
Abstract
Mammalian target of rapamycin (mTOR) is a master regulator of cell growth and metabolism, which is activated in response to intra- and extracellular signals, including nutrients, growth factors, and cellular energy levels. The frequent dysregulation of mTOR signaling in cancer makes it an attractive therapeutic target, and several types of mTOR inhibitors have been developed. Nanoparticle-based mTOR modulators are predicted to target various cancers and deliver as well as release drugs in a controlled manner, resulting in enhanced bioavailability and reduced side effects. This mini-review is focused on the molecular mechanism of nanoparticle-based mTOR modulator action as well as the current development of mTOR inhibitors using nanoparticles. Understanding the biological function of nanoparticle-based mTOR modulators will contribute to the development of efficient nano-therapeutics for the treatment of cancers.
Introduction
The serine/threonine kinase mammalian target of rapamycin (mTOR) is a master regulator of cell growth that integrates cellular responses to growth factors, nutrient availability, and other diverse environmental signals.Citation1 Several proteins upstream or downstream of mTOR, as well as mTOR itself, have been reported to be either overexpressed or mutated in a number of cancers.Citation2 The hyperactivity of mTOR signaling pathway has been observed to be associated with the phosphatidylinositol 3-kinase (PI3K)/Akt pathway in many human cancers.Citation1 Indeed, mTOR has been identified as a potential target for the development of molecular therapies to treat cancer.
This mini-review summarizes our current understanding of mTOR regulation, as well as the development of novel mTOR inhibitors. New strategies using nanotechnology to overcome the disadvantages of existing mTOR inhibitors, such as drug resistance, and to enhance the efficacy of current mTOR inhibitor-based therapies will be discussed.
Mammalian Target of Rapamycin
mTORC1 and mTORC2
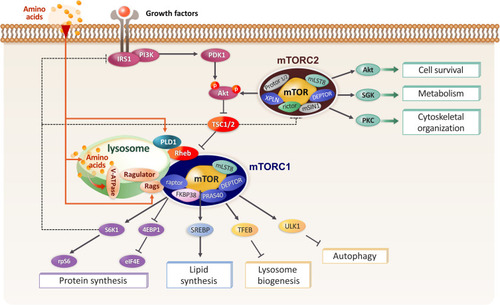
mTOR, a member of the phosphatidylinositol-3-kinase-related protein kinase (PIKK) family, is a serine/threonine kinase and there are two biochemically and functionally distinct complexes, namely, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) ().Citation3,Citation4 mTORC1 consists of mTOR, regulatory-associated protein of mTOR (raptor), mammalian lethal with SEC13 protein 8 (mLST8), DEP domain-containing mTOR interacting protein (DEPTOR), and proline-rich Akt substrate 40 (PRAS40).Citation5 mTORC1 controls cell growth, cell proliferation, and metabolic homeostasis through the integration of multiple extracellular and intracellular signals including nutrients, intracellular energy status, oxygen level, and mitogens.Citation6 Ribosomal protein S6 kinase 1 (S6K1) and eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1) are the downstream targets of mTORC1, which regulate protein translation through the ribosomal protein S6 and eukaryotic translation initiation factor 4E (eIF4E), respectively.Citation7,Citation8 mTORC1 controls the expression and maturation process of the sterol regulatory element-binding protein 1/2 (SREBP1/2) transcription factors, which regulate the expression of fatty acid and cholesterol synthesis-related genes.Citation9 mTORC1 also regulates SREBP by controlling the nuclear localization of Lipin-1, a phosphatidic acid phosphataseCitation10 (). Rapamycin forms a complex with the 12 kDa FK506-binding protein FKBP12 and binds the FRB domain of mTOR in a highly specific manner, leading to the allosteric blockage of mTORC1 through the inhibition of substrate recruitment.Citation11 The tuberous sclerosis 1 (TSC1)/TSC2 complex serves as a molecular hub, integrating upstream signals such as intracellular oxygen levels, growth factors, and energy sensing pathways to regulate mTORC1 activity. TSC1/2 negatively regulates Ras homolog enriched in brain (Rheb), functioning as a GTPase activating protein (GAP)Citation12 (). mTORC2 comprises rapamycin-insensitive companion of mTOR (rictor), mLST8, DEPTOR, mammalian stress-activated protein kinase interacting protein (mSIN1), protein observed with rictor-1 (Protor-1), Protor-2, and exchange factor found in platelet, leukemic, and neuronal tissues (XPLN).Citation13,Citation14 Even though mTORC2 is activated by growth factors, the regulation of mTORC2 is not fully understood. mTORC2 stimulates Akt, serum and glucocorticoid inducible kinase (SGK), and PKC, thus regulating cell survival, metabolism, and the reorganization of actin cytoskeletonCitation15 (). Despite the absence of a direct inhibitory effect of rapamycin on mTORC2, prolonged rapamycin treatment impairs mTORC2 activity, most likely through irreversible mTOR sequestration.Citation16
Figure 1 Diagram showing mTORC1 and mTORC2 signaling pathways. Growth factors activate mTOR complex 1 (mTORC1) through IRS1/PI3K-PDK1-Akt by regulating the tuberous sclerosis complex (TSC)1/2. TSC functions as a GTPase activator protein (GAP) for the small G-protein Rheb, an upstream positive regulator of mTORC1. Amino acids signaling causes mTORC1 translocation to the lysosomes, where Rheb resides, via the Rag GTPases–Ragulator complex. S6K1-rpS6 and 4EBP1-eIF4E are well-known downstream targets of mTORC1 and are responsible for the translation pathway. mTORC1 also regulates lipid synthesis through SREBP and inhibits autophagy by phosphorylating TFEB and ULK1. mTORC2 controls cell metabolism, cell survival, and cytoskeleton rearrangement by activating Akt, SGK1, and PKC. Akt activity is regulated by both PDK1 and mTORC2. Dotted lines indicate feedback mechanisms.
The Crosstalk Between mTORC1 and mTORC2
The activity of mTORC1 is associated with that of mTORC2 through the regulation of Akt phosphorylation (). While Akt indirectly activates mTORC1 via the phosphorylation and inhibition of TSC1/2, TSC1/2 positively regulates mTORC2 through physical association with mTORC2 to indirectly regulate Akt.Citation17 S6K1 and mTOR block insulin receptor substrate-1 (IRS-1) by phosphorylating it at multiple sites, inducing its degradation, altering its localization, and resulting in the inhibition of PI3K/Akt activation (, dotted line).Citation18 Furthermore, S6K1 directly phosphorylates rictor, thus negatively regulating Akt phosphorylation at Ser473 (, dotted line).Citation19 mTORC2- and phosphoinositide dependent kinase 1 (PDK1)-phosphatidylinositol 3 kinase (PI3K)-mediated Akt phosphorylation at Ser473 and Thr308, respectively, are required for the maximal activity of Akt.Citation20 These findings demonstrate a complicated relationship between mTOR and PDK1 and partially explain the role of mTORC2 in oncogenesis, suggesting that a careful interpretation is required for cancer therapy using rapamycin or other mTOR inhibitors.
mTORC1 Activation on the Lysosome
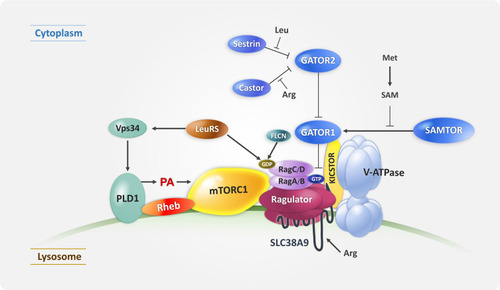
Upon amino acid signaling, mTORC1 translocates to the lysosomes where Rheb resides in a Rags/Ragulator-dependent mannerCitation21,Citation22 (). Four Rag GTPases, RagA or B/RagC or D, form a heterodimer and their nucleotide-bound states are regulated by amino acid sufficiency.Citation21 In the absence of amino acids, the guanine nucleotide exchange factor (GEF) activity of Ragulator on the lysosomal membrane is inhibited by its interaction with the vacuolar H+-ATPase (V-ATPase); however, in the presence of amino acids, the GEF activity of Ragulator is fully activated by a conformational change in the V-ATPase–Ragulator complex.Citation23 Amino acid signaling also leads to the translocation of phospholipase D1 (PLD1) on lysosomes under the regulation of leucyl-tRNA synthetase (LeuRS) and Vps34 through the interaction of phosphatidyl inositol 3-phosphate (PtdIns(3)P) with the PX domain of PLD1, resulting in the production of phosphatidic acids (PA) and further activation of the mTORC1 on the lysosomes.Citation24,Citation25 The nucleotide states of the RagC/D and RagA/B GTPases are regulated by LeuRS, FLCN, GATOR1, GATOR2, and KICSTOR.Citation26–Citation29 LeuRS functions as an amino acid sensor for both RagC/D GTPases and Vps34-PLD1.Citation25 In addition, the following amino acid-specific sensors have been identified: SLC38A9 as lysosomal arginine sensor and Sestrin2 and CASTOR1 as cytosolic leucine and arginine sensors, respectively.Citation30–Citation32 Most recently, SAMTOR was recognized as an S-adenosylmethionine sensor in amino acid-induced mTOR signaling.Citation33
Figure 2 Diagram showing the components of mTORC1 upstream signaling on the lysosome mTORC1 is regulated by amino acid sensors and several multiprotein complexes which regulate Rag GTPases. LeuRS/Vps34/PLD1 also activates mTORC1 through PA during amino acid stimulation (see “mTORC1 Activation on the Lysosome” section).
mTOR as a Repressor of Autophagy
mTOR responds to the amino acids from the cytosol, as well as from the lysosome (the products of protein degradation). Absence of amino acids in the cytosol inhibits mTOR signaling and initiates autophagy to increase amino acid levels through protein degradation in the lysosome.Citation34 mTORC1 directly phosphorylates unc-51-like autophagy-activating kinase1 (ULK1) and ATG13, two key early effectors in the initiation of autophagy, inhibiting the induction of autophagy and consequently preventing a futile cycle between mTORC1-stimulated mass accumulation and autophagic degradation ().Citation1 mTORC1 also phosphorylates UVRAG to enhance the association with RUBICON and subsequently decrease the interaction with HOPS complex, a component of late endosomes and lysosomes, blocking autophagosome and endosome maturation.Citation35 In addition, mTORC1 regulates the expression of genes involved in lysosomal biogenesis by controlling nuclear translocation of both transcription factor EB (TFEB) and the related transcription factor E3 (TFE3)Citation36,Citation37 (). During long-term starvation, mTORC1 regulates ribophagy, a selective autophagy of ribosome, through the autophagy receptor nuclear fragile X mental retardation interacting protein 1 (NUFITP1).Citation38 In this context, ribosome functions as a reservoir of amino acids and nucleotides to maintain cell viability, suggesting that the communication between lysosome and mTORC1 is essential for cell survival in the nutrient-deprived conditions.
The Role of mTOR in Metabolism
The functional regulation of mTOR pathways is more complex at the organismal level. mTOR pathways sense the nutrition and energy status and regulate tissue-specific catabolic or anabolic cascades.Citation1 mTOR signaling enhances adipogenesis, a process to form adipose tissues, the energy storage site in mammals.Citation9 In skeletal muscles, insulin activates mTORC2-Akt signaling to induce glucose uptake and storage of glucose as glycogen, as well as mTORC1 to incorporate amino acids into the muscle biomass.Citation1 In the liver, mTORC1 also regulates the production of ketone bodies, the energy storage sources for peripheral tissues during fasting.Citation9 In obese rodents, hyperactive mTORC1/S6K1 induces IRS-1 phosphorylation at serine residues and consequent IRS-1 degradation, leading to the impairment of PI3K-Akt signaling. Subsequently, insulin resistance results in reduced glucose uptake in skeletal muscles, increased gluconeogenesis in the liver, enhanced FFA release by the adipocytes, and ectopic fat deposition and lipotoxicity in adipose tissue.Citation9 In summary, mTOR activity is tightly associated with metabolic dynamics, while its imbalance results in metabolic dysregulation and the development of metabolic diseases such as obesity, nonalcoholic fatty liver disease, insulin resistance, and type 2 diabetes.
mTOR in Cancer
The Role of mTOR in Cancer
As mTOR maintains a balance between cell growth and cell division, dysregulated mTOR signaling has been reported in many cancers.Citation39 An abnormal increase in PI3K/mTOR signaling is observed in a variety of cell lines and murine xenograft models, and plays a role in tumor growth, angiogenesis, and metastasis.Citation40 Furthermore, a constitutively active mTOR mutant has been identified in several human cancer cells,Citation2 and the upstream and downstream regulators of mTOR have been found to be altered in many human cancer cases.Citation41
PI3K catalytic activity is also frequently dysregulated in human cancer.Citation42 The PI3K/Akt-dependent activation of mTORC1 can be altered by the loss or inactivation of tumor suppressors, including p53, LKB1, phosphatase and tensin homolog deleted on chromosome 10 (PTEN), and TSC1/2, resulting in the promotion of tumorigenesis through increased mTORC1 signaling.Citation43 LKB1 is a positive regulator of AMP kinase (AMPK), which is activated by low intracellular energy levels and inhibits mTORC1 activation by regulating TSC1/2.Citation44 Mutations in LKB1 are linked to Peutz-Jeghers syndromeCitation45 and lung cancer,Citation46 and lead to constitutive activation of mTORC1 under low intracellular energy conditions. PTEN mutations, including silencing and deletions, have been found in glioblastoma, hepatocellular carcinoma, lung carcinoma, melanoma, endometrial carcinoma, and prostate cancer, where they cause the constitutive activation of mTOR signaling.Citation47–Citation49 Mutations in TSC2, a negative regulator of mTORC1, lead to tuberous sclerosis syndrome,Citation50,Citation51 which is related to well-vascularized hamartomas and an increased risk of renal cell carcinoma (RCC).Citation52,Citation53 Rheb1 is overexpressed in patients with acute myeloid leukemia (AML) and its expression level is associated with the median survival of patients.Citation54 At the same time, mTOR mutations in FAT and kinase domains identified in human kidney cancer increase mTOR kinase activity in a Rheb-independent manner, decrease nutrient dependency, and augment cell size.Citation55 Various mutations in the FAT domain of mTOR in RCC decrease the binding of endogenous inhibitors, such as DEPTOR and PRAS40, leading to increased mTORC1 and mTORC2 activities.Citation56 Furthermore, amplification of rictor or increase in mTORC2 activity is observed in numerous cancers, including those of breast, stomach, liver, brain, lung, and tongue,Citation57–Citation59 whereas mTOR overexpression is rarely seen in human cancers.
Increased levels and/or phosphorylation of mTOR downstream effectors in human malignancies are linked to tumor aggressiveness and poor prognosis.Citation60,Citation61 For example, S6K1 is overexpressed in lung and ovarian cancers,Citation62 and is amplified in some breast carcinomas as well.Citation63 mTORC1 positively regulates eIF4E, a critical rate-limiting initiation factor of cap-dependent translation. The phosphorylation of 4EBP1 by mTORC1 causes its dissociation from eIF4E, allowing it to initiate the translation of its target mRNAs. The levels of eIF4E are elevated in many human tumors, such as breast, colon, late stage head and neck carcinoma, non-Hodgkin’s lymphomas, and chronic and acute myelogenous leukemia.Citation64–Citation66 In addition, 4EBP1 phosphorylation has been shown to correlate with chemoresistance in ovarian cancer.Citation67 The elevated mTOR activity observed in different cancers emphasizes the importance of targeting mTOR in anticancer therapy.
mTOR as an Effector in the Tumor Microenvironment
Recent cancer immunotherapies targeting immune checkpoint inhibitors have achieved remarkable clinical success.Citation68 Different types of immune cells play roles in either the promotion or suppression of tumor progression, indicating the importance of controlling the immune cells in the tumor microenvironment. Immune checkpoints, the physiological immunoinhibitory and regulatory components of the immune system, which include programmed-death 1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), prevent immune activation to maintain immune homeostasis, enhance self-tolerance, and restrict autoimmunity. Hence, the modulation of the tumor microenvironment by the immune checkpoint inhibitors may be one of the most promising approaches for treating cancers. mTOR controls multiple T-cell fates and functions in adaptive immune cells,Citation69 which are necessary for proper T cell function and immune homeostasis.Citation70 In addition, mTOR activation is related to PD-L1 expression in human lung adenocarcinomas and squamous cell carcinomas, which leads to immune escape.Citation71 Furthermore, treatment with rapamycin decreases PD-L1 expression in PTEN-mutant triple-negative breast cancer cell lines in vitro.Citation72 Conversely, co-treatment with an immune checkpoint inhibitor and sirolimus (mTOR inhibitor) maintains anticancer efficacy while improving allograft tolerance in patients with melanoma by decreasing the number of cytotoxic T-cells and increasing the number of eosinophils (eosinophilia) and Treg cells in the peripheral blood, and by increasing the number of IFN-γ+ CD4+ T cells and serum IFN-γ levels.Citation73 Therefore, the immune-modulatory function of mTOR needs to be considered in cancer therapy when using immune checkpoint inhibitors.
mTOR Inhibitors as Anticancer Drugs
Rapamycin and Its Analogs as First-Generation mTOR Inhibitors
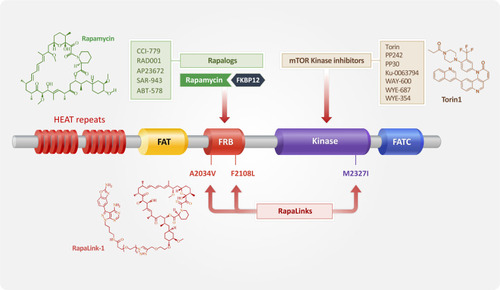
The field of study of mTOR signaling originated from studies on rapamycin, a macrolide obtained from the bacterium Streptomyces hygroscopicus.Citation11 Rapamycin was first identified as an anti-fungal and a potent immunosuppressive agent.Citation74 The molecular target of rapamycin (TOR) was determined later in a screen of yeast mutants, which were able to proliferate in the presence of rapamycin.Citation75 Subsequently, a mammalian TOR (mTOR) was identified by its affinity to FKBP12, an FK506-binding protein.Citation76,Citation77 The complex of rapamycin and FKBP12 binds to the FKBP-rapamycin binding (FRB) domain in the C-terminus of mTOR and allosterically inhibits mTORC1 downstream signaling ().Citation78 mTORC2 is not inhibited by the rapamycin–FKBP12 complex directly; however, prolonged treatment with rapamycin can prevent the newly-synthesized mTOR from associating with rictor, resulting in inhibition of Akt phosphorylation at Ser473.Citation79 At the same time, rapamycin decreases the negative feedback inhibition of PI3K signaling via S6K1, leading to upregulation of survival effector Akt. In addition, the effect of rapamycin on 4EBP1 is limited and is cell type dependent.Citation80 High resolution cryo-electron microscopy revealed that the rapamycin–FKBP12 complex reduces access of substrates to the active site of mTOR, indicating that the FRB domain functions as a gatekeeper to the kinase active site.Citation80
Rapamycin inhibits the growth of a wide range of cancers, including rhabdomyosarcoma, neuroblastoma, glioblastoma, small cell lung carcinoma, osteosarcoma, pancreatic cancer, leukemia, B-cell lymphoma, as well as breast and colon cancer-derived cell lines.Citation81 Several rapamycin derivatives have been developed with similar mechanisms of action, but improved pharmacokinetic and solubility properties compared to those of rapamycin: CCI-779 (Wyeth, Madison, NJ, USA),) RAD001 (Novartis, Basel, Switzerland), AP23572 (ARIAD, Cambridge, MA, USA), 32-deoxorapamycin (SAR943), and ABT-578 (Abbott Park, IL, USA). The rapamycin analogs CCI-779 and RAD001 show a prolonged bioactivity compared to rapamycin. These rapamycin analogs have been evaluated in cancer clinical trials and have shown anti-proliferative activity against a wide range of cancers.Citation82,Citation83 Phase II trials with RAD001 have reached an objective response rate of 47% for Hodgkin lymphoma, 30% for non-Hodgkin lymphoma, and 12% for breast cancers,Citation84–Citation86 whereas phase II/III studies with CCI-779 achieved an objective response rate of 4 to 14% for endometrial cancer and 22% for mantle-cell lymphoma.Citation87,Citation88
These findings indicate that rapamycin and a wide range of its analogs can be considered for anticancer therapy. Although rapamycin and the first-generation rapamycin analogs, temsirolimus and everolimus, have shown modest success in clinical trials, the role of rapamycin in inhibiting mTORC1 activity is questionable as rapamycin does not block all mTORC1 functionsCitation89 and, in addition, reduces the negative feedback inhibition of PI3K signaling via S6K1, which has been shown to lead to upregulation of the survival effector Akt in many cancer cells and clinical samples,Citation90 which raise concerns regarding the attenuation of its therapeutic effect. At the same time, mTORC1 functions as a downstream effector of PI3K/Akt and promotes tumorigenesis,Citation91 indicating that rapamycin therapy could be an attractive therapy when Akt is hyperactivated.
ATP Analog mTOR Inhibitors as a Second-Generation mTOR Inhibitor
A different class of mTOR inhibitors that block the mTOR catalytic site has been developed to address the failure of rapamycin to control all functions of mTOR. Examples of second generation of mTOR inhibitors include Torin,Citation92 PP242, pp30,Citation89 Ku-0063794,Citation93 WAY-600, WYE-687, and WYE-354.Citation94 These novel mTOR inhibitors are ATP analogs that compete with ATP for binding to the kinase domain of mTOR, leading to the inhibition of mTOR kinase activity (). While rapamycin and its analogs, known as allosteric mTOR inhibitors, inhibit only mTORC1, ATP analog inhibitors block both mTORC1 and mTORC2, inhibiting all target activities of mTOR, including Akt phosphorylation at Ser473, and causing a more effective inhibition of cell proliferation.Citation89,Citation92 Additionally, the phosphorylation of 4EBP1 at both rapamycin sensitive Ser65 and Thr70, and rapamycin resistant Thr37/46, is suppressed by ATP analog inhibitors; this suppression is associated with the effective targeting of the initiation of translation. Therefore, ATP analog inhibitors provide many therapeutic advantages and are superior to rapamycin and its analogs. Notably, these inhibitors have a much lower half-maximal inhibitory concentration (IC50) values for mTOR than those for PI3K.Citation80
Figure 3 Diagram showing the mTOR structural domain targets for three generations of mTOR inhibitors. mTOR consists of several structural domains: HEAT repeats, FAT (for FRAP, ATM, TRAP), FRB (FK506 binding protein 12 (FKBP12)–rapamycin binding), kinase, and FATC (for C-terminal FAT) domains. As the name implies, the FRB domain is responsible for the binding of mTOR to FKBP12 and rapamycin. FAT, kinase, and FATC domains are required for maintaining phosphatidylinositol 3-kinase-related kinases (PIKKs) activity. Rapalogs, the first-generation mTOR inhibitors, decrease mTOR activity by interacting with the FRB domain of mTOR. The second-generation mTOR inhibitors competes with ATP for binding to the kinase domain of mTOR. RapaLink, the third-generation mTOR inhibitor, was developed to overcome the limitations of previous mTOR inhibitors. FRB domain mutations (mTORA2034V and mTORF2108L) and a kinase domain mutation (mTORM2327I) contribute to the resistance of mTOR to rapalogs and mTOR kinase inhibitors, respectively.
Most cancers can survive in hypoxic and energy-poor environments by strongly enhancing glycolysis. The glycolysis-preferential metabolism of cancer depends partially on the Akt-dependent activation of glucose transporter 1 (GLUT1). As ATP analog inhibitors suppress the PI3K-induced activation of Akt and the Akt-dependent accumulation of GLUT1, they reduce glycolysis in cancers more efficiently compared to rapamycin.Citation94 This may explain the improved anticancer effects of ATP analogs in xenograft tumors.Citation94 Furthermore, ATP analogs also inhibit lipid biosynthetic processes, thereby regulating Akt-dependent SREBP stabilization.Citation13
Dual PI3K-mTOR Inhibitors
Even though the results of preclinical trials investigating ATP analogs are encouraging, it has been revealed that cancers can become resistant to ATP analogs through the mTORC1-driven activation of PI3K- and PDK-induced phosphorylation of Akt at Thr308. Even in the absence of Akt phosphorylation at Ser473, the phosphorylation of Akt at Thr308 activates mild substrate-dependent activity, reducing the efficacy of ATP analogs. The newly developed dual mTOR/PI3K inhibitors are expected to solve these problems.Citation43 These dual inhibitors also suppress PI3K effectors through the mTORC1-induced IRS feedback, potentiating their anticancer effects.Citation43 However, the dual inhibitors are only effective in cells with hyperactive PI3K signaling and not in cells with K-Ras hyperactivation.Citation95 Concomitant inhibition of PI3K/mTOR and mitogen-activated protein kinase (MAPK) may be required in Ras-driven tumorigenesis.Citation96,Citation97 NVPBEZ235 (Novartis), a dual inhibitor of PI3K/mTOR, is currently being tested in several Phase I/II trials for the treatment of advanced solid tumors and metastatic breast cancer.Citation11,Citation98 The combination of NVPBEZ235 and AZD6244, a MAPK/extracellular signal-regulated kinase (ERK) kinase (MEK) inhibitor, showed a significant tumor regression in the K-Ras transgenic mouse model.Citation96
RapaLink as a Third-Generation mTOR Inhibitor
It has been reported that clinically relevant mutations in mTOR enhance the catalytic activity of mTOR and consequently decrease the efficacy of mTOR inhibitors and dual PI3K/mTOR inhibitors in cancer cells.Citation2,Citation99 In addition, single amino acid substitution (A2034V and F2108L) in the FRB-FKBP12-rapamycin binding domain confers rapamycin resistance, disrupting the interaction of the FRB domain with the rapamycin–FKBP12 complex.Citation2 Moreover, a somatic mutation in the kinase domain at the M2327 position induces resistance to the ATP competitive inhibitor AZD8055, resulting in the hyperactivation of mTOR through an allosteric mechanism.Citation99 To overcome drug resistance caused by mutations in the FRB domain or kinase domain, the third generation of mTOR inhibitors, RapaLink, was developed (). These inhibitors combine the high affinity for the FRB domain of mTORC1 with the effective kinase inhibition of mTOR kinase inhibitor MLN0128 through a unique juxtaposition of two-drug binding pockets.Citation99 In addition, although the mTOR kinase inhibitor MLN0128 is more potent than rapamycin, as it can inhibit 4EBP1 more effectively, it has a short retention time in vivo, and a decreased in vivo activity compared to rapamycin.Citation100 Additionally, RapaLink has an enhanced efficacy towards 4EBP1 compared to rapamycin and is able to bind to FKBP12, which improves its retention time in vivo.Citation100 Moreover, the binding of FKBP12 to mTOR is increased in RapaLink treated cells, suggesting that the dual binding of FKBP12 to both RapaLink and mTOR functions to augment the efficacy of RapaLink. Therefore, RapaLink functions effectively in both wild type cells and in cells with resistance to rapalogs or mTOR kinase inhibitors to hinder tumor growth and signaling.
Nanoparticles
Even though mTOR inhibitors have been investigated as potential cancer therapies against a wide range of tumors, their clinical development has been impeded by their pharmacokinetic limitations and the efficiency of their delivery to the target tissues. The increasing interest in nanoparticle-based drug delivery systems is aimed at improving the current cancer therapies, as these systems are able to overcome multiple biological barriers and release their therapeutic load in the optimal dose range.
The Characteristics of Nanoparticles
Nanomedicine is a multifaceted field that employs nanotechnology for diagnosis, therapy, and other medical applications.Citation101–Citation104 The definition of term “nanoparticle” is currently expanding to include structures of up to several hundred nanometers, which are formed by the precise assembly of individual componentsCitation105. Nanomaterials have a large surface area to volume ratio compared to the larger scale materials, providing them with distinct physicochemical properties. Following the success of lipid-based vesicular drug delivery nanoparticles, many different nanoparticle compositions have been developed, including polymeric micelles, dendrimers, drug conjugates, and polypeptide- and polysaccharide-based nanoparticles. Among them, Genexol-PM (methoxy-PEG-poly (D, L-lactide) Taxol), a polymeric micellar nanoparticle, was the first to be evaluated in phase II clinical trials.
Compared to a single drug, using nanoparticle drug delivery systems offers several advantages: improved efficacy, bioavailability, solubility, and stability of drugs.Citation106 Nanocarriers have the potential to enhance the effectiveness of chemical entities by optimizing their pharmacokinetic and biochemical properties. Therefore, nanocarriers allow patients to endure higher doses of drugs without experiencing side effects. Recently, nanocarriers have accelerated the development of multifunctional systems for targeted drug delivery and combined therapies and systems for simultaneous therapeutic and diagnostic applications.
The Targeting Efficacy of Nanoparticles
Higher nanoparticle delivery efficiency increases the interaction of nanoparticles with the tumor tissue. In addition to small size and susceptibility to modifications, nanoparticles can passively target solid tumors due to their enhanced permeability and retention (EPR) effect.Citation107,Citation108 The abnormalities of tumor vasculature and hypervascularization can be attributed to changes in vascular architecture and extensive production of vascular permeability factors, which stimulate extravasation within the tumor tissues, lacking lymphatic drainage.Citation107
Notably, the median efficiency of injected nanoparticles delivery was reported to be only 0.7% across 117 studies between 2006 and 2016, despite recent advances in nanotechnology.Citation109,Citation110,Citation111 Most administered nanoparticles do not reach their targeted tissues or organs due to their accumulation and sequestration in the liver and spleen (if >6 nm) or elimination through the kidney (if <6 nm).Citation112 Mononuclear phagocytic system (MPS) in liver and spleen recognize nanoparticles as foreign substances.Citation113 Therefore, to increase the delivery efficiency, it is necessary to prevent blood clearance of nanoparticles by liver, spleen, or kidney, allowing accumulation in the target tissues.Citation113
After nanoparticles enter the liver, they transverse through the hepatic sinusoid and then may be sequestered by the Kupffer cells, the tissue resident macrophages in liver sinusoids.Citation112 Kupffer cells are important for the targeting efficiency of nanoparticles; they phagocytose and destroy foreign materials and pathogens in blood, and are involved in erythrocytes recycling. Kupffer cells function as the first layer of innate immunity and the surface charge, ligand chemistry, and size of nanoparticles severely affect the uptake and the retention of nanoparticles.Citation112 Hence, in order to reach to the targeted organ, many applications were developed to block the interaction and the removal of nanoparticles by Kupffer cells ().Citation112 Once the nanoparticles transcytose through the hepatocytes, nanoparticles may travel through hepatic ducts, accumulate inside the gallbladder, or enter into the common bile duct, and then are excreted into the duodenum of small intestines.Citation112
Table 1 Summary of Applications to Prevent Nanoparticle Uptake by Mononuclear Phagocyte System (MPS)
However, physiological barriers such as tumor penetration, tumor heterogeneity, relative hypoxia, and endosomal escape may affect EPR effectiveness, necessitating the development of active targeting strategies.Citation129 The surface of nanoparticles can be modified with different ligands such as antibodies, aptamers, peptides, or small molecules, which enable them to recognize tumor-specific antigens and thus, actively target cancers and remain at the target site.Citation130 Nanoparticle-cell interactions are determined by the protein layer (corona; opsonin) that forms around nanoparticles in the plasma, inducing the subsequent internalization of nanoparticles by defined pathways, such as the endo-lysosomal pathway.Citation131 The protein corona may mask the targeting ligands, reducing the uptake of nanoparticles in the cell membrane and the targeting capacity of surface functionalized nanoparticles.Citation42 Moreover, protein corona elicits conformational changes on the surface of nanoparticles, leading to changes in accessibility and subsequent cellular signaling.Citation132 The presence of a protein corona on the nanoparticle surface may introduce undesirable interactions between nanoparticles and the immune system, resulting in immunostimulation or immunosuppression.Citation133 Therefore, the new challenges now are to predict protein corona on the surfaces of nanoparticles, or to limit the interactions of nanoparticles with serum proteins and the immune system using smaller antibody fragments (eg, scFv, Fab, F(ab')2, etc.), or other homing molecules (eg, aptamers, natural ligands, etc), functionalizing nanoparticles with self-markers, respectively.Citation133
Nanoparticles as Regulators of mTOR Signaling
The Effect of Nanoparticles on mTOR Signaling
Cancer cell lysosomes show an increased hydrolase activity during lysosomal biogenesis, leading to the destabilization of lysosomes.Citation134 In addition, lysosomes play a critical role in amino acid-induced mTORC1 activationCitation135 (). Most nanoparticles accumulate in the acidic vesicular organelles, such as endosomes and lysosomes,Citation136,Citation137 which contain hydrolytic enzymes that induce the degradation of nanoparticles. Some nanoparticles affect either the stability of lysosomes or autophagy, leading to cell death.Citation138 Therefore, targeting lysosomes using nanoparticles may present an effective strategy for anticancer therapy via the regulation of mTOR signaling ().
Table 2 Modulation of mTOR Signaling by Nanoparticles
Silica nanoparticles (Nano-SiO2) have been widely used because of biocompatibility, ease of modification, and large surface area.Citation143 Nano-SiO2 specifically localize in the lysosomes and induces autophagy in endothelial cells, as demonstrated by an increase in autophagy ultrastructures and LC3-I/II conversion.Citation140 Furthermore, Nano-SiO2 blocks the PI3K/Akt/mTOR pathway, leading to endothelial cell dysfunction.Citation139
Various modifications of nanoparticle surfaces have different effects on the stability of lysosomes. The sequestration of protons by unsaturated amino groups on the surface of nanoparticles can block the V-ATPase proton pumps and retain water in lysosomes. This “proton sponge effect” may lead to the swelling of lysosomes, followed by the leakage of lysosomal contents, lysosome rupture, and finally apoptotic cell death.Citation144 However, a specific modification with a carboxyl group, such as the modification of polystyrene nanoparticles with a carboxyl group (PS-COOH), does not affect lysosomal stability, whereas modification of polystyrene nanoparticles with an amino group (PS-NH2) leads to the elevation of pH levels in the acidic vesicular organelles and an impaired lysosomal function.Citation142,Citation145 Two kinds of functionalization of nanoparticles regulate mTOR signaling differently: PS-COOH induces mTOR activation, while PS-NH2 inhibits mTOR signaling; this indicates that nanoparticle surface modifications are important for the regulation of mTOR activity.Citation142 However, since the modulation of mTOR action by nanoparticles has low specificity, conjugating selective mTOR inhibitors with nanoparticles could be a promising approach to increase their therapeutic effects.Citation146
Rapamycin–Nanoparticle Conjugates
Polymer-drug conjugates are one of the most dominant classes of therapeutic products based on nanotechnology due to their higher pharmaceutical efficacy compared to the conjugated drugs.Citation103 These conjugates of therapeutic products with nanoparticles have been developed to improve the efficacies of previously approved drugs, rather than designing entirely new ones.Citation147,Citation148 A rapalog sirolimus (Rapamune, Pfizer, New York City, NY, USA), only available as an oral formulation, has a low bioavailability, since it is too hydrophobic for the preparation as an injectable formulation.Citation149 The low bioavailability of sirolimus restricts its usage to low-dosage treatments, such as those used for immunosuppression in renal and liver transplant recipients.Citation150 Polymeric nanoparticles (PNPs) containing sirolimus have been shown to last in blood and disperse easily in physiological media without the loss of free sirolimus effect against the cancer cells.Citation147 Furthermore, the PNP-sirolimus combination therapy has an improved radiotherapeutic effect compared to sirolimus only, leading to drastic inhibition of tumor growth.Citation147
Although nanoparticle conjugation has resolved issues associated with single-rapamycin cancer therapy, its use is still significantly restricted due to low bioavailability and resistance profiles. Therefore, co-delivery of rapamycin with other drugs is an efficient strategy for solving problems related to single-rapamycin therapy and improving the anticancer effects. Nanoparticles such as poly (lactic-co-glycolic acid) (PLGA) nanoparticles serve as carriers encapsulating the active ingredient being used for the targeted treatment of cancers. Recently, Rapa-loaded PLGA nanoparticles (Rapa-PLGA-NPs) coated with P80 (Rapa-PLGA-P80-NPs) showed anti-glioma activity in vitro.Citation151
Co-delivery of rapamycin- and piperine-loaded polymeric nanoparticles improved the oral bioavailability and efficacy of rapamycin and piperine.Citation152 Piperine (PIP) is a natural alkaloid, well known for the enhanced intestinal permeability as a P-glycoprotein (P-gp) inhibitor, as well as mild anticancer activity.Citation153–Citation155 Multidrug resistance (MDR) in tumors is caused by P-gp, an efflux transporter, leading to low drug bioavailability.Citation152 Using poly (D, L-lactide-co-glycolide) (PLGA) nanoparticles, the co-delivery of rapamycin and piperine reduced their doses and improved their bioavailability through the inhibition of P-gp efflux and increased uptake by the breast cancer tissues.Citation152
The use of cisplatin is restricted by systemic toxicities coupled with drug resistance, even though cisplatin has been widely used against a broad spectrum of solid neoplasms.Citation156 Encapsulation of cisplatin together with rapamycin inside PLGA nanoparticles led to the synergistic effect of the two drugs through the alteration of the tumor microenvironment.Citation157 In many solid tumors, an increased interstitial fluid pressure (IFP) builds a barrier to trans capillary transport, resulting in inefficient uptake of therapeutic agents.Citation158 Therefore, remodeling the tumor microenvironment might be an effective strategy for the sensitization of tumor cells to drug treatment.Citation158
Concluding Remarks and Future Directions
In the last decade, considerable progress has been made in understanding mTOR signaling and the potential of mTOR-targeted anticancer therapies; however, significant aspects of mTOR regulation need to be further clarified. The growth inhibition characteristics of rapamycin and its analogs stimulated investigations of their anticancer properties. Despite several promising findings in preclinical studies, investigations of rapalogs in cell lines and patients have shown a limited clinical impact, including selective inhibition of mTORC1 and activation of feedback loops. One of the impediments to progress in mTOR inhibition therapy is the lack of predictive biomarkers to determine its effectiveness and lack of information regarding the mechanisms of cancer unresponsiveness to this therapy. Additionally, the direct mTOR kinase inhibitors, ATP analogs, are likely to have off-target effects on other related kinases, causing an increase in toxicity. These limitations in the use of current mTOR kinase inhibitors prompted the improvement of the efficacy of these drugs using nanoparticles.
The development of nanocarriers containing mTOR inhibitors aims to optimize drug delivery to the target tissues. Although rapamycin–nanoparticle conjugates showed improved bioavailability compared to sirolimus alone (see 6.2), the molecular mechanisms of nanoparticle-induced modulation of mTOR signaling need to be investigated to maintain the selective therapeutic effect on cancers. Also, the interplay between the lysosome-mTORC1 and nanoparticles in cancer cells warrants further investigation, since both mTORC1 activation and nanoparticle accumulation occur at the lysosome. Further development of nanocarriers for the second and third generations of mTOR inhibitors will enable the emergence of cancer therapeutics targeting mTOR with high efficacy. Therefore, it is of high importance to investigate a combination of selective nanoparticles and mTOR inhibitors to treat specific cancers.
Acknowledgment
This work was supported by grants from the National Research Foundation (NRF) of Korea funded by the Korean government (Ministry of Science and ICT; NRF-2018R1A2B6004513) and by the Gachon University research fund of 2018 (GCU 2018-0312).
Disclosure
The author reports no conflicts of interest in this work.
References
- Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. 2020;21(4):183–203. doi:10.1038/s41580-019-0199-y31937935
- Sato T, Nakashima A, Guo L, Coffman K, Tamanoi F. Single amino-acid changes that confer constitutive activation of mTOR are discovered in human cancer. Oncogene. 2010;29(18):2746–2752. doi:10.1038/onc.2010.2820190810
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124(3):471–484. doi:10.1016/j.cell.2006.01.01616469695
- Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17(6):596–603. doi:10.1016/j.ceb.2005.09.00916226444
- Zoncu R, Efeyan A, Sabatini D. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12(1):21–35.21157483
- Bar-Peled L, Sabatini DM. Regulation of mTORC1 by amino acids. Trends Cell Biol. 2014;24(7):400–406. doi:10.1016/j.tcb.2014.03.00324698685
- Rui L. A link between protein translation and body weight. J Clin Invest. 2007;117(2):310–313. doi:10.1172/JCI3128917273554
- Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10(5):307–318. doi:10.1038/nrm267219339977
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. doi:10.1016/j.cell.2012.03.01722500797
- Peterson TR, Sengupta SS, Harris TE, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146(3):408–420. doi:10.1016/j.cell.2011.06.03421816276
- Benjamin D, Colombi M, Moroni C, Hall MN. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat Rev Drug Discov. 2011;10(11):868–880. doi:10.1038/nrd353122037041
- Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol. 2003;13(15):1259–1268. doi:10.1016/S0960-9822(03)00506-212906785
- Laplante M, Sabatini DM. An emerging role of mTOR in lipid biosynthesis. Curr Biol. 2009;19(22):R1046–52. doi:10.1016/j.cub.2009.09.05819948145
- Khanna N, Fang Y, Yoon MS, Chen J. XPLN is an endogenous inhibitor of mTORC2. Proc Natl Acad Sci U S A. 2013;110(40):15979–15984. doi:10.1073/pnas.131043411024043828
- Cybulski N, Hall MN. TOR complex 2: a signaling pathway of its own. Trends Biochem Sci. 2009;34(12):620–627. doi:10.1016/j.tibs.2009.09.00419875293
- Sarbassov DD, Ali SM, Sengupta S, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22(2):159–168. doi:10.1016/j.molcel.2006.03.02916603397
- Huang J, Dibble CC, Matsuzaki M, Manning BD. The TSC1-TSC2 complex is required for proper activation of mTOR complex 2. Mol Cell Biol. 2008;28(12):4104–4115. doi:10.1128/MCB.00289-0818411301
- Efeyan A, Sabatini DM. mTOR and cancer: many loops in one pathway. Curr Opin Cell Biol. 2010;22(2):169–176. doi:10.1016/j.ceb.2009.10.00719945836
- Dibble CC, Asara JM, Manning BD. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol Cell Biol. 2009;29(21):5657–5670. doi:10.1128/MCB.00735-0919720745
- Polak P, Hall MN. mTORC2 caught in a sinful Akt. Dev Cell. 2006;11(4):433–434. doi:10.1016/j.devcel.2006.09.00517011481
- Sancak Y, Peterson TR, Shaul YD, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320(5882):1496–1501. doi:10.1126/science.115753518497260
- Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141(2):290–303. doi:10.1016/j.cell.2010.02.02420381137
- Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011;334(6056):678–683. doi:10.1126/science.120705622053050
- Yoon MS, Du G, Backer JM, Frohman MA, Chen J. Class III PI-3-kinase activates phospholipase D in an amino acid-sensing mTORC1 pathway. J Cell Biol. 2011;195(3):435–447. doi:10.1083/jcb.20110703322024166
- Yoon MS, Son K, Arauz E, Han JM, Kim S, Chen J. Leucyl-tRNA synthetase activates Vps34 in amino acid-sensing mTORC1 signaling. Cell Rep. 2016;16(6):1510–1517. doi:10.1016/j.celrep.2016.07.00827477288
- Tsun ZY, Bar-Peled L, Chantranupong L, et al. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol Cell. 2013;52(4):495–505. doi:10.1016/j.molcel.2013.09.01624095279
- Wolfson RL, Chantranupong L, Wyant GA, et al. KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature. 2017;543(7645):438–442. doi:10.1038/nature2142328199306
- Chantranupong L, Wolfson RL, Orozco JM, et al. The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep. 2014;9(1):1–8. doi:10.1016/j.celrep.2014.09.01425263562
- Han JM, Jeong SJ, Park MC, et al. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell. 2012;149(2):410–424. doi:10.1016/j.cell.2012.02.04422424946
- Castellano BM, Thelen AM, Moldavski O, et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science. 2017;355(6331):1306–1311. doi:10.1126/science.aag141728336668
- Chantranupong L, Scaria SM, Saxton RA, et al. The CASTOR proteins are arginine sensors for the mTORC1 pathway. Cell. 2016;165(1):153–164. doi:10.1016/j.cell.2016.02.03526972053
- Wolfson RL, Chantranupong L, Saxton RA, et al. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science. 2016;351(6268):43–48. doi:10.1126/science.aab267426449471
- Gu X, Orozco JM, Saxton RA, et al. SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science. 2017;358(6364):813–818. doi:10.1126/science.aao326529123071
- Condon KJ, Sabatini DM. Nutrient regulation of mTORC1 at a glance. J Cell Sci. 2019;132:21. doi:10.1242/jcs.222570
- Kim YM, Jung CH, Seo M, et al. mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol Cell. 2015;57(2):207–218. doi:10.1016/j.molcel.2014.11.01325533187
- Roczniak-Ferguson A, Petit CS, Froehlich F, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012;5(228):ra42. doi:10.1126/scisignal.200279022692423
- Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8(6):903–914. doi:10.4161/auto.1965322576015
- Wyant GA, Abu-Remaileh M, Frenkel EM, et al. NUFIP1 is a ribosome receptor for starvation-induced ribophagy. Science. 2018;360(6390):751–758. doi:10.1126/science.aar266329700228
- Chiang GG, Abraham RT. Targeting the mTOR signaling network in cancer. Trends Mol Med. 2007;13(10):433–442. doi:10.1016/j.molmed.2007.08.00117905659
- Faivre S, Kroemer G, Raymond E. Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov. 2006;5(8):671–688. doi:10.1038/nrd206216883305
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18(16):1926–1945. doi:10.1101/gad.121270415314020
- Hua H, Kong Q, Zhang H, Wang J, Luo T, Jiang Y. Targeting mTOR for cancer therapy. J Hematol Oncol. 2019;12(1):71. doi:10.1186/s13045-019-0754-131277692
- Wander SA, Hennessy BT, Slingerland JM. Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. J Clin Invest. 2011;121(4):1231–1241. doi:10.1172/JCI4414521490404
- Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9(8):563–575. doi:10.1038/nrc267619629071
- Corradetti MN, Inoki K, Bardeesy N, DePinho RA, Guan KL. Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. 2004;18(13):1533–1538. doi:10.1101/gad.119910415231735
- Carretero J, Medina PP, Blanco R, et al. Dysfunctional AMPK activity, signalling through mTOR and survival in response to energetic stress in LKB1-deficient lung cancer. Oncogene. 2007;26(11):1616–1625. doi:10.1038/sj.onc.120995116953221
- Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275(5308):1943–1947. doi:10.1126/science.275.5308.19439072974
- Risinger JI, Hayes AK, Berchuck A, Barrett JC. PTEN/MMAC1 mutations in endometrial cancers. Cancer Res. 1997;57(21):4736–4738.9354433
- Zhou J, Wulfkuhle J, Zhang H, et al. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc Natl Acad Sci U S A. 2007;104(41):16158–16163. doi:10.1073/pnas.070259610417911267
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–590. doi:10.1016/S0092-8674(03)00929-214651849
- Liu L, Cash TP, Jones RG, Keith B, Thompson CB, Simon MC. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol Cell. 2006;21(4):521–531. doi:10.1016/j.molcel.2006.01.01016483933
- Yeung RS, Xiao GH, Jin F, Lee WC, Testa JR, Knudson AG. Predisposition to renal carcinoma in the Eker rat is determined by germ-line mutation of the tuberous sclerosis 2 (TSC2) gene. Proc Natl Acad Sci U S A. 1994;91(24):11413–11416. doi:10.1073/pnas.91.24.114137972075
- Kwiatkowski DJ. Tuberous sclerosis: from tubers to mTOR. Ann Hum Genet. 2003;67(Pt 1):87–96. doi:10.1046/j.1469-1809.2003.00012.x12556239
- Gao Y, Gao J, Li M, et al. Rheb1 promotes tumor progression through mTORC1 in MLL-AF9-initiated murine acute myeloid leukemia. J Hematol Oncol. 2016;9:36. doi:10.1186/s13045-016-0264-327071307
- Xu J, Pham CG, Albanese SK, et al. Mechanistically distinct cancer-associated mTOR activation clusters predict sensitivity to rapamycin. J Clin Invest. 2016;126(9):3526–3540. doi:10.1172/JCI8612027482884
- Ghosh AP, Marshall CB, Coric T, et al. Point mutations of the mTOR-RHEB pathway in renal cell carcinoma. Oncotarget. 2015;6(20):17895–17910. doi:10.18632/oncotarget.496326255626
- El Shamieh S, Saleh F, Moussa S, Kattan J, Farhat F. RICTOR gene amplification is correlated with metastasis and therapeutic resistance in triple-negative breast cancer. Pharmacogenomics. 2018;19(9):757–760. doi:10.2217/pgs-2018-001929790419
- Kim ST, Kim SY, Klempner SJ, et al. Rapamycin-insensitive companion of mTOR (RICTOR) amplification defines a subset of advanced gastric cancer and is sensitive to AZD2014-mediated mTORC1/2 inhibition. Ann Oncol. 2017;28(3):547–554. doi:10.1093/annonc/mdw66928028034
- Gkountakos A, Pilotto S, Mafficini A, et al. Unmasking the impact of Rictor in cancer: novel insights of mTORC2 complex. Carcinogenesis. 2018;39(8):971–980. doi:10.1093/carcin/bgy08629955840
- De Benedetti A, Graff JR. eIF-4E expression and its role in malignancies and metastases. Oncogene. 2004;23(18):3189–3199. doi:10.1038/sj.onc.120754515094768
- Nakamura JL, Garcia E, Pieper RO. S6K1 plays a key role in glial transformation. Cancer Res. 2008;68(16):6516–6523. doi:10.1158/0008-5472.CAN-07-618818701474
- Barlund M, Forozan F, Kononen J, et al. Detecting activation of ribosomal protein S6 kinase by complementary DNA and tissue microarray analysis. J Natl Cancer Inst. 2000;92(15):1252–1259. doi:10.1093/jnci/92.15.125210922410
- Couch FJ, Wang XY, Wu GJ, Qian J, Jenkins RB, James CD. Localization of PS6K to chromosomal region 17q23 and determination of its amplification in breast cancer. Cancer Res. 1999;59(7):1408–1411.10197603
- Bjornsti MA, Houghton PJ. Lost in translation: dysregulation of cap-dependent translation and cancer. Cancer Cell. 2004;5(6):519–523. doi:10.1016/j.ccr.2004.05.02715193254
- Ruggero D, Montanaro L, Ma L, et al. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat Med. 2004;10(5):484–486. doi:10.1038/nm104215098029
- Haydon MS, Googe JD, Sorrells DS, Ghali GE, Li BD. Progression of eIF4e gene amplification and overexpression in benign and malignant tumors of the head and neck. Cancer. 2000;88(12):2803–2810. doi:10.1002/1097-0142(20000615)88:12<2803::AID-CNCR20>3.0.CO;2-510870064
- No JH, Jeon YT, Park IA, et al. Activation of mTOR signaling pathway associated with adverse prognostic factors of epithelial ovarian cancer. Gynecol Oncol. 2011;121(1):8–12. doi:10.1016/j.ygyno.2010.12.36421276607
- O’Donnell JS, Massi D, Teng MWL, Mandala M. PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Semin Cancer Biol. 2018;48:91–103. doi:10.1016/j.semcancer.2017.04.01528467889
- Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol. 2012;12(5):325–338. doi:10.1038/nri319822517423
- Zeng H, Chi H. mTOR signaling in the differentiation and function of regulatory and effector T cells. Curr Opin Immunol. 2017;46:103–111. doi:10.1016/j.coi.2017.04.00528535458
- Lastwika KJ, Wilson W 3rd, Li QK, et al. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res. 2016;76(2):227–238. doi:10.1158/0008-5472.CAN-14-336226637667
- Mittendorf EA, Philips AV, Meric-Bernstam F, et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol Res. 2014;2(4):361–370. doi:10.1158/2326-6066.CIR-13-012724764583
- Esfahani K, Al-Aubodah T-A, Thebault P, et al. Targeting the mTOR pathway uncouples the efficacy and toxicity of PD-1 blockade in renal transplantation. Nat Commun. 2019;10(1):4712. doi:10.1038/s41467-019-12628-131624262
- Vezina C, Kudelski A, Sehgal SN. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J Antibiot (Tokyo). 1975;28(10):721–726. doi:10.7164/antibiotics.28.7211102508
- Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253(5022):905–909. doi:10.1126/science.17150941715094
- Sabers CJ, Martin MM, Brunn GJ, et al. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J Biol Chem. 1995;270(2):815–822. doi:10.1074/jbc.270.2.8157822316
- Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell. 1994;78(1):35–43. doi:10.1016/0092-8674(94)90570-37518356
- Chen J, Zheng XF, Brown EJ, Schreiber SL. Identification of an 11-kDa FKBP12-rapamycin-binding domain within the 289-kDa FKBP12-rapamycin-associated protein and characterization of a critical serine residue. Proc Natl Acad Sci U S A. 1995;92(11):4947–4951. doi:10.1073/pnas.92.11.49477539137
- Guertin DA, Sabatini DM. An expanding role for mTOR in cancer. Trends Mol Med. 2005;11(8):353–361. doi:10.1016/j.molmed.2005.06.00716002336
- Xie J, Wang X, Proud CG. mTOR inhibitors in cancer therapy. F1000Res. 2016;5(5):2078. doi:10.12688/f1000research.9207.1
- Huang S, Houghton PJ. Inhibitors of mammalian target of rapamycin as novel antitumor agents: from bench to clinic. Curr Opin Investig Drugs. 2002;3(2):295–304.
- Dancey JE. Therapeutic targets: MTOR and related pathways. Cancer Biol Ther. 2006;5(9):1065–1073. doi:10.4161/cbt.5.9.317516969122
- Wolpin BM, Hezel AF, Abrams T, et al. Oral mTOR inhibitor everolimus in patients with gemcitabine-refractory metastatic pancreatic cancer. J Clin Oncol. 2009;27(2):193–198. doi:10.1200/JCO.2008.18.951419047305
- Johnston PB, Inwards DJ, Colgan JP, et al. A Phase II trial of the oral mTOR inhibitor everolimus in relapsed Hodgkin lymphoma. Am J Hematol. 2010;85(5):320–324. doi:10.1002/ajh.2166420229590
- Witzig TE, Reeder CB, LaPlant BR, et al. A phase II trial of the oral mTOR inhibitor everolimus in relapsed aggressive lymphoma. Leukemia. 2011;25(2):341–347. doi:10.1038/leu.2010.22621135857
- Ellard SL, Clemons M, Gelmon KA, et al. Randomized phase II study comparing two schedules of everolimus in patients with recurrent/metastatic breast cancer: NCIC Clinical Trials Group IND.163. J Clin Oncol. 2009;27(27):4536–4541. doi:10.1200/JCO.2008.21.303319687332
- Oza AM, Elit L, Tsao MS, et al. Phase II study of temsirolimus in women with recurrent or metastatic endometrial cancer: a trial of the NCIC clinical trials group. J Clin Oncol. 2011;29(24):3278–3285. doi:10.1200/JCO.2010.34.157821788564
- Hess G, Herbrecht R, Romaguera J, et al. Phase III study to evaluate temsirolimus compared with investigator’s choice therapy for the treatment of relapsed or refractory mantle cell lymphoma. J Clin Oncol. 2009;27(23):3822–3829. doi:10.1200/JCO.2008.20.797719581539
- Feldman ME, Apsel B, Uotila A, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7(2):e38. doi:10.1371/journal.pbio.100003819209957
- O’Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66(3):1500–1508. doi:10.1158/0008-5472.CAN-05-292516452206
- Skeen JE, Bhaskar PT, Chen CC, et al. Akt deficiency impairs normal cell proliferation and suppresses oncogenesis in a p53-independent and mTORC1-dependent manner. Cancer Cell. 2006;10(4):269–280. doi:10.1016/j.ccr.2006.08.02217045205
- Thoreen CC, Kang SA, Chang JW, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284(12):8023–8032. doi:10.1074/jbc.M90030120019150980
- Garcia-Martinez JM, Moran J, Clarke RG, et al. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR). Biochem J. 2009;421(1):29–42. doi:10.1042/BJ2009048919402821
- Yu K, Toral-Barza L, Shi C, et al. Biochemical, cellular, and in vivo activity of novel ATP-competitive and selective inhibitors of the mammalian target of rapamycin. Cancer Res. 2009;69(15):6232–6240. doi:10.1158/0008-5472.CAN-09-029919584280
- Brachmann S, Fritsch C, Maira SM, Garcia-Echeverria C. PI3K and mTOR inhibitors: a new generation of targeted anticancer agents. Curr Opin Cell Biol. 2009;21(2):194–198. doi:10.1016/j.ceb.2008.12.01119201591
- Engelman JA, Chen L, Tan X, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14(12):1351–1356. doi:10.1038/nm.189019029981
- Ihle NT, Lemos R Jr, Wipf P, et al. Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is a dominant predictor for resistance. Cancer Res. 2009;69(1):143–150. doi:10.1158/0008-5472.CAN-07-665619117997
- Baumann P, Mandl-Weber S, Oduncu F, Schmidmaier R. The novel orally bioavailable inhibitor of phosphoinositol-3-kinase and mammalian target of rapamycin, NVP-BEZ235, inhibits growth and proliferation in multiple myeloma. Exp Cell Res. 2009;315(3):485–497. doi:10.1016/j.yexcr.2008.11.00719071109
- Rodrik-Outmezguine VS, Okaniwa M, Yao Z, et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature. 2016;534(7606):272–276. doi:10.1038/nature1796327279227
- Fan Q, Aksoy O, Wong RA, et al. A kinase inhibitor targeted to mTORC1 drives regression in glioblastoma. Cancer Cell. 2017;31(3):424–435. doi:10.1016/j.ccell.2017.01.01428292440
- Sprintz M. Editorial: nanotechnology for advanced therapy and diagnosis. Biomed Microdevices. 2004;6(2):101–103. doi:10.1023/B:BMMD.0000031745.64314.2e15320630
- Wang X, Yang L, Chen ZG, Shin DM. Application of nanotechnology in cancer therapy and imaging. CA Cancer J Clin. 2008;58(2):97–110. doi:10.3322/CA.2007.000318227410
- Farokhzad OC, Langer R. Impact of nanotechnology on drug delivery. ACS Nano. 2009;3(1):16–20. doi:10.1021/nn900002m19206243
- Majumdar D, Peng XH, Shin DM. The medicinal chemistry of theragnostics, multimodality imaging and applications of nanotechnology in cancer. Curr Top Med Chem. 2010;10(12):1211–1226. doi:10.2174/15680261079138417120388107
- Ferrari M. Cancer nanotechnology: opportunities and challenges. Nat Rev Cancer. 2005;5(3):161–171. doi:10.1038/nrc156615738981
- Alexis F. Nano-polypharmacy to treat tumors: coencapsulation of drug combinations using nanoparticle technology. Mol Ther. 2014;22(7):1239–1240. doi:10.1038/mt.2014.9624981439
- Greish K. Enhanced permeability and retention of macromolecular drugs in solid tumors: a royal gate for targeted anticancer nanomedicines. J Drug Target. 2007;15(7–8):457–464. doi:10.1080/1061186070153958417671892
- Hare JI, Lammers T, Ashford MB, Puri S, Storm G, Barry ST. Challenges and strategies in anti-cancer nanomedicine development: an industry perspective. Adv Drug Deliv Rev. 2017;108:25–38. doi:10.1016/j.addr.2016.04.02527137110
- Torrice M. Does nanomedicine have a delivery problem? ACS Cent Sci. 2016;2(7):434–437. doi:10.1021/acscentsci.6b0019027504489
- Park K. Facing the truth about nanotechnology in drug delivery. ACS Nano. 2013;7(9):7442–7447. doi:10.1021/nn404501g24490875
- Wilhelm S, Tavares AJ, Dai Q, et al. Analysis of nanoparticle delivery to tumours. Nat Rev Mater. 2016;1(5):16014. doi:10.1038/natrevmats.2016.14
- Zhang YN, Poon W, Tavares AJ, McGilvray ID, Chan WCW. Nanoparticle-liver interactions: cellular uptake and hepatobiliary elimination. J Control Release. 2016;240:332–348. doi:10.1016/j.jconrel.2016.01.02026774224
- Feliu N, Docter D, Heine M, et al. In vivo degeneration and the fate of inorganic nanoparticles. Chem Soc Rev. 2016;45(9):2440–2457. doi:10.1039/C5CS00699F26862602
- Geng Y, Dalhaimer P, Cai S, et al. Shape effects of filaments versus spherical particles in flow and drug delivery. Nat Nanotechnol. 2007;2(4):249–255. doi:10.1038/nnano.2007.7018654271
- Champion JA, Mitragotri S. Shape induced inhibition of phagocytosis of polymer particles. Pharm Res. 2009;26(1):244–249.18548338
- He C, Hu Y, Yin L, Tang C, Yin C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31(13):3657–3666. doi:10.1016/j.biomaterials.2010.01.06520138662
- Beningo KA, Wang YL. Fc-receptor-mediated phagocytosis is regulated by mechanical properties of the target. J Cell Sci. 2002;115(4):849–856.11865040
- Merkel TJ, Jones SW, Herlihy KP, et al. Using mechanobiological mimicry of red blood cells to extend circulation times of hydrogel microparticles. Proc Natl Acad Sci. 2011;108(2):586–591. doi:10.1073/pnas.101001310821220299
- Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4(2):145–160. doi:10.1038/nrd163215688077
- Liu T, Choi H, Zhou R, Chen IW. RES blockade: a strategy for boosting efficiency of nanoparticle drug. Nano Today. 2015;10(1):11–21. doi:10.1016/j.nantod.2014.12.003
- Lehenkari PP, Kellinsalmi M, Näpänkangas JP, et al. Further insight into mechanism of action of clodronate: inhibition of mitochondrial ADP/ATP translocase by a nonhydrolyzable, adenine-containing metabolite. Mol Pharmacol. 2002;61(5):1255–1262. doi:10.1124/mol.61.5.125511961144
- Hu Q, van Rooijen N, Liu D. Effect of macrophage elimination using liposome-encapsulated dichloromethylene diphosphonate on tissue distribution of liposomes. J Liposome Res. 1996;6(4):681–698. doi:10.3109/08982109609039921
- Ohara Y, Oda T, Yamada K, et al. Effective delivery of chemotherapeutic nanoparticles by depleting host Kupffer cells. Int J Cancer. 2012;131(10):2402–2410. doi:10.1002/ijc.2750222362271
- Simberg D, Duza T, Park JH, et al. Biomimetic amplification of nanoparticle homing to tumors. Proc Natl Acad Sci. 2007;104(3):932–936. doi:10.1073/pnas.061029810417215365
- Parodi A, Quattrocchi N, van de Ven AL, et al. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat Nanotechnol. 2013;8(1):61–68. doi:10.1038/nnano.2012.21223241654
- Choi M-R, Stanton-Maxey KJ, Stanley JK, et al. A cellular trojan horse for delivery of therapeutic nanoparticles into tumors. Nano Lett. 2007;7(12):3759–3765. doi:10.1021/nl072209h17979310
- Choi J, Kim H-Y, Ju EJ, et al. Use of macrophages to deliver therapeutic and imaging contrast agents to tumors. Biomaterials. 2012;33(16):4195–4203. doi:10.1016/j.biomaterials.2012.02.02222398206
- Doshi N, Zahr AS, Bhaskar S, Lahann J, Mitragotri S. Red blood cell-mimicking synthetic biomaterial particles. Proc Natl Acad Sci. 2009;106(51):21495–21499. doi:10.1073/pnas.090712710620018694
- Navya PN, Kaphle A, Srinivas SP, Bhargava SK, Rotello VM, Daima HK. Current trends and challenges in cancer management and therapy using designer nanomaterials. Nano Convergence. 2019;6(1):23.31304563
- Hua S, de Matos MBC, Metselaar JM, Storm G, Trends C. Challenges in the clinical translation of nanoparticulate nanomedicines: pathways for translational development and commercialization. Front Pharmacol. 2018;9:790. doi:10.3389/fphar.2018.0079030065653
- Bertoli F, Garry D, Monopoli MP, Salvati A, Dawson KA. The intracellular destiny of the protein corona: a study on its cellular internalization and evolution. ACS Nano. 2016;10(11):10471–10479. doi:10.1021/acsnano.6b0641127797479
- Lunova M, Prokhorov A, Jirsa M, et al. Nanoparticle core stability and surface functionalization drive the mTOR signaling pathway in hepatocellular cell lines. Sci Rep. 2017;7(1):16049. doi:10.1038/s41598-017-16447-629167516
- Rosenblum D, Joshi N, Tao W, Karp JM, Peer D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat Commun. 2018;9(1):1410. doi:10.1038/s41467-018-03705-y29650952
- Kallunki T, Olsen OD, Jaattela M. Cancer-associated lysosomal changes: friends or foes? Oncogene. 2013;32(16):1995–2004. doi:10.1038/onc.2012.29222777359
- Lunova M, Smolkova B, Lynnyk A, et al. Targeting the mTOR signaling pathway utilizing nanoparticles: a critical overview. Cancers (Basel). 2019;11:1. doi:10.3390/cancers11010082
- Parveen S, Misra R, Sahoo SK. Nanoparticles: a boon to drug delivery, therapeutics, diagnostics and imaging. Nanomedicine. 2012;8(2):147–166. doi:10.1016/j.nano.2011.05.01621703993
- Nel AE, Madler L, Velegol D, et al. Understanding biophysicochemical interactions at the nano-bio interface. Nat Mater. 2009;8(7):543–557. doi:10.1038/nmat244219525947
- Stern ST, Adiseshaiah PP, Crist RM. Autophagy and lysosomal dysfunction as emerging mechanisms of nanomaterial toxicity. Part Fibre Toxicol. 2012;9(1):20. doi:10.1186/1743-8977-9-2022697169
- Duan J, Yu Y, Yu Y, et al. Silica nanoparticles induce autophagy and endothelial dysfunction via the PI3K/Akt/mTOR signaling pathway. Int J Nanomedicine. 2014;9:5131–5141. doi:10.2147/IJN.S7107425395850
- Al-Rawi M, Diabate S, Weiss C. Uptake and intracellular localization of submicron and nano-sized SiO(2) particles in HeLa cells. Arch Toxicol. 2011;85(7):813–826. doi:10.1007/s00204-010-0642-521240478
- Chiu HW, Xia T, Lee YH, Chen CW, Tsai JC, Wang YJ. Cationic polystyrene nanospheres induce autophagic cell death through the induction of endoplasmic reticulum stress. Nanoscale. 2015;7(2):736–746. doi:10.1039/C4NR05509H25429417
- Loos C, Syrovets T, Musyanovych A, Mailander V, Landfester K, Simmet T. Amino-functionalized nanoparticles as inhibitors of mTOR and inducers of cell cycle arrest in leukemia cells. Biomaterials. 2014;35(6):1944–1953. doi:10.1016/j.biomaterials.2013.11.05624331713
- Halas NJ. Nanoscience under glass: the versatile chemistry of silica nanostructures. ACS Nano. 2008;2(2):179–183. doi:10.1021/nn800052e19206616
- Xia T, Kovochich M, Liong M, Zink JI, Nel AE. Cationic polystyrene nanosphere toxicity depends on cell-specific endocytic and mitochondrial injury pathways. ACS Nano. 2008;2(1):85–96. doi:10.1021/nn700256c19206551
- Loos C, Syrovets T, Musyanovych A, et al. Functionalized polystyrene nanoparticles as a platform for studying bio-nano interactions. Beilstein J Nanotechnol. 2014;5:2403–2412. doi:10.3762/bjnano.5.25025671136
- Hulea L, Markovic Z, Topisirovic I, Simmet T, Trajkovic V. Biomedical potential of mTOR modulation by nanoparticles. Trends Biotechnol. 2016;34(5):349–353. doi:10.1016/j.tibtech.2016.01.00526900005
- Woo HN, Chung HK, Ju EJ, et al. Preclinical evaluation of injectable sirolimus formulated with polymeric nanoparticle for cancer therapy. Int J Nanomedicine. 2012;7:2197–2208. doi:10.2147/IJN.S2948022619555
- Gonzalez-Angulo AM, Meric-Bernstam F, Chawla S, et al. Weekly nab-Rapamycin in patients with advanced nonhematologic malignancies: final results of a Phase I trial. Clin Cancer Res. 2013;19(19):5474–5484. doi:10.1158/1078-0432.CCR-12-311024089446
- Dias VC, Yatscoff RW. An in vitro method for predicting in vivo oral bioavailability of novel immunosuppressive drugs. Clin Biochem. 1996;29(1):43–49. doi:10.1016/0009-9120(95)02015-28929823
- Maramattom BV, Wijdicks EF. Sirolimus may not cause neurotoxicity in kidney and liver transplant recipients. Neurology. 2004;63(10):1958–1959. doi:10.1212/01.WNL.0000144351.63740.8115557524
- Escalona-Rayo O, Fuentes-Vázquez P, Jardon-Xicotencatl S, García-Tovar CG, Mendoza-Elvira S, Quintanar-Guerrero D. Rapamycin-loaded polysorbate 80-coated PLGA nanoparticles: optimization of formulation variables and in vitro anti-glioma assessment. J Drug Deliv Sci Technol. 2019;52:488–499. doi:10.1016/j.jddst.2019.05.026
- Katiyar SS, Muntimadugu E, Rafeeqi TA, Domb AJ, Khan W. Co-delivery of rapamycin- and piperine-loaded polymeric nanoparticles for breast cancer treatment. Drug Deliv. 2015;1–9. doi:10.3109/10717544.2015.1039667
- Bezerra DP, Castro FO, Alves AP, et al. In vivo growth-inhibition of Sarcoma 180 by piplartine and piperine, two alkaloid amides from piper. Braz J Med Biol Res. 2006;39(6):801–807. doi:10.1590/S0100-879X200600060001416751987
- Shoba G, Joy D, Joseph T, Majeed M, Rajendran R, Srinivas PS. Influence of piperine on the pharmacokinetics of curcumin in animals and human volunteers. Planta Med. 1998;64(4):353–356. doi:10.1055/s-2006-9574509619120
- Bhardwaj RK, Glaeser H, Becquemont L, Klotz U, Gupta SK, Fromm MF. Piperine, a major constituent of black pepper, inhibits human P-glycoprotein and CYP3A4. J Pharmacol Exp Ther. 2002;302(2):645–650. doi:10.1124/jpet.102.03472812130727
- Gregg RW, Molepo JM, Monpetit VJ, et al. Cisplatin neurotoxicity: the relationship between dosage, time, and platinum concentration in neurologic tissues, and morphologic evidence of toxicity. J Clin Oncol. 1992;10(5):795–803. doi:10.1200/JCO.1992.10.5.7951569451
- Guo S, Lin CM, Xu Z, Miao L, Wang Y, Huang L. Co-delivery of cisplatin and rapamycin for enhanced anticancer therapy through synergistic effects and microenvironment modulation. ACS Nano. 2014;8(5):4996–5009. doi:10.1021/nn501081524720540
- Heldin CH, Rubin K, Pietras K, Ostman A. High interstitial fluid pressure - an obstacle in cancer therapy. Nat Rev Cancer. 2004;4(10):806–813. doi:10.1038/nrc145615510161