Abstract
Oral anticoagulant therapy could be advanced using lipid-based nanoparticulate systems. This study examined lipid nanocapsules for their oral absorption potential as the first step in developing oral fondaparinux (Fp) novel carriers. Using phase inversion method and cationic surfactants such as hexadecyltrimethyl ammonium bromide (CTAB) or stearylamine (SA), cationic lipid nanocapsules (cLNCs), loaded with Fp on their surface, were prepared and characterized (zeta potential, size and Fp association efficiency and content). In vivo studies were conducted after single oral increasing doses of Fp-loaded cLNCs (0.5 to 5 mg/kg of Fp) in rats and the concentration of Fp in the plasma was measured by anti-factor Xa activity assay. The monodisperse, (~50 nm), positively charged Fp-cLNCs with high drug loadings demonstrated linear pharmacokinetic profiles of the drug with an increased oral absolute bioavailability (up to ~21%) compatible with therapeutic anticoagulant effect (>0.2 μg/mL).
Video abstract
Point your SmartPhone at the code above. If you have a QR code reader the video abstract will appear. Or use:
Introduction
Anticoagulants, being a mainstay of cardiovascular therapy, are widely used for prevention and treatment of venous thromboembolism, management of acute coronary syndromes and stroke prevention in patients with atrial fibrillation or mechanical heart valves.Citation1,Citation2 The traditional anticoagulants: unfractionated heparin (UFH); low molecular weight heparins (LMWH); and the synthetic pentasaccharide anti-factor Xa, fondaparinux (Fp), are administered only via intravenous (IV) or subcutaneous (SC) injection since they are not efficiently absorbed through the gastrointestinal tract when taken orally.Citation3 Oral anticoagulation is still commonly achieved by administration of vitamin K antagonists (VKA) that are associated with untoward side effects due to their narrow therapeutic range and interaction potential.Citation4 For the new oral direct factor Xa inhibitors and direct thrombin inhibitors,Citation1,Citation5 various challenges still remain such as decreasing the incidence of bleeding side effects.Citation4,Citation6,Citation7 There is thus a rationale to develop an oral formulation of Fp.
Different oral strategies have proved to be effective as noninvasive delivery systems. The use of polymers of high molecular weight with mucoadhesion properties such as chitosan derivatives or thiolated polymers alone or with glutathione demonstrated a powerful capacity to increase UFH and recently Fp oral absorption.Citation8–Citation10 However, potentially, safety concerns have always been considered with these compounds.Citation11 Polymeric drug delivery systems based on hydrogels, nanoparticles, microspheres,Citation12–Citation14 as well as lipid-based drug delivery systems (eg, microemulsions, liposomes, and solid lipid nanoparticles) have been developed and employed for oral macromolecular drug delivery in general and specifically for UFH or LMWH delivery. They seem to provide high transfer of the drug across the epithelium mucosa and protect against enzymatic degradation. The role of lipid dosage forms in intestinal absorption was extensively discussed.Citation15–Citation17 One important issue with the lipid-based particles (conventional liposomes and microemulsions) is that, generally, they lack efficiency to entrap hydrophilic macromolecular drugs and have low stability in the GI tract.Citation18,Citation19
Lipid nanocapsules (LNCs), recently developed, combine the advantages of colloidal particles and lipid delivery systems. They are prepared by a solvent-free, simple phase inversion technique and are composed of rigid surfactant shell surrounding an oily liquid core.Citation20 They have shown potential for the delivery of many drugs by different routes, mainly parenteralCitation21 as well as pulmonaryCitation22 and more interestingly, the oral route.Citation23 Their gastrointestinal stabilityCitation24 and their P-gp inhibiting properties are attributed to Solutol® HS15 (BASF, Ludwigshafen, Germany), which is a key excipient in their composition.Citation25 Stability of nanocapsules and P-gp inhibition facilitate intracellular uptake of paclitaxel, an anticancer drug, resulting in its oral absorptionCitation23 proven to be mainly via active endocytic process.Citation26 The safety of these new nanocapsules has been demonstrated.Citation27
Fondaparinux (Arixtra®, GlaxoSmithKline, London, UK) shares all the pharmacological and biological advantages of LMWHs over UFH. Being an entirely synthetic molecule, with high affinity to antithrombin (AT), Fp catalyzes AT-mediated inactivation of factor Xa.Citation28 It displays a rapid onset of action and long terminal half-life that permits once-daily administration.Citation29 It lacks antifactor IIa activity, which has been associated with bleeding in Hp and other derivative drugs in clinical trials.Citation29–Citation31 Fp does not require dose adjustment for age or gender, has predictable effects over the defined therapeutic dose range, and does not require routine laboratory monitoring.Citation32 Due to its safety/efficacy profile, Fp should be an ideal agent for home therapy.Citation30,Citation32–Citation34 Until nowadays, it is not clear even if dabigatran and/or rivaroxaban, are more effective than Fp due to the lack of studies comparing them.Citation31
On the basis of the above mentioned considerations, it was sought, in this study, to combine the advantages of LNCs as oral delivery systems with the benefit of the presence of acquired positive charge on their surfaces, to both associate the anionic hydrophilic macromolecule Fp and to increase electrostatic interactions with the intestinal mucosa. With this goal in mind, Fp-loaded cationic lipid nanocapsules (cLNCs) were prepared and were characterized in vivo. Then, the impact of this delivery system on the pharmacokinetics of the drug after oral administration in rats was investigated.
Material and methods
Preparation of lipid nanocapsules
The cLNCs were prepared by the phase inversion method previously describedCitation20 but modified to incorporate cationic surfactants: They are composed of a liquid oily core surrounded by a cohesive interface and dispersed in an aqueous medium. The oily phase is composed of 5.11% w/w of the lipophilic Labrafac® WL 1349 (caprylic-capric acid triglycerides; European Pharmacopeia, IVth, 2002, Gattefossé S.A., Saint-Priest, France). The aqueous phase consists of 0.42% w/w NaCl (Prolabo; Fontenay-sous-Bois, France) and 14.04% w/w water (Milli RO® System, Millipore, Paris, France). Two surfactants are used for LNC preparation: 0.39%–0.7% w/w Lipoïd® S75-3 (a soybean lecithin at 69% of phosphatidylcholine, Lipoïd GMBH Ludwigshafen, Germany) and 4.72% w/w Solutol® HS 15 (a mixture of free polyethylene glycol 660 and polyethylene glycol 660 hydroxystearate, European Pharmacopeia, IVth, 2002, BASF, Ludwigshafen, Germany). Briefly, the emulsion is first prepared by mixing all the components under magnetic stirring at room temperature, with a progressive heating to 85°C followed by a progressive cooling from 85°C to 60°C at a rate of 4°C/minute. Three temperature cycles are applied to obtain the inversion process defined by a temperature range which depends on the composition of the mixture.Citation35 An irreversible shock is then induced by dilution with cold deionized water (2°C) added to the mixture (3.5-fold by dilution) leading to the formation of stable nanocapsules.Citation20 Afterwards slow magnetic stirring is applied for 5 minutes, followed by filtration (0.2 μm syringe filter, Sartorius, Gottingen, Germany).
Cationic surfactants (hexadecyltrimethyl ammonium bromide (CTAB) or stearylamine (SA) (Sigma Aldrich, St Quentin-Fallavier, and Yvelines, France)) having a molecular weight of 364.45 and 269.52 g/mol respectively, were added from the beginning to the other components in different concentrations (0.125% to 1% w/v of the final volume). The concentration of the cationic surfactant was chosen based on an adsorption studyCitation36 and on the desired amount of drug to be associated with the nanocarriers; higher cationic surfactant concentrations were used for higher drug loadings. Lipoid® concentration was also changed in all formulations, and its ratio with the cationic surfactants differed being the least with SA (1:1.5) (). Different volumes of Fp solution (Arixtra® 10 mg/0.8 mL, GlaxoSmithKline, Evreux, France) were rapidly injected at the phase inversion zone at a temperature close to 70°C, at the third cycle before the final dilution with cold water. A final Fp concentration ranging from 90 to 1517 μg/mL was obtained in cationic lipid nanocapsules dispersions (Fp-cLNCs). Blank cLNCs with CTAB and SA were also prepared as controls.
Table 1 Variation in composition of cationic lipid nanocapsule formulations
In vitro characterisation
Particle size and zeta potential measurements
The cLNCs were analyzed for their average particle size (PS) and the polydispersity index (PI) by photon correlation spectroscopy (PCS) using a Malvern Zetasizer® Nano ZS Series DTS 1060 (Malvern Instruments S.A., Worcestershire, UK) at a fixed angle (173°) at 25°C furnished by a 4 mW He–Ne_laser at 633 nm. The polydispersity index (PDI) was used as a measurement of the size distribution (a value of PDI ≤ 0.2 indicates a unimodal size distribution). A 1:60 v/v dilution of the nanoparticle suspensions in deionized water was achieved in order to enable measurements.
PCS data are presented as the mean (volume) intensity-particle size distribution, and standard deviation (SD) of the mean of particle size measurements performed in triplicate was calculated. The samples were kept in polystyrene vials. The shelf stability of Fp-cLNCs at 4°C with respect to particle size was investigated for up to 4 months.
The zeta (ζ) potential and electrophoretic mobility measurements of the different nanoparticles were performed using the same Malvern instrument equipped with an AZ-4 cell and were based on the laser Doppler effect. Zeta potential measurements were made also after similar dilutions (1:60 v/v) in deionized water at 25°C, with a dielectric constant of 79, refractive index of 1.33, viscosity of 0.89 cP, cell voltage 150 V and current of 5 mA. Data from the analysis of ζ potential are presented as mean and SD of triplicate runs.
Drug association efficiency and drug payload
The amount of Fp unentrapped within the cLNCs was determined with a modified Azure A colorimetric method.Citation37 The amount of free dye in the external LNC suspension was indirectly proportional to the amount of unentrapped drug. Typically, aliquots (500 μL) of cLNCs samples were reacted with 1.5 mL of Azure A (Sigma Aldrich, St Quentin-Fallavier, Yvelines, France) solution (0.04 mg/mL) at room temperature followed by ultrafiltration at 4000 rpm for 30 minutes using (Millipore® Centricon YM-100 filters, Millipore Corporation, Billerica, MA) to separate the purple complex of Azure with free Fp as well as the Fp-cLNCs. The excess blue dye solution in the filtrate was then measured in triplicate at 633 nm by UV spectroscopy in reference to a standard calibration performed with different concentrations of Fp solution and empty cLNCs dispersions. The error of determination of Fp does not exceed 0.04 μg/mL, and the detection limit is 4 μg/mL. The drug association efficiency was expressed as the percentage of Fp associated with respect to the total drug content.
Prior to using the indirect technique for routine assay purposes, the correlation of our results with the biological anti-Xa activity assay was done to prove that association with cLNCs did not affect its anticoagulant effect.
In vivo experiments
Experiments were carried out according to the French legislation on animal experiments. Male Wistar rats (mean weight 325.8 ± 14.1 g) were fasted for 12 hours. They were anesthetized with isoflurane (0.5%) and divided into 4 groups: two groups received Fp (200 μg/kg) in isotonic solution by IV bolus injection via the tail vein or by SC injection (dorsal), the two remaining groups received orally Fp solution or Fp-loaded cLNCs suspensions as a single dose with different Fp concentrations. Oral administration was through an oral gavage that was carefully passed down the esophagus into the stomach using a stainless steel catheter with a blunt end to avoid lesions. The Fp control solution and Fp-loaded cLNCs suspensions were prepared in a sodium bicarbonate buffer (1.5 g/100 mL, pH 8.2) to neutralize the gastric acidity as described elsewhere.Citation38 The total orally administered volume of the Fp-loaded cLNCs suspensions was 1.5 to 2.5 mL. The dose amounts were 0.5, 2 and 5 mg/kg. Five rats were included in each group at the beginning of the study: some died during the study due to the sampling procedure (n final = 3–5). Blood (950 μL) was collected serially by capillary intracardiac sampling into pediatric citrated tubes containing a constant volume of sodium citrate (0.109 M). Blood sampling was started at 2 minutes in the case of IV or SC injection and after 15 minutes in the case of oral administration followed by sampling at 30 minutes, 1 hour, 2, 4, 8 and 24 hours. Plasma was collected after centrifugation (4000 rpm, 15 minutes, 4°C) and stored at −20°C until analysis using the chromogenic assay.
Measurement of anti-Xa activity (biological assay)
The anti-Xa chromogenic assay was performed using the Biophen Heparin 6 kit (HYPHEN BioMed, Neuville- sur-Oise, France) on an automated analyzer (STA-R®, STAGO, Asnières-sur-Seine, France) according to the protocol supplied by the manufacturer. In case of results over range, samples were diluted with human normal plasma to obtain Fp concentrations below 0.7 μg/mL.
The anti-Xa activity was measured from the calibration curve of absorbance versus Fp standard concentrations (μg/mL) with STA-fondaparinux calibrator and STA-fondaparinux control kits (Diagnostica Stago, Asnières-sur-Seine, France) according to the protocol supplied by the manufacturer. This assay had a coefficient of variation <7% and a limit of quantification of 0.01 μg/mL. The fondaparinux concentration was directly deduced from a calibration curve.
Determination of pharmacokinetic parameters (PK)
Noncompartmental analysis (EquivTest,™ Statistical Solutions Ltd, Saugus, MA) was performed for Fp absorption profiles. The area under the plasma concentration versus time curve (AUC0→∞) was calculated by the trapezoidal method. The absolute bioavailability BA (%) of oral Fp was calculated as the ratio of the AUC after oral and after IV administration with a correction for the difference in dose. Using the plasma concentration time curves after oral and IV administration, relevant PK parameters including mean residence time (MRT), half-life, volume of distribution, total clearance were calculated using the appropriate equations derived from noncompartmental analysis.
Statistical analysis
All studies were repeated a minimum of three times and parameters determined at least in triplicate. Results were reported as means ± SD (SD = standard deviation). Statistical significance was analyzed using the Student’s t-test or Anova (Excel®, Microsoft Corporation, Redmond WA,). Differences between experimental groups were considered significant when the P-value was less than 0.05.
Results
In vitro characterization
Particle size, zeta potential measurements and association efficiency
The mean diameter of Fp-loaded cLNCs prepared with different cationic surfactant ranged from 45.2 ± 2.4 nm to 60.1 ± 2.3 nm with a mean of 53.2 ± 8.9 nm and 47.0 ± 0.8 nm for Fp-CTAB-LNCs and Fp-SA-LNCs respectively, whereas that for the empty cLNCs was 49.8 ± 2.6 nm. A low PDI was shown for all the formulations (<0.22) (). The particle size increased with the increase in drug loading showing the highest (60.1 ± 2.3 nm) in case of Fp-cLNCs-C4 but remained unchanged with Fp-SA-LNCs formulations. CTAB concentration range was (0.12%–0.36% w/w) of total cLNCs weight. Higher CTAB and SA concentrations were needed for higher drug concentrations as shown in . The cLNCs sizes remained almost unchanged after 4 months storage at 4°C. Ratios of initial size versus size after storage for Fp-CTAB-LNCs and Fp-SA-LNCs were as follows: 0.72 ± 0.02 and 0.80 ± 0.06, respectively.
Table 2 In vitro characterization of the cLNC
The overall charge of the cLNCs was positive as shown from ζ potential data, ranging from (3.9 ± 0.4 to 42.9 ±0.5 mV), showing the highest positive value with Fp-cLNCs-C3 and the lowest values shown with SA formulations.
Association efficiency for the anionic drug was high for all cLNCs, ranging from 80% to 100% with a mean of 93.5% ± 6.0% and the highest values (100%) with Fp-cLNCs-C1 and Fp-cLNCs-C3.
In vivo study
Pharmacokinetics of Fp-loaded lipid nanocapsules
Fondaparinux plasma concentration after intravenous administration of a 200 μg/kg dose is shown in . A SC injection of a dose equal to the IV dose resulted in an anti-Xa level of 0.836 μg/mL within 60 minutes and 115% ± 17% bioavailability (). Fp is totally absorbed by the SC route:Citation29 The maximum plasma concentration (Cmax) is reached between 1 and 3 hours.
Table 3 Pharmacokinetic parameters following oral administration of Fp-loaded CTAB-LNC and SA-LNC in comparison with a bolus IV injection
Figure 1 Anti Xa activity in plasma versus time of fondaparinux in rats, after IV bolus injection and SC injection of Fp (200 μg/kg, n = 4).
Abbreviations: MEC, minimum effective therapeutic concentration; IV, intravenous; SC, subcutaneous; μg, microgram; mL, milliliter.
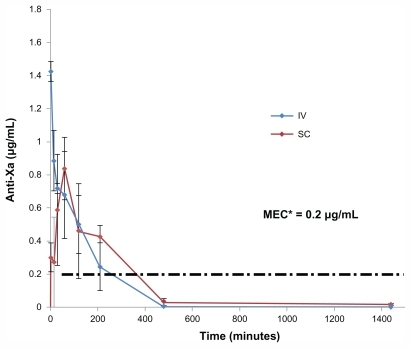
Oral formulations with CTAB-LNCs had a fast onset of action (30–60 minutes). On the contrary, SA-LNCs showed a much slower action rate (Tmax 90–210 minutes, P < 0.05) with the anticoagulant effect lasting up to 16 hours ().
Figure 2 Anti Xa activity in plasma versus time of fondaparinux in rats after oral administration of LNCs formulations (A) = CTAB-LNCs, (B) = SA-LNCs, different Fp doses (0.5, 2 or 5 mg/kg).
Notes: Data are shown as the mean concentration, and error bars represent the SEM (n = 3–5).
Abbreviations: IU, international unit; mL, milliliter; mg, milligram; kg, kilogram; CTAB, hexadecyltrimethyl ammonium bromide; LNC, lipid nanocapsule; SA, stearylamine; Fp, fondaparinux; SEM, standard error mean.
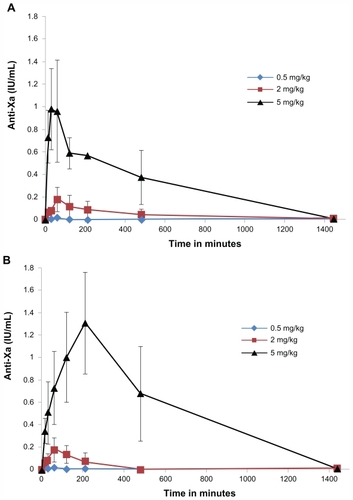
The oral administration of Fp solution alone (0.5, 2 mg/kg) did not affect the anti-Xa level in rat plasma (all samples were found to be below the limit of quantification). All failed to attain the therapeuticCitation39 anti-factor Xa levels in rats (0.2 μg/mL).Citation40
Oral administration of Fp-loaded cLNCs at a 2 mg/kg dose produced a slight increase in the peak plasma concentration of Fp (0.18 μg/mL) with both formulations and reached higher values with higher dose (5 mg/kg), up to 0.98 ± 0.33 μg/mL with CTAB-LNCs and more significantly with SA-LNCs (1.31 ± 0.37 μg/mL, P < 0.05).
The area under the anti-Xa activity in rat plasma versus time curve (AUC0–∞h) at the dose of 2 mg/kg with CTAB-LNCs and SA-LNCs formula was 78.7 μg.minutes/mL and 42.75 μg.minutes/mL respectively. The lowest dose (0.5 mg/kg) did not affect AUC0–∞h but the effect was significantly higher with the highest doses especially with SA-LNCs (5 mg/kg) (730.89 ± 214.8 μg. minutes/mL, P < 0.05) compared to 138.21 μg · minutes/mL for the IV dose (200 μg/kg). The extent of increase of the absolute bioavailability is shown in .
Figure 3 Effect of Fp dose in different loaded cLNCs on the absolute bioavailability.
Notes: Mean ± SD, normalized for the dose difference between oral and iv dose. (n = 3–5). *P < 0.05 and **P < 0.001.
Abbreviations: CTAB, hexadecyltrimethyl ammonium bromide; SA, stearylamine; LNC, lipid nanocapsules; μg, microgram; kg, kilogram; SD, standard deviation.
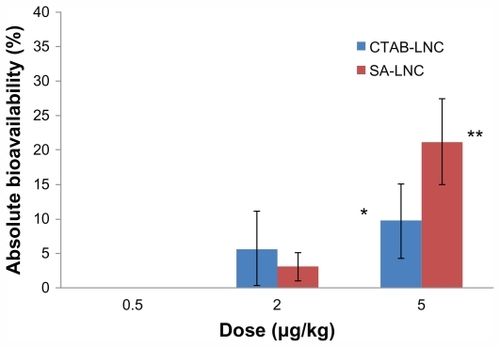
In summary, cLNCs significantly increased oral bioavailability of Fp and improved its PK profile in a dose-dependent fashion. It is obvious from the above results that the Fp-loaded cLNCs increased the oral bioavailability of the pentasaccharide significantly with a 5 mg/kg oral dose CTAB formulation (9.75% ± 5.4%, P < 0.05) and more with SA-LNCs (21.18% ± 6.2%, P < 0.05) but non significantly with lower oral doses (2 mg/kg) of the same formulations. Furthermore, the MRT and half-life increased up to three times after oral administration of the different Fp-cLNCs formulations compared to 70.49 minutes and 108.17 minutes respectively after IV administration of Fp solution ().
The computation of PK parameters with a non-compartmental model for the different groups showed high variations in each group from the mean, especially at the highest doses of Fp-loaded CTAB-LNCs and SA-LNCs (5 mg/kg). Data from SA-LNCs (5 mg/kg) were shown to be less variable with SEM <30% and n = 5. Variability is well recognized in a number of studies conducted in vivo.Citation23,Citation41
Discussion
Heparin derivatives (UFH, LMWH, Fp) cannot be administered by the oral route due to a very low bioavailability. New oral direct factor Xa inhibitors have reached the market but do not display the same safety profile as heparin derivatives, especially on the liver. New lipid nanocapsules (LNCs) have shown a great potential to improve the bioavailability of drugs such as paclitaxelCitation23 or Sn38,Citation42 but these drug delivery systems easily entrap only lipophilic and amphiphilic drugs.Citation43 In the present work we studied the possibility of using LNCs to improve the bioavailability of oral Fp, which is a negatively charged hydrophilic molecule. The challenge was to find a way to associate Fp to LNCs and then to obtain therapeutic anti-Xa activity after oral administration of the new formulations.
To successfully entrap Fp, introduction of a positively charged molecule into the structure of LNCs was conducted in order to form ion pairs with the polyanionic Fp at the water/oil interface. Using CTAB or SA in the primary emulsion did not prevent the phase inversion phenomenon, which is a key step in producing LNCs. In another parallel study, the LNC surface was positively charged using a layer-by-layer approach with lipochitosan as the cationic polymer.Citation44 Other authors reported similar strategies for other anionic molecules.Citation45
Blank LNCs have an average diameter (~50 nm) based on the respective proportions of their components.Citation35 CTAB- LNCs average particle diameter was increased slightly (~60 nm) with an increase in drug loading (). This could be attributed to the association of the drug on the surface of the cLNCs. The positively charged quaternary ammonium groups (QA) of CTAB could be mainly directed toward the external aqueous phase, thus leading to the increase in the mean diameter, with the increase in drug loading. In SA formulations, the particle size remained more or less constant (47 nm) although the proportions of total surfactants were higher than that in CTAB formulations (5.88%–7.27% w/w compared to 5.41% w/w respectively) and even more if molecular weight is taken into account. This could be due to reduced interaction of Fp with the primary amine groups of SA as reflected by lower association efficiency (84%–90%).
The ζ potential was changed for the cLNCs when compared to blank LNCs (~ − 1 mV), being more positive in the case of CTAB-LNCs (+32.0 ± 1.7 mV) than in the case of SA-LNCs (+11.9 ± 0.5 mV). This is due to the presence of the cationic agent in its ionized form at the oil/water interface.Citation46 The magnitude of the ζ potential decreased in a Fp concentration-dependent manner due to neutralization. Whether Fp was located on the surface of the cLNCs or in the core has to be further investigated. The steric effect of SA or CTAB or Fp chains at the surface of the complex can significantly improve stability and prevent aggregation of the cLNCs even when the ζ potential is less than 10 mV (eg, SA-LNCs). Similar phenomena were also observed by other groups.Citation47
The toxicity of CTAB and SA is well defined (rat LD50 oral 410 and 1395 mg/kg respectively) and their concentration can be kept to a minimum in the formulations (0.45 and 0.92 mg/kg LNC) respectively, corresponding to doses of 30 and 90 mg/kg in rats. This is lower than a tenth of the LD50. Moreover, although CTAB was chosen as the initial surfactant, the work with SA clearly shows that CTAB can be replaced, if necessary, by safer single-tailed cationic lipid moieties without reducing the potency of the delivery system. However, the toxicity of these new cationic nanocapsules remains to be determined, as it was previously assessed with standard LNCs containing only Solutol HS15 as a surfactant.Citation27
Fondaparinux, like Hp, is presumed unstable under acidic enzymatic conditions.Citation48 Few studies support the observations that Hps are absorbed by the gastrointestinal tract but they do not reach therapeutic concentration.Citation49 Active transport is involved and is pH-dependent, preferably around pH = 7.Citation50 Some in vivo studies of UFH and LMWHs were carried out using in situ intestinal administration.Citation51,Citation52 Subsequently, most were carried out by oral gavage where promising results were obtainedCitation53 and some even extended to clinical trials.Citation54 In the present study, cLNCs have been prepared using bicarbonate buffer, resulting in dispersions having a final mean pH of 8.5 ± 0.5 for CTAB and SA formulations. Bicarbonate was also added to the control formulation (free Fp). They were administered in doses that are somewhat higher and much higher than the IV or SC doses for Fp, to compensate for the detrimental effects of the oral route (eg, degradation and dilution).Citation55 Fp reached therapeutic concentration in plasma only if it was associated to LNCs (), but not if administered as free Fp at a dose of 5 mg/kg, thus highlighting the role of cLNCs in facilitating Fp absorption and stability.
Linear PKs were reported in the literature for Hp, LMWH and Fp after SC injection.Citation29 Fp-CTAB-LNCs and Fp-SA-LNCs gave the same responses for low oral doses of 2 mg/kg: Cmax and percentage bioavailability did not differ (P > 0.05) although CTAB formulations were more positive (30 mV) compared to SA formulations (11 mV). In the high dose group (5 mg/kg), a small positive charge on the SA-LNCs formulation (3 mV) gave a significantly better pharmacokinetics profile than that of CTAB-LNCs formulations (42 mV) (), and further resulted in a significant increase in AUC (17-fold, P < 0.05). Bioavailability also significantly increased (up to 21%) (). These results suggest that the influence of surface charge on bioavailability is complex for Fp-LNCs. In fact, a high positive charge is often claimed to improve nanoparticle absorption but could on the other hand limit mucus diffusion.Citation56
The pharmacokinetic parameters, such as half-life and mean residence time (MRT), were longer for Fp-cLNCs formulations after oral administration compared to Fp control solutions (oral and IV) but clearance was not significantly different (). These results suggest a long circulating time in vivo with sustained release of the drug resulting in better bioavailability while the elimination of the drug remained constant whatever the formulation or route of administration used. This could be helpful in providing a sustained anti-Xa effect. However, further studies are necessary to explore any possible permeation of intact LNCs across the GI membrane or its distribution to lipid tissues acting as storage compartment in order to explain the observed PK parameters.
In a recent study using another oral delivery system as a permeation enhancer for fondaparinux, based on thiolated polycarbophil (PCP-Cys) and glutathione (GSH) combined with sodium decanoate, only 4.4% bioavailability was reported using tablets and/or gel formulations containing doses of 5 mg/kg Fp.Citation10 The bioavailability found with cLNCs was higher (up to 21%).
Is oral Fp better than the new generation of oral anti-factor Xa and IIa inhibitors such as dabigatran etexilate (DTI), a promising novel, small molecule that specifically and reversibly inhibits thrombin?Citation57 Interestingly, Fp-loaded cLNCs gave an anti-factor Xa effect (Cmax >0.1 μg/mL) within 2 hours (Tmax) with sustained release properties. Could oral Fp compete with these agents, having the same advantages and including: no requirement for anticoagulant monitoring; a low drug–drug interaction potential; and the possibility of use in both acute and chronic settings. Further studies and comparisons should be carried out to provide a better therapeutic outcome.
Conclusion
This study is considered an extension of the applicability of lipid nanocapsules as an oral delivery system. In the present case, lipid nanocapsule technology was used for encapsulation of a high therapeutic value drug, fondaparinux. Fp-loaded cLNCs demonstrated potential for a formulation increasing both the oral bioavailability and residence time of the indirect anti-factor Xa inhibitor. Oral Fp could thus be envisioned as an alternative to currently used subcutaneous delivery. This first proof of concept of enhanced oral bioavailability of fondaparinux by nanocarriers may be of great clinical use. This new simple formulation strategy for an oral treatment may replace the currently used injectable anticoagulants, to improve patient acceptability and therapeutic effects, facilitating the development of chronic treatment schedules without the bleeding side effects and the coagulation monitoring required with vitamin K antagonist therapy.
Acknowledgments
Alyaa Ramadan acknowledges all funding sources that supported this work as well as all institutional or corporate affiliations of the authors. This includes the contribution of the University of Angers, INSERM U646 and Angers University Hospital for financial and personal support for the conduction of the study as well as the University of Alexandria, Egypt to the joint-supervision grant.
Disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
- De CaterinaRHustedSWallentinLAnticoagulants in heart disease: current status and perspectivesEur Heart J200728788091317426080
- WeitzDSWeitzJIUpdate on heparin: what do we need to know?J Thromb Thrombolysis201029219920719882363
- MotlekarNAYouanBBThe quest for non-invasive delivery of bioactive macromolecules: a focus on heparinsJ Control Release200611329110116777255
- WeitzJINew oral anticoagulants in developmentThromb Haemost20101031627020062934
- GarciaDLibbyECrowtherMAThe new oral anticoagulantsBlood20101151152019880491
- MavrakanasTBounameauxHThe potential role of new oral anti-coagulants in the prevention and treatment of thromboembolismPharmacol Ther20111301465821185864
- TurpieAGDirect Factor Xa and direct thrombin inhibitors: a clinical trial updateCurr Opin Drug Discov Devel2009124497508
- ThanouMHendersonSKydonieusAElsonCN-sulfonato-N,O-carboxymethylchitosan: a novel polymeric absorption enhancer for the oral delivery of macromoleculesJ Control Release2007117217117817184870
- Bernkop-SchnurchAKastCEGuggiDPermeation enhancing polymers in oral delivery of hydrophilic macromolecules: thiomer/GSH systemsJ Control Release20039329510314636716
- VetterAPereraGLeithnerKKlimaGBernkop-SchnurchADevelopment and in vivo bioavailability study of an oral fondaparinux delivery systemEur J Pharm Sci2010413–448949720705133
- Khafagyel-SMorishitaMOnukiYTakayamaKCurrent challenges in non-invasive insulin delivery systems: a comparative reviewAdv Drug Deliv Rev200759151521154617881081
- LamprechtAUbrichNMaincentPOral low molecular weight heparin delivery by microparticles from complex coacervationEur J Pharm Biopharm200767363263817540548
- JiaoYUbrichNMarchand-ArvierMIn vitro and in vivo evaluation of oral heparin-loaded polymeric nanoparticles in rabbitsCirculation2002105223023511790706
- HoffartVUbrichNLamprechtAMicroencapsulation of low molecular weight heparin into polymeric particles designed with biodegradable and nonbiodegradable polycationic polymersDrug Deliv20031011712554358
- PorterCJPoutonCWCuineJFCharmanWNEnhancing intestinal drug solubilisation using lipid-based delivery systemsAdv Drug Deliv Rev200860667369118155801
- MartinsSSarmentoBFerreiraDCSoutoEBLipid-based colloidal carriers for peptide and protein delivery – liposomes versus lipid nanoparticlesInt J Nanomedicine20072459560718203427
- ChakrabortySShuklaDMishraBSinghSLipid – an emerging platform for oral delivery of drugs with poor bioavailabilityEur J Pharm Biopharm200973111519505572
- GotoTMorishitaMNishimuraKNovel mucosal insulin delivery systems based on fusogenic liposomesPharm Res200623238439116382280
- TakeuchiHMatsuiYYamamotoHKawashimaYMucoadhesive properties of carbopol or chitosan-coated liposomes and their effectiveness in the oral administration of calcitonin to ratsJ Control Release2003862–323524212526820
- HeurtaultBSaulnierPPechBProustJEBenoitJPA novel phase inversion-based process for the preparation of lipid nanocarriersPharm Res200219687588012134960
- LacoeuilleFHindreFMoalFIn vivo evaluation of lipid nanocapsules as a promising colloidal carrier for paclitaxelInt J Pharm20073441–214314917646066
- HureauxJLagarceFGagnadouxFLipid nanocapsules: ready-to-use nanovectors for the aerosol delivery of paclitaxelEur J Pharm Biopharm200973223924619560538
- PeltierSOgerJMLagarceFCouetWBenoitJPEnhanced oral paclitaxel bioavailability after administration of paclitaxel-loaded lipid nanocapsulesPharm Res20062361243125016715372
- RogerELagarceFBenoitJPThe gastrointestinal stability of lipid nanocapsulesInt J Pharm2009379226026519524655
- CoonJSKnudsonWClodfelterKLuBWeinsteinRSSolutol HS 15, nontoxic polyoxyethylene esters of 12-hydroxystearic acid, reverses multidrug resistanceCancer Res19915138979021988130
- RogerELagarceFGarcionEBenoitJPLipid nanocarriers improve paclitaxel transport throughout human intestinal epithelial cells by using vesicle-mediated transcytosisJ Control Release2009140217418119699246
- HureauxJLagarceFGagnadouxFToxicological study and efficacy of blank and paclitaxel-loaded lipid nanocapsules after i.v. administration in micePharm Res201027342143020054705
- TurpieAGErikssonBILassenMRBauerKAFondaparinux, the first selective factor Xa inhibitorCurr Opin Hematol200310532733212913785
- DonatFDuretJPSantoniAThe pharmacokinetics of fondaparinux sodium in healthy volunteersClin Pharmacokinet200241Suppl 21912383039
- SchieleFFondaparinux and acute coronary syndromes: update on the OASIS 5–6 studiesVasc Health Risk Manag2010617918720407625
- SantiRContinoLGattoSLevisAFondaparinux in major orthopedic surgical patientsIntern Emerg Med20105216720195927
- BauerKAHawkinsDWPetersPCFondaparinux, a synthetic pentasaccharide: the first in a new class of antithrombotic agents – the selective factor Xa inhibitorsCardiovasc Drug Rev2002201375212070533
- RosencherNBellamyLArnaoutLShould new oral antico-agulants replace low-molecular-weight heparin for thromboprophylaxis in orthopaedic surgery?Arch Cardiovasc Dis2009102432733319427610
- FrankRDSchabbauerGHolscherTThe synthetic pentasaccharide fondaparinux reduces coagulation, inflammation and neutrophil accumulation in kidney ischemia-reperfusion injuryJ Thromb Haemost20053353154015748244
- HeurtaultBSaulnierPPechBThe influence of lipid nanocapsule composition on their size distributionEur J Pharm Sci2003181556112554073
- RawatAMajumderQHAhsanFInhalable large porous microspheres of low molecular weight heparin: in vitro and in vivo evaluationJ Control Release2008128322423218471921
- KhokhlovVYSelemenevVFKhokhlovaONShcherbininRLHeparin determination by spectrophotometric and photocolorimetric methodsPharm Chem J1999338450451
- MotlekarNASrivenugopalKSWachtelMSYouanBBModulation of gastrointestinal permeability of low-molecular-weight heparin by L-arginine: in-vivo and in-vitro evaluationJ Pharm Pharmacol200658559159816640827
- KlaefflingCPiechottkaGDaemgen-von BrevernGDevelopment and clinical evaluation of two chromogenic substrate methods for monitoring fondaparinux sodiumTher Drug Monit200628337538116778722
- BianchiniPBergonziniGLParmaBOsimaBRelationship between plasma antifactor Xa activity and the antithrombotic activity of heparins of different molecular massHaemostasis19952562882988586320
- StangierJClinical pharmacokinetics and pharmacodynamics of the oral direct thrombin inhibitor dabigatran etexilateClin Pharmacokinet200847528529518399711
- RogerELagarceFBenoitJPDevelopment and characterization of a novel lipid nanocapsule formulation of Sn38 for oral administrationEur J Pharm Biopharm201179118118821303693
- HuynhNTPassiraniCSaulnierPBenoitJPLipid nanocapsules: a new platform for nanomedicineInt J Pharm2009379220120919409468
- HirsjarviSQiaoYRoyereABibetteJBenoitJPLayer-by-layer surface modification of lipid nanocapsulesEur J Pharm Biopharm201076220020720656028
- MeissnerYUbrichNEl GhazouaniFMaincentPLamprechtALow molecular weight heparin loaded pH-sensitive microparticlesInt J Pharm20073351–214715317150317
- PeiraECarlottiMETrottaCCavalliRTrottaMPositively charged microemulsions for topical applicationInt J Pharm20083461–211912317618070
- BaeKHMoonCWLeeYParkTGIntracellular delivery of heparin complexed with chitosan-g-poly(ethylene glycol) for inducing apoptosisPharm Res20092619310018777202
- MoriSMatsuuraARama PrasadYVTakadaKStudies on the intestinal absorption of low molecular weight heparin using saturated fatty acids and their derivatives as an absorption enhancer in ratsBiol Pharm Bull200427341842114993814
- MalkovDWangHZDinhSGomez-OrellanaIPathway of oral absorption of heparin with sodium N-[8-(2-hydroxybenzoyl)amino] caprylatePharm Res20021981180118412240944
- MoazedBHiebertLMMovement of heparins across rat gastric mucosa is dependent on molecular weight and pHPharm Res200926118919518937058
- Rama PrasadYVMinamimotoTYoshikawaYIn situ intestinal absorption studies on low molecular weight heparin in rats using labrasol as absorption enhancerInt J Pharm20042711–222523215129989
- Scala-BertolaJRabiskovaMLecompteTBonneauxFMaincentPGranules in the improvement of oral heparin bioavailabilityInt J Pharm20093741–2121619446753
- MousaSAZhangFAljadaAPharmacokinetics and pharmaco-dynamics of oral heparin solid dosage form in healthy human subjectsJ Clin Pharmacol200747121508152018048572
- BerkowitzSDMarderVJKosuticGBaughmanRAOral heparin administration with a novel drug delivery agent (SNAC) in healthy volunteers and patients undergoing elective total hip arthroplastyJ Thromb Haemost2003191914191912941031
- HoffartVLamprechtAMaincentPLecompteTVigneronCUbrichNOral bioavailability of a low molecular weight heparin using a polymeric delivery systemJ Control Release20061131384216697485
- RogerELagarceFGarcionEBenoitJPBiopharmaceutical parameters to consider in order to alter the fate of nanocarriers after oral deliveryNanomedicine (Lond)20105228730620148639
- StangierJRathgenKStahleHGansserDRothWThe pharmacokinetics, pharmacodynamics and tolerability of dabigatran etexilate, a new oral direct thrombin inhibitor, in healthy male subjectsBr J Clin Pharmacol200764329230317506785