembled or cross-linked polymer micelles from PEG-p(Asp-Hyd) block copolymers to which drugs were conjugated through acid-labile hydrazone bonds. With recent interest in MTM for several novel treatment approaches, a compatible delivery system with ability to selectively deliver the drug to the area of interest would be advantageous. Both the SK and SDK analogs offer higher anticancer activity compared with MTM and thus were investigated with respect to a compatible delivery system. SK and SDK were successfully loaded into polymer micelles and demonstrated a pH-dependent release profile, accelerating drug release at pH 5.0. By cross-linking the micelles, the size of the particles was increased without significantly affecting the drug loading. Both the self-assembled and cross-linked structures were able to deliver the drugs in the biologically active form and efficiently kill the human nonsmall cell lung cancer cell line (A549). The cross-linked micelles interestingly improved the cytotoxicity of MTM analogs, even compared with the free drugs. The ability to prepare delivery platforms without losing any biological activity will streamline the development of therapeutic options, and these results conclude that nanoparticulate formulations offer prime candidates to pave the path for SK, SDK, and other MTM analogs to be explored further with animal models and potentially in the clinical field.
Acknowledgments
This research was supported in part by the National Institutes of Health (grant CA 091901 to JR) and the Kentucky Lung Cancer Research Program (to YB). DS acknowledges financial support from a NCI-CNTC postdoctoral traineeship, and the project described was supported by grant number 5R25CA153954 from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Disclosure
The authors report no conflicts of interest in the work. The University of Kentucky has determined that it has a legal ownership interest in this intellectual property based on material use of university resources and the appropriate protection for this intellectual property will be pursued.
References
- MillerDMPolanskyDAThomasSDMithramycin selectively inhibits transcription of G-C containing DNAAm J Med Sci198729453883942962490
- SastryMFialaRPatelDJSolution structure of mithramycin dimers bound to partially overlapping sites on DNAJ Mol Biol199525156746897666419
- SastryMPatelDJSolution structure of the mithramycin dimer-DNA complexBiochemistry19933226658866048329387
- BlumeSWSnyderRCRayRThomasSKollerCAMillerDMMithramycin inhibits SP1 binding and selectively inhibits transcriptional activity of the dihydrofolate reductase gene in vitro and in vivoJ Clin Invest1991885161316211834700
- SnyderRCRayRBlumeSMillerDMMithramycin blocks transcriptional initiation of the c-myc P1 and P2 promotersBiochemistry19913017429042971827033
- SafeSAbdelrahimMSp transcription factor family and its role in cancerEur J Cancer200541162438244816209919
- AbdelrahimMSmithR3rdBurghardtRSafeSRole of Sp proteins in regulation of vascular endothelial growth factor expression and proliferation of pancreatic cancer cellsCancer Res200464186740674915374992
- BlackARBlackJDAzizkhan-CliffordJSp1 and kruppel-like factor family of transcription factors in cell growth regulation and cancerJ Cell Physiol2001188214316011424081
- KennedyBJMetabolic and toxic effects of mithramycin during tumor therapyAm J Med19704944945034320120
- BrownJHKennedyBJMithramycin in the treatment of disseminated testicular neoplasmsN Engl J Med196527211111814224214
- KollerCAMillerDMPreliminary observations on the therapy of the myeloid blast phase of chronic granulocytic leukemia with plicamycin and hydroxyureaN Engl J Med198631523143314382431313
- RyanWGMithramycin for Paget’s disease of boneN Engl J Med19702832111715474359
- RyanWGSchwartzTBNorthropGExperiences in the treatment of Paget’s disease of bone with mithramycinJAMA19702137115311575468261
- RemsingLLGarcia-BernardoJGonzalezAKetopremithramycins and ketomithramycins, four new aureolic acid-type compounds obtained upon inactivation of two genes involved in the biosynthesis of the deoxysugar moieties of the antitumor drug mithramycin by Streptomyces argillaceus, reveal novel insights into post-PKS tailoring steps of the mithramycin biosynthetic pathwayJ Am Chem Soc200212481606161411853433
- AlbertiniVJainAVignatiSNovel GC-rich DNA-binding compound produced by a genetically engineered mutant of the mithramycin producer Streptomyces argillaceus exhibits improved transcriptional repressor activity: implications for cancer therapyNucleic Acids Res20063461721173416571899
- RemsingLLGonzálezAMNur-e-AlamMMithramycin SK, a novel antitumor drug with improved therapeutic index, mithramycin SA, and demycarosyl-mithramycin SK: three new products generated in the mithramycin producer Streptomyces argillaceus through combinatorial biosynthesisJ Am Chem Soc2003125195745575312733914
- TakakuraYFujitaTHashidaMSezakiHDisposition characteristics of macromolecules in tumor-bearing micePharm Res1990743393461694582
- van VlerkenLVyasTAmijiMPoly(ethylene glycol)-modified Nanocarriers for tumor-targeted and intracellular deliveryPharm Res20072481405141417393074
- SmartTLomasHMassignaniMFlores-MerinoMVPerezLRBattagliaGBlock copolymer nanostructuresNano Today200833–43846
- BaeYKataokaKIntelligent polymeric micelles from functional poly(ethylene glycol)-poly(amino acid) block copolymersAdv Drug Deliv Rev2009611076878419422866
- ChenHKimSLiLWangSParkKChengJXRelease of hydrophobic molecules from polymer micelles into cell membranes revealed by Forster resonance energy transfer imagingProc Natl Acad Sci U S A2008105186596660118445654
- LeeHJBaeYCross-linked nanoassemblies from poly(ethylene glycol)-poly(aspartate) block copolymers as stable supramolecular templates for particulate drug deliveryBiomacromolecules20111272686269621644544
- BeckerMLRemsenEEPanDWooleyKLPeptide-derivatized shell-cross- linked nanoparticles. 1. Synthesis and characterizationBioconjug Chem200415469970915264856
- WestKROttoSReversible covalent chemistry in drug deliveryCurr Drug Discov Technol20052312316016472224
- JonesATGumbletonMDuncanRUnderstanding endocytic pathways and intracellular trafficking: a prerequisite for effective design of advanced drug delivery systemsAdv Drug Deliv Rev200355111353135714597135
- Vander HeidenMGCantleyLCThompsonCBUnderstanding the Warburg effect: the metabolic requirements of cell proliferationScience200932459301029103319460998
- GilliesRJRobeyIGatenbyRACauses and consequences of increased glucose metabolism of cancersJ Nucl Med200849Suppl 24S42S
- BaeYNishiyamaNKataokaKIn vivo antitumor activity of the folate-conjugated pH-sensitive polymeric micelle selectively releasing adriamycin in the intracellular acidic compartmentsBioconjug Chem20071841131113917488066
- HowardMDPontaAEckmanAJayMBaeYPolymer micelles with hydrazone-ester dual linkers for tunable release of dexamethasonePharm Res201128102435244621614636
- PontaABaeYPEG-poly(amino acid) block copolymer micelles for tunable drug releasePharm Res201027112330234220372992
- AlaniAWBaeYRaoDAKwonGSPolymeric micelles for the pH-dependent controlled, continuous low dose release of paclitaxelBiomaterials20103171765177219959225
- JinH-JKanthasamyAAnantharamVRanaAKanthasamyAGTranscriptional regulation of pro-apoptotic protein kinase Cδ: implications for oxidative stress-induced neuronal cell deathJ Biol Chem2011286198401985921467032
- SeznecJSilkenstedtBNaumannUTherapeutic effects of the Sp1 inhibitor mithramycin A in glioblastomaJ Neurooncol201110136537720556479
- SleimanSFLangleyBCBassoMMithramycin is a gene-selective Sp1 inhibitor that identifies a biological intersection between cancer and neurodegenerationJ Neurosci2011316858687021543616
- GaoYJiaZKongXCombining betulinic acid and mithramycin A effectively suppresses pancreatic cancer by inhibiting proliferation, invasion, and angiogenesisCancer Res2011715182519321673052
- TagashiraMKitagawaTIsonishiSOkamotoAOchiaiKOhtakeYMithramycin represses MDR1 gene expression in vitro, modulating multidrug resistanceBiol Pharm Bull200023892692910963297
- TagashiraMKitagawaTNozatoNTwo novel C-glycosides of aureolic acid repress transcription of the MDR1 geneChem Pharm Bull (Tokyo)200048457557810783084
- GroharPJWoldemichaelGMGriffinLBIdentification of an Inhibitor of the EWS-FLI1 oncogenic transcription factor by high-throughput screeningJ Natl Cancer Inst201110396297821653923
- GibsonMNur-e-alamMLipataFOliveiraMARohrJCharacterization of kinetics and products of the Baeyer-Villiger oxygenase MtmOIV, the key enzyme of the biosynthetic pathway toward the natural product anticancer drug mithramycin from Streptomyces argillaceusJ Am Chem Soc200512750175941759516351075
- FrezardFGarnier-SuillerotADemicheliCEncapsulation of mithramycin in liposomes in response to a transmembrane gradient of calcium ionsJ Incl Phenom Macrocycl Chem19972815162