 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Poly(propylene adipate)-block-poly(ɛ-caprolactone) copolymers were synthesized using a combination of polycondensation and ring-opening polymerization of ɛ-caprolactone in the presence of poly(propylene adipate). Gel permeation chromatography was used for molecular weight determination, whereas hydrogen-1 nuclear magnetic resonance and carbon-13 nuclear magnetic resonance spectroscopy were employed for copolymer characterization and composition evaluation. The copolymers were found to be block while their composition was similar to the feeding ratio. They formed semicrystalline structures, while only poly(ɛ-caprolactone) formed crystals, as shown by wide angle X-ray diffraction. Differential scanning calorimetry data suggest that the melting point and heat of fusion of copolymers decreased by increasing the poly(propylene adipate) amount. The synthesized polymers exhibited low cytotoxicity and were used to encapsulate desferrioxamine, an iron-chelating drug. The desferrioxamine nanoparticles were self-assembled into core shell structures, had mean particle size <250 nm, and the drug remained in crystalline form. Further studies revealed that the dissolution rate was mainly related to the melting temperature, as well as to the degree of crystallinity of copolymers.
Introduction
Biodegradable synthetic polymers, consisting mainly of aliphatic polyesters, are extensively used as biomaterials for medical applications ranging from drug delivery to tissue or bone substitution.Citation1–Citation3 Among them, poly(ɛ-caprolactone) (PCL) has attracted a lot of attention due to its favorable physicochemical properties. This semicrystalline polymer is a flexible material with low melting temperature (Tm = 60°C), low glass transition temperature (Tg = −60°C), appropriate mechanical properties, and good solubility in most solvents. However, the high crystallinity and hydrophobicity of PCL decreases its compatibility with soft tissues and lowers its in vivo biodegradability, compromising its potential application as matrix for drug release systems. A common approach to overcome this obstacle is copolymerization or blending PCL with polymers that have a higher biodegradation rates, leading to materials that decompose faster compared to neat PCL.Citation4,Citation5 It was previously shown that poly(propylene adipate) (PPAd), a semicrystalline polyester with melting point close to body temperature, degrades much faster than PCL.Citation6,Citation7
Nanomedicine offers promising tools to combat human diseases. The principle aim of using nanoparticles in pharmaceutical delivery systems is to enable targeted and efficient delivery of the appropriate levels of therapeutic or diagnostic agents to the target sites with reduced side effects to the patient.Citation8 In the last decade numerous studies have been published on the encapsulation of drugs, antitumor agents, and genes into polymeric matrices. In brief, Shah et alCitation9 prepared amphiphilic biodegradable core-shell nanoparticles using copolymers of hydrophobic polyhydroxyalkanoate and hydrophilic methoxy poly(ethylene glycol) (mPEG) segments and thymoquinone as a model hydrophobic drug by emulsification-solvent evaporation technique. Chen et alCitation10 synthesized biodegradable tricomponent graft copolymers, chitosan-PCL-PEG, using paclitaxel and rutin as model drugs for the evaluation of the controlled released capacity of copolymers. Wang et alCitation11 prepared cationic biodegradable poly(lactic-co-glycolic acid)-polyethyleneimine, poly(lactic-co-glycolic acid)-chitosan and mPEG-poly(lactic acid) (PLA)/polyethyleneimine, mPEG-PLA-chitosan nanoparticles as a small interfering ribonucleic acid delivery system for hepatitis B. Li et alCitation12 used polyethyleneimine as basic unit to construct a new star-shaped copolymer which was equipped with MC11 peptide as a “navigator” to mediate targeted gene delivery. Gao et alCitation13 prepared dual functional nanoparticles composed of PEG-foliate acid polymers for the chemotherapy drug doxorubicin. Miao et alCitation14 synthesized an amphiphilic copolymer with hyper-branched poly(amine-ester)-co-PLA to generate nanoparticles for the controlled delivery of anti-tumor agents.
Desferrioxamine (DFO) is a low-molecular weight natural siderophore isolated from Streptomyces pilosus. It is a linear trihydroxamic acid that forms a kinetically and thermodynamically stable complex with ferric oxide, ferrioxamine. The log of the formation constant of ferrioxamine is 30.6.Citation15 Mesylate salt is commonly used as a therapeutic agent (Desferal®) for the removal of excess iron in iron overload states, such as in β-thalassemia.Citation16 However, it has a short half-life and is poorly absorbed by the gastrointestinal tract. As a result, treatment with this drug requires prolonged parenteral infusion, at least 4–5 days each week, which renders compliance difficult; furthermore this procedure is uncomfortable for the patients. The encapsulation of DFO in nanoparticles for its slow and controlled release into patients’ blood could provide an alternative means for administration of the drug, which bypasses these issues. In published studies, liposomes have been employed for subcutaneousCitation17,Citation18 and intravenous administrationCitation19 of DFO. Poly(lactic-co-glycolic acid) microspheres containing DFO have also been prepared using the double emulsion technique for intravenous administration.Citation20 The conjugation of DFO to either a PEG-containing copolymerCitation21 or to already formed nanoparticlesCitation22 has also been described.
In the present study, the synthesis of novel biocompatible polymers and their employment for preparation of DFO-nanoparticles is reported.
Materials and methods
Adipic acid (purum >99.5%) was purchased from Fluka (Sigma-Aldrich Corporation, St Louis, MO); 1,3-propanediol (purum >99.6%), ɛ-caprolactone (ɛ-CL; purum 99%), and titanium(IV) butoxide (Ti[OBu]4; purum 97%), used as catalyst, were purchased from Aldrich Chemical Company (Sigma- Aldrich). ɛ-CL was distilled at 94°C–95°C under reduced pressure at about 5 mmHg prior to use in order to remove the inhibitor contained within. Polyphosphoric acid, used as heat stabilizer, was supplied from Fluka. Ferric chloride hexahydrate was purchased from Sigma-Aldrich. Cholic acid sodium salt (purum 99%) was purchased from Acros Organics (Geel, Belgium). Dichloromethane (purum ≥99.8%) was purchased from Merck KGaA (Darmstadt, Germany).
Synthesis of polyesters
Synthesis of PPAd was performed following the two-stage melt polycondensation method (esterification and polycondensation) in a glass batch reactor, similar to the procedure described in a previous paper.Citation23 Synthesis of PCL was performed by bulk polymerization of ɛ-CL in a 250 mL round-bottomed flask equipped with a mechanical stirrer and vacuum apparatus. The initiator was added as a solution in toluene at a final concentration of 1 × 10−4 mol Ti(OBu)4/mol ɛ-CL. The polymerization mixture was purged with nitrogen three times. The reaction was carried out at 180°C for 2 hours. Unreacted monomer was removed through distillation by applying a vacuum (≈5 Pa) over a period of 15 minutes. Polymerization was stopped by fast cooling to room temperature. Synthesis of PPAd-block-PCL copolymers, at mass ratios 10/90, 20/80, 30/70, 55/45, 65/35, and 85/15 weight/weight (w/w), was performed. PPAd was added into the same apparatus used for PCL synthesis and the proper amounts of ɛ-CL monomer were added, as well as Ti(OBu)4 (1 × 10−4 mol Ti[OBu]4/mol ɛ-CL). Polymerization took place at 180°C under nitrogen flow for 60 minutes while the stirring rate was kept stable at 500 rpm. Unreacted monomer was removed through distillation by applying vacuum (≈5 Pa), slowly to avoid excessive foaming, over a period of 15 minutes. The polyesters were removed from reaction apparatus and purified (to remove neat PPAd) by repeated dissolution in chloroform and precipitation in cold methanol (three times).
Polymer characterization
Molecular weight determinations were performed by gel permeation chromatography (GPC) using a high temperature GPC system, model 150C, equipped with differential refractometer as detector and three Ultrastyragel™ (103, 104, 105 Å) columns (Waters Corporation, Milford, MA). Tetrahydrofuran was used as the eluent (1 mL/minute) and the measurements were performed at 35°C. Calibration was performed using polystyrene standards with a narrow molecular weight distribution.
Hydrogen-1 nuclear magnetic resonance (1H NMR) and carbon-13 NMR (13C NMR) spectra of polyesters were obtained using a spectrometer (Bruker Daltonics, Billerica, MA) operating at a frequency of 400 MHz and 75.5 MHz, respectively. Deuterated chloroform was used as solvent in order to prepare solutions of 5% weight/volume and the spectra were internally referenced to tetramethylsilane.
Wide angle X-ray diffraction (WAXD) analysis was performed on randomly oriented samples, scanned over the internal of 5°–55° 2θ, using a MiniFlex™ II diffractometer (Rigaku Americas Corporation, The Woodlands, TX), with Bragg–Brentano geometry (θ, 2θ) and nickel-filtered copper Kα radiation.
Differential scanning calorimetry (DSC) was performed using a Pyris™ Diamond DSC (PerkinElmer, Inc, Waltham, MA), calibrated with indium and zinc standards. Samples of 5 ± 0.1 mg were used, sealed in aluminum pans, and heated until 50°C above the melting point at heating rate 20°C/minute under nitrogen atmosphere. The samples were held at that temperature for 5 minutes in order to erase any thermal history and then cooled to −75°C at cooling rate 200°C/minute in order to prevent crystallization. The samples were heated again at the same conditions and from this second scan Tg, Tm, and heat of fusion were recorded.
Cytotoxicity of copolymers
The cytotoxicity of copolymers was evaluated by measuring the viability of human umbilical vein endothelial cells (HUVEC) in the presence of different polymer concentrations and comparing results with those obtained from PLA. HUVEC were grown routinely in Roswell Park Memorial Institute-1640 medium supplemented with 15% fetal bovine serum, 15 mg endothelial cell growth supplement, 100 U/mL penicillin, 100 μg/mL streptomycin, 50 μg/mL gentamicin, and 2.5 μg/mL amphotericin B. The cultures were maintained at 37°C, 5% carbon dioxide, and 100% humidity. Cell viability was determined by means of 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay.Citation24 HUVEC cells were seeded in 24-well plates at a density of 30,000 cells per well in 500 μL cell culture medium 199 with Earle’s salts supplemented with 15% fetal bovine serum with stable glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin, 50 μg/mL gentamicin, 0.5 μg/mL amphotericin B, 0.15 mg/mL endothelial cell growth supplement, and 5 U/mL heparin. Twenty-four hours after plating, different amounts of the nanoparticles prepared (suspended in water) were added in the wells. After 24 hours of incubation at 37°C, 50 μL of 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide solution (5 mg/mL in phosphate buffered saline, pH 7.4) was added into each well and plates were incubated at 37°C for 2 hours. The medium was withdrawn and 200 μL acidified isopropanol (0.33 mL hydrochloride in 100 mL isopropanol) was added in each well and agitated thoroughly to dissolve the formazan crystals. The solution was transferred to 96-well plates and immediately read by a microplate reader (Bio-Rad Laboratories, Inc, Hercules, CA) at 490 nm. The experiments were performed in triplicate. Cytocompatibility of polymers was expressed as percentage cell viability, calculated as the ratio between the number of cells treated with the nanoparticles and that of nontreated cells (control).
Preparation of nanoparticles
Polymer nanoparticles loaded with DFO mesylate were prepared by a modified double emulsion method. DFO (5 mg) was dissolved in water (0.5 mL) and the solution was emulsified in a polymer solution in dichloromethane (typically 50 mg polymer in 2 mL solvent) using probe sonication (model UP50H; Hielscher Ultrasonics GmbH, Teltow, Germany) at 15 W for 1 minute. The emulsion was transferred to an aqueous solution of sodium cholate (6 mL, 12 mM) and the mixture was probe sonicated at 15 W for 2 minutes. The water-in-oil-in-water emulsion formed was gently stirred at room temperature in a fume hood until the evaporation of the organic solvent was complete. The nanoparticles were purified by centrifugation and reconstitution of the precipitate in fresh water and filtered through a 1.2 μ filter (Millex-AP Filter Unit; Millipore, Billerica, MA). Nanoparticles were maintained at room temperature under vacuum to remove the remaining water traces.
Characterization of nanoparticles
The size and zeta potential of the nanoparticles were determined using photon correlation spectroscopy and micro-electrophoresis, respectively, in a Malvern Zetasizer 4 (ZET 5000; Malvern Instruments, Malvern, Worcestershire, United Kingdom) (five runs per sample). The zeta potential of the nanoparticles was measured in phosphate buffered saline (0.01 M, pH 7.4).
The morphology and crystallinity of prepared nanoparticles was examined using scanning electron microscopy and WAXD respectively, as described previously.
High-resolution transmission electron microscopy was performed using a JEOL 2011 electron microscope, which operates at (200 kV) and has a point resolution of 0.194 nm. This microscope was equipped with a KeenView G2 CCD camera for digital recording of the images, accompanied by the appropriate software for image processing (iTEM; Olympic Soft Imaging Solutions Corporation, Lakewood, CO).
Determination of DFO concentration
Nanoparticle yield, drug loading content, and drug entrapment efficiency were determined as described in a previous study.Citation23 The drug content of nanoparticles was determined using a direct procedure. Lyophilized samples of nan oparticles (1 mg) were dissolved in a mixture of chloroform/ferric chloride (20 mg/mL) 50/50% volume/volume and the solutions were assayed for drug content by measuring their absorbance at 430 nm.
In vitro dissolution
Dissolution studies were conducted using United States Pharmacopeia I basket method (50 rpm, 37°C, and 500 mL dissolution medium). Drug-loaded nanoparticle suspensions corresponding to 2.5 mg of drug were placed in a dialysis cellulose membrane bag (D9402-100FT; Sigma-Aldrich), with molecular weight cut-off 12,400, tied, placed into the baskets, and transferred to 500 mL phosphate buffer (pH 7.4) containing 2 mL of ferric chloride solution 15 mg/mL in concentration. Samples (2 mL) were withdrawn at predetermined intervals and filtered through 0.45 μm nylon filters (Whatman, Maidstone, Kent, United Kingdom). Concentration of the sample was determined using high-performance liquid chromatography according to a validated procedure developed in the authors’ lab.Citation25
Results and discussion
Polyester synthesis
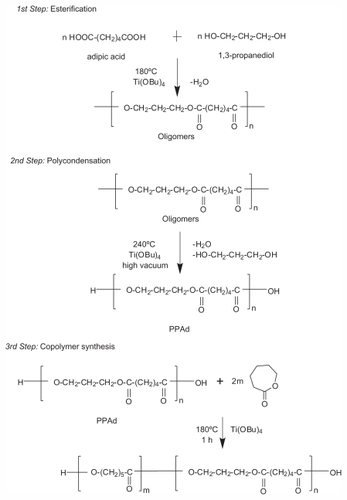
PPAd-block-PCL copolymers were synthesized following a combination of melt polycondensation and ring-opening polymerization, as described in . Initially, the aliphatic polyester PPAd was synthesized following the two-stage melt polycondensation method (esterification and polycondensation). At the first (esterification) stage, PPAd oligomers were synthesized and water was removed as byproduct. At the second (polycondensation) stage, the temperature increased in order to augment the molecular weight of the polyester. Water and 1,3-propanediol byproducts were removed by distillation to a graduated cylinder under reduced pressure. 1,3-propanediol was added in small excess ensuring that most of prepared macromolecules acquired hydroxyl end groups. This was very important for the next stage and the preparation of copolymers between PPAd and ɛ-CL monomer by ring-opening polymerization, since these groups can initiate the polymerization of ɛ-CL. PPAd-block-PCL copolyesters, in concentrations 10, 20, 30, 55, 65, and 85 weight percent (wt%) for PPAd were synthesized. To avoid extended transesterification reactions between PPAd and PCL at this stage, the reaction time was limited to 1 hour.
Figure 1 Schematic presentation of the chemical reactions during the preparation of poly(propylene adipate)-block-poly(ɛ-caprolactone) copolymers.
Abbreviation: PPAd, poly(propylene adipate).
Polyester characterization
PPAd prepared in the first stages had an average number molecular weight of 13,700 g/mol and an average weight molecular weight of 28,900 g/mol, as determined by the use of GPC, which is highly satisfactory for an aliphatic polyester. Slightly lower was the molecular weight of neat PCL. The intrinsic viscosities of the prepared copolymers ranged between 0.4–0.57 dL/g and the recorded molecular weights between 11,000–19,000 g/mol (). All samples exhibited unimodal distribution in GPC (data not shown), ie, only a single broad peak, an indication that only one type of polyester at each sample was formed. Oligomers of both polymers were not recorded.
Table 1 Theoretical and calculated compositions, intrinsic viscosities, and molecular weights of prepared polymers
In order to characterize the copolymerization process and to establish the composition of synthesized copolymers, 1H NMR and 13C NMR spectra were recorded. The 1H NMR spectra of PCL, PPAd, and their PPAd/PCL 85/15 w/w copolymer as an example are shown in .
Figure 2 Hydrogen-1 nuclear magnetic resonance (1H NMR) spectra of poly–(ɛ-caprolactone) (PCL), poly(propylene adipate) (PPAd), and PPAd/PCL 85/15 weight/weight copolymer (A) and 1H NMR spectra of PPAd/PCL 85/15 weight/weight copolymer at the region 3.98–4.16 ppm (B).
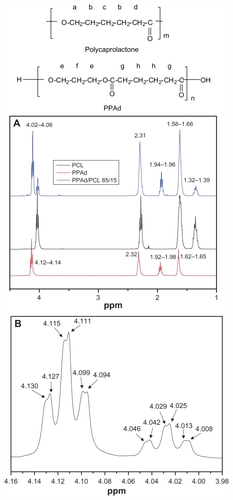
Typical signals were obtained for PCL: a multiple peak at 1.32–1.39 attributed to methylene group c, an eightfold peak at 1.58–1.66 attributed to methylene group b, a triple peak at 2.26–2.31 owing to methylene d next to carbonyl group, and a triple peak at 4.02–4.06 due to methylene group a standing next to oxygen atom. These peaks are in accordance with previous studies.Citation26–Citation28 The 1H NMR spectra of PPAd show a triple peak at 4.12–4.14 corresponding to methylene group e, a quintuple peak at 1.92–1.98 corresponding to methylene group f, a single peak at 2.32 owing to methylene group g, and a quintuple peak at 1.62–1.65 attributed to methylene group h. These peaks have also been documented in previous studies.Citation26 1H NMR spectra of copolymers exhibited the characteristics of both PCL and PPAd. Analogous were 1H NMR spectra for all copolymers synthesized (data not shown). A closer look at the copolymer’s spectra at the region 3.98–4.16, ie, at the region corresponding to methylene groups of the neat polymers standing next to oxygen atom, shows a double scission of all the six peaks (). These scissions can be attributed to cross-interactions of methylene groups, indicating an efficient conjugation between PCL and PPAd.Citation26
The percentage concentration compositions of the copolymers were calculated by using (1) the peaks attributed to methylene groups c and f corresponding to PCL and PPAd respectively for 1H NMR and (2) the peaks attributed to diol carbons bonded to the ester oxygen for 13C NMR. All results are presented in . Evidently, the calculated molar ratios are almost identical with the theoretical ones, taking into account the feeding ratios, providing evidence for the appropriateness of the procedure used.
depicts the 13C NMR spectra of neat polymers PCL and PPAd and the spectra of their copolymer PPAd/PCL 65/35 w/w. These spectra can be used to evaluate the nature of the prepared copolymers.
Figure 3 Carbon-13 nuclear magnetic resonance (13C NMR) spectrum of poly(ɛ-caprolactone) (PCL), poly(propylene adipate) (PPAd), and their copolymer PPAd/PCL 65/35 weight/weight (A) and 13C-NMR spectrum at region 173 ppm of PPAd/PCL 65/35 weight/weight (B).
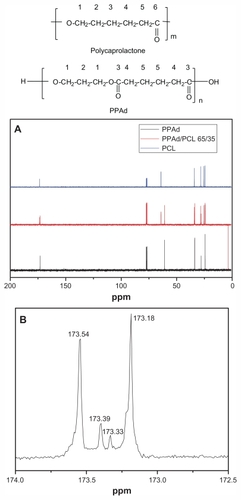
For PCL, the signal at 64.14 ppm corresponds to carbon next to oxygen. The signal at 28.33, 25.23, and 24.57 ppm corresponds to methylene carbons C2, C3, and C4, respectively, while the signal at 34.10 ppm indicates a carbon next to carbonyl group (C5). Finally the signal at 173.56 ppm corresponds to the carbonyl carbon. For PPAd the signal at 60.92 corresponds to the carbons next to oxygen, while the signals at 27.97 ppm and 24.31 ppm refer to methylene carbons C2 and C5, respectively. There are two carbons next to carbonyl groups (C4), which gives a signal at 33.78 ppm. Finally, the signal of the carbonyl carbon (C3) appears at 173.19 ppm.
As seen from spectra of PPAd/PCL 65/35 w/w copolymer, the signals of both homopolymers are present at the same position. A closer look at region 173 ppm shows the formation of two new smaller peaks between those corresponding to neat polymers (). The signals of the copolymer at this region can be identified as follows: (1) the signal at 173.54 ppm is attributed to carbonyl carbon of PCL moiety (CL-CL), (2) the new signal at 173.39 ppm corresponds to carbonyl carbon of PCL which is bonded to carbonyl carbon of PPAd (CL-Ad), (3) the second new signal at 173.33 ppm corresponds to carbonyl carbon of PPAd which is bonded to carbonyl carbon of PCL (Ad-CL), and (4) the signal at 173.18 is attributed to carbonyl carbon of PPAd moiety (Ad-Ad).
Spectra of all the synthesized copolymers exhibited these two peaks at this specific region and their intensity varied analogous to the proportion of neat polymers to its sample. The variability of the intensity was caused by the different chemical environment that carbonyl carbons were exposed to. Integration of these four peaks can be adopted to characterize the chemical microstructures of the synthesized copolymers.Citation29
The total areas of the four peaks were normalized to one for each copolymer, and the percentage values of each peak area are given in under the subtitles CL-CL, CL-Ad, Ad-CL, Ad-Ad. From normalized areas three possible triad sequences (CL-CL-CL, CL-Ad-CL or Ad-CL-Ad, and Ad-Ad-Ad) of copolymers were calculated and presented also in . The percentage concentration of the synthesized copolymers was then calculated using the following equations:
Table 2 Composition, probabilities of triads, randomness factor, and the average number sequence lengths of synthesized copolyesters
The agreement of copolymer compositions as calculated by 13C NMR is very similar to that calculated by 1H NMR () and is very close to the feeding ratio. The randomness β of the copolymers is calculated by the equation:
When β = 1 the copolymers take a random distribution, the value β = 0 represents blends, and when β < 1 the copolymers take a block distribution. The values calculated for all the synthesized copolymers range from 0.12 to 0.20 (), indicating that block copolymers have indeed been synthesized, since randomness β is very close to zero.
By using the possible triad sequences, the number of different lengths of PCL and PPAd segments in macromolecule chain can be calculated, which is named as average number sequence length. PPCL and PPPAd are a segment sequence number distribution function that expresses the probability of PCL and PPAd segments in copolymers, respectively. In order to calculate the average number sequence length of PCL and PPAd to its copolymer, the following equations were used:
Respective values are presented in . As can be observed, average number sequence length value increased proportionally to neat polymer concentration to the copolymer. Thus, in copolymers with high PPAd content the calculated triad sequence distribution of PPAd block is higher as the content of PCL lowers. The opposite occurs when PCL dominates in the molar ratio. Compared to the corresponding triads between PPAd and PCL blocks, it can be seen that PCL formed triads with higher length than PPAd.
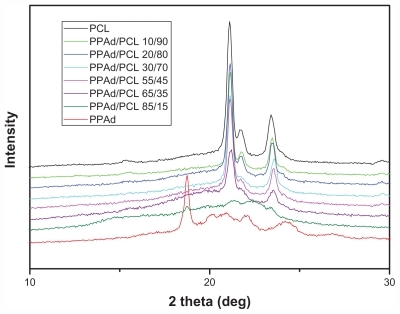
shows X-ray diffraction patterns from synthesized polyesters. Neat PCL is a polymer with high degree of crystallinity with characteristic peaks at 2θ = 21.11°, 21.53°, and 23.23°. PPAd showed peaks at 2θ = 18.77°, 20.15°, 20.89°, 22.07°, and 24.31°. The intensity at 2θ = 18.77° is high but the other peaks were very broad. This indicates that PPAd is highly amorphous and has lower degree of crystallinity than PCL. A parallel study conducted in the authors’ lab involving DSC crystallization experiments verified that crystallization rates and degree of crystallinity decreased with increasing PPAd content. Concerning the copolymers, it can be seen that only the characteristic peaks of PCL were recorded and the relative intensities became shorter. This is an indication that PPAd blocks cannot be crystallized and only in sample PPAd/PCL 85/15 w/w a weak peak at 2θ = 18.77° was shown. The degree of crystallinity of all samples was calculated from the recorded patterns and is presented in . Neat PCL has the highest degree of crystallinity (55.1%) while in the copolymers degree of crystallinity decreased with increasing PPAd content. This can be justified by the calculated average number sequence length for the two segments. PCL sequence length was lower in samples with high PPAd content; consequently PCL chains cannot organize easily in order to form big crystals.
Table 3 Glass transition temperature, melting temperature, heat of fusion values, and degree of crystallinity of poly(ɛ-caprolactone), poly(propylene adipate), and their copolymers
Figure 4 X-ray diffraction patterns for synthesized polyesters.
Abbreviations: PCL, poly(ɛ-caprolactone); PPAd, poly(propylene adipate).
Tg and Tm values of neat polymers and synthesized block copolymers were studied by DSC. Thermographs from the second scan after quenching are shown in .
Figure 5 Differential scanning calorimetric thermograms of neat polymers and synthesized copolymers.
Abbreviations: PCL, poly(ɛ-caprolactone); PPAd, poly(propylene adipate).
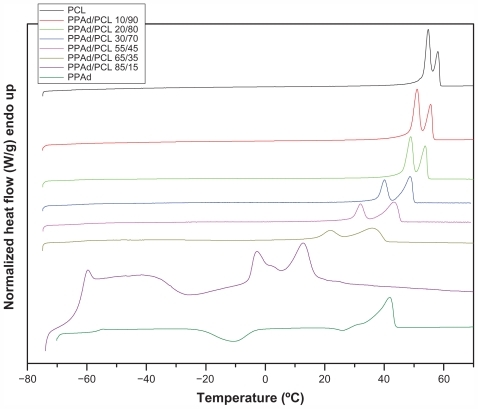
One Tg was recorded in all samples and the values are presented in . PCL showed Tg = −68.2°C and PPAd Tg = −56.1°C. All other copolymers showed intermediate values and Tg values increased as PPAd content increased. Values of Tm and heat of fusion are also presented in . The heat of fusion of copolymers reduced gradually with increasing PPAd content, in good agreement with the calculated values of degree of crystallinity using WAXD patterns. Furthermore, all synthesized copolymers showed a double peak at melting point. According to 13C NMR study, all synthesized copolymers are block; thus, by taking into account that both PCL and PPAd are semicrystalline materials, as well as the block length of each segment, two peaks were expected to emerge. However, two melting points were also recorded in PCL homopolymer. The double melting peak corresponds either to the formation of two different crystals, after quenching from its melt, or to the formation of block copolymers with each melting point corresponding to a different polymer block.Citation30 PCL shows high crystallinity and considering that polymers crystallize as metastable lamellae, it is possible for PCL to form two different types of crystals. This could also occur in copolymers. Furthermore, data from WAXD patterns suggest that only PCL can be crystallized in copolymers and PPAd remains in amorphous form. Thus, as in neat PCL, these melting points should be attributed to the formation of different type of crystals or to crystals with different perfection. This observation is very typical to similar polyesters.Citation31
Polymer cytotoxicity
The PPAd-block-PCL copolymers exhibited low toxicity against HUVEC, with appreciable cytotoxicity (higher than 20% reduction of cell viability) being observed only after exposing the cells at high nanoparticle concentrations, ie, 800 μg/mL or higher (). In terms of polymer toxicity to HUVEC, the PPAd-block-PCL copolymers appear comparable, if not less toxic, than PCL, a well-characterized biocompatible polymer that is widely used in biomedical applications.
Figure 6 Viability of human umbilical vein endothelial cells as a function of copolymer type and concentration (cell incubation time: 24 hours).
Abbreviations: PCL, poly(ɛ-caprolactone); PPAd, poly(propylene adipate).
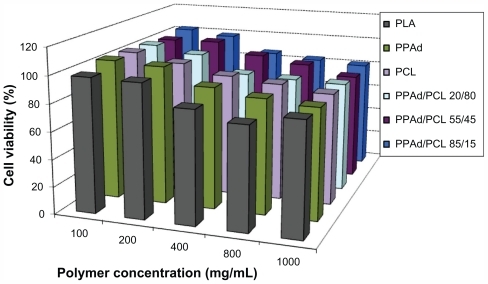
Apart from PLA and PCL, which are already extensively employed for drug delivery, PPAd and its copolymers with PCL have similar cytotoxicity with poly(propylene succinate) (PPSu) and poly(ethylene succinate); the latter were reported to be suitable drug carriers in previous studies.Citation32,Citation33 Based on polymer toxicity on HUVEC, the biocompatibility of the studied copolyesters was comparable to the biocompatibility of PLA, which is also widely used as drug carrier.Citation34 The new aliphatic polyesters reported here have a potential for drug delivery applications, by analogy to PCL or PLA.
DFO/copolymer nanoparticle characteristics
Block copolymers and neat polymers were used for nanoencapsulation of DFO. The size and zeta potential of the prepared nanoparticles are shown in . All prepared nanoparticles had average diameters less than 250 nm and showed a unimodal size distribution. Low values of poly-dispersity index for all polymer carriers suggest a relatively narrow size distribution.Citation35 The copolymers with higher PPAd content, as well as neat PPAd, tended to yield nanoparticles with higher size, possibly due to the lower degree of crystallinity of these copolymers. The analysis of zeta potential, which is the electric potential at the plane of shear, is a useful marker to predict the physical storage stability of colloidal systems. Zeta potential values higher than 30 mV show good stability during shelf-life.Citation35 In the present study, DFO-loaded polymers presented zeta potential between −28.21 and −34.60 mV. This range suggests that both suspensions might classify as physically stable, since particle aggregation is not likely to occur under these conditions of electrostatic repulsion between the particles.
Table 4 Characteristics of prepared desferrioxamine-loaded nanoparticles such as average diameter, polydispersity index, zeta potential, nanoparticles yield (%), drug loading (%), and entrapment efficiency (%)
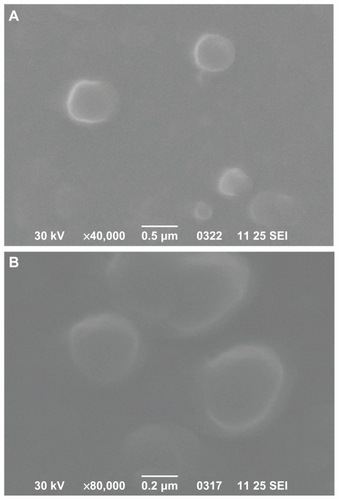
Scanning electron microscopy was used to characterize the morphology of prepared nanoparticles. shows the image of DFO-loaded PCL and PPAd/PCL 65/35 w/w polymers. These two images are representative for all nanoparticles prepared. It can be seen that the drug-loaded nanoparticles have a discrete spherical shape with a size varying between 100 nm and 300 nm, consistent with dynamic light scattering measurements.
Figure 7 Scanning electron microscope photos from desferrioxamine-loaded poly(ɛ-caprolactone) (A) and poly(propylene adipate)/poly(ɛ-caprolactone) 65/35 weight/weight (B) nanoparticles.
Abbreviation: SEI, secondary electron imaging.
High-resolution transmission electron microscopy photo-micrographs were obtained to study the detailed nanoparticle morphology (). DFO-loaded PPAd/PCL 30/70 w/w polymer nanoparticles had a circular shape and core-shell morphology. Core-shell nanoparticles were previously generated from PCL by using similar aliphatic PPSu/PCL block copolymers.Citation21 These nanoparticles are self-organized into core-shell structures, driven by differences in hydrophobicity and, mainly, physical state (amorphous versus crystalline, respectively) between the PPSu and PCL segments. More specifically, PCL moieties, which are mainly crystalline, create the nanoparticle shell during solvent evaporation, while the PPSu moieties are amorphous and form the core. The same also happened in the nanoparticles described herein. PCL corresponding to the crystalline phase of copolymers arranged perimetrically to the nanoparticle forming the shell, while PPAd, corresponding to the amorphous phase, stood in the middle of nanoparticle forming the core. The dark spots in the right part of nanoparticle may be attributed to the drug entrapped in nanoparticles.
Figure 8 High-resolution transmission electron microscope photos from prepared nanoparticles poly(propylene adipate)/poly(ɛ-caprolactone) 30/70 weight/weight.

also presents nanoparticle yields, drug loading, and entrapment efficiency. Nanoparticle recovery was <27% for all preparations, ie, ~70% of the polymer was totally wasted during the nanoparticle preparation procedure. Residual mass loss can be attributed to agglomerates that are formed during preparation and either cannot be reconstituted or are retained by filter. Furthermore, mass loss may also be associated with the hydrophilic character of DFO, ie, the hydrophilic drug prefers to remain in the aqueous phase during preparation rather than transferred to the organic phase. Drug entrapment values ranged between 36% and 99% without any specific sequence. Interestingly, PPAd/PCL 85/15 w/w and 65/35 w/w showed the highest drug entrapment value, possibly because they have the lowest degree of crystallinity.
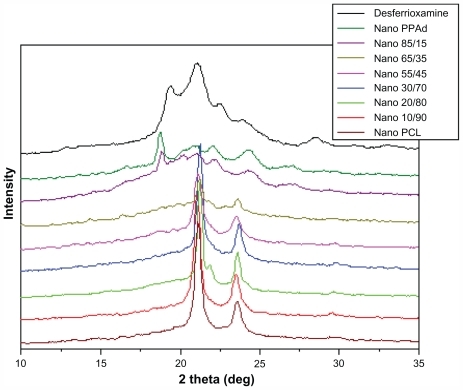
In order to study the physical state of the nanoparticle-encapsulated drug, X-ray diffraction patterns were recorded (). DFO exhibited characteristic peaks at 2θ = 19.32°, 21.03°, 22.56°, 23.98°, and 28.46°. Due to overlap with peaks of neat polymers, especially PCL, it is difficult to conclude whether these represent patterns of nanocomposites. Some peaks with small intensity were recorded at 19.38° in neat PPAd and its copolymer containing 85 wt% PPAd. This may suggest that the drug is entrapped mainly in these polymers in crystalline phase rather than in amorphous state. Since the size of nanoparticles was <250 nm, it can be concluded that the drug is entrapped in polymer nanoparticles in the form of nanocrystals. Nevertheless, in the neat PCL as well as in other copolymers such crystalline peaks were not observed, consistently with the drug being in amorphous state.
Figure 9 X-ray diffraction patterns of desferrioxamine mesylate and drug-loaded nanoparticles.
Abbreviations: PCL, poly(ɛ-caprolactone); PPAd, poly(propylene adipate).
Dissolution study
For the dissolution tests the nanoparticles were added in a membrane and phosphate buffer media under sink conditions. The recorded dissolution profiles are presented in .
Figure 10 (A) Dissolution profiles of desferrioxamine from prepared nanoparticles. The release rate in copolymer containing 85 weight percent poly(propylene adipate) (PPAd) was not possible to study due to the softness of the material. (B) Illustration of possible distribution patterns of drug in the self-assembly PPAd/poly(ɛ-caprolactone) (PCL) nanoparticles based high-resolution transmission electron microscope micrographs and its possible influence on early drug release from the nanoparticles.
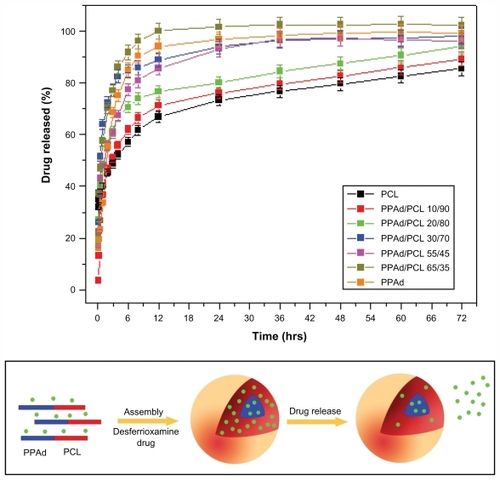
PPAd/PCL 65/35 w/w and PPAd released DFO more rapidly than all the others and the dissolution of DFO reached 100% within 12 hours. In the other copolymers, as the amount of PCL increased, the release rate became gradually slower and neat PCL exhibited the slowest release rate. Furthermore, the release rate was not stable over time. The release was very fast at early time intervals (until 10–12 hours), while it gradually became slower and controlled afterwards. In the case of hydrophilic and very soluble drugs like DFO, high release rates reflect the absorbance of higher amounts of the drug on the nanoparticle surface, which results in rapid release at initial stages (). Subsequently, the diffusion of the drug from the inert part of nanoparticles is rate-limiting and thus the drug release rate decreases. The rapid release rate of DFO at initial dissolution times can be attributed to the high solubility of the drug. The nanoparticle size may also be a crucial factorCitation36 as drugs tend to be released more rapidly from higher size nanoparticles. Nonetheless, PPAd and its copolymer with 65 and 55 wt% PPAd exhibited different release rates despite having almost identical particle sizes. Similar results were obtained with neat PCL and its copolymers with 90, 80, and 70 wt% PCL content. These nanoparticles had similar particle size (170–180 nm) but different release rates. Thus, the particle sizes in the copolymers do not appear to have any major role on the rate of DFO release.
Except the mean particle size, drug release is also affected by polymer properties, ie, degree of crystallinity and Tm, as well as by drug characteristics within the nanoparticles, ie, whether it is in crystalline or amorphous state.Citation36 Such behavior in release rates of samples could be explained by taking into account the melting point of used polymers for the preparation of nanoparticles. As shown in a previous study, the release rate was higher in polyesters with low melting points.Citation36 The release rate of such nanoparticles is mainly taking place via a diffusion process. The closer the melting temperature of polymer to that of dissolution medium, the faster the dissolution rate of the drug. This is because the macromolecular chains of polymers with lower melting points have higher mobility in the dissolution temperature conditions and therefore higher amounts of drug can be released. By comparing the release rates of DFO from the copolymers, it can be seen that those with low melting points exhibit the highest release rates. Conversely, PCL and the copolymers with the highest melting points exhibit the lowest release rates. It seems that the release of DFO is gradually reduced as the melting point of copolymers increases. Polymer erosion via hydrolysis could also affect the release rate since at 37°C the copolymers with low melting points were hydrolyzed much faster than the corresponding copolymers with higher melting points. However, consistently with hydrolysis studies of copolymers with melting points higher than 37°C, the mass loss ranged between 1–2.2 wt% without any significant trend. Thus, the effect of copolymer erosion is negligible to DFO release rates.
Another factor that has to be considered is the crystallinity of the used carriers. PCL, which has the highest crystallinity from all studied polyesters, manifests the lowest drug release. In addition, copolymers with high PCL content (70 wt%–90 wt%) and high degree of crystallinity have the lowest release rates. It appears that the drug forms nanocrystals within the nanoparticles and thus it is difficult for the dissolution medium to move it across the microchannel structures of the polyester matrices in high crystalline form.
Conclusion
A procedure of melt polycondensation was used for the preparation of PPAd and ring-opening polymerization of ɛ-CL in the presence of PPAd, to synthesize PPAd-block-PCL copolymers. This was verified by 13C NMR studies. Block length of each polymeric part in copolymers largely depends on the molar ratio between PPAd/ɛ-CL. DSC study revealed that an increase in PPAd content is associated with concomitant decrease in the melting point and heat of fusion of copolymers. WAXD measurements showed that PCL has higher crystallinity than PPAd, while in copolymers only the PCL moiety was able to form crystals. An exception was in PPAd-block-PLC 85/15 w/w copolymer, where both parts can crystallize, but in low magnitude.
The synthesized copolymers exhibited low cytotoxicity, which renders them potentially suitable for nanoencapsulation of drugs. This hypothesis was tested by nanoencapsulating DFO, a known iron-chelating drug. All the prepared nanoparticles had mean particle size lower than 250 nm and WAXD study showed that DFO was entrapped mainly in crystalline state. High-resolution transmission electron microscopy revealed that nanoparticles attained core-shell morphology. The drug release from the copolymers was mainly controlled by their melting temperatures and their degree of crystallinity. Dissolution studies showed that the release of DFO from nanoparticles exhibited variable rates. At initial stages the drug was released rapidly, due to its high solubility, while after 10–12 hours a controlled release was recorded. These dual kinetics meet pharmacological criteria for iron chelation therapy: in iron overload states, a transient bolus of high DFO doses would be required to remove excessive iron. However, after equilibrium is reached, a sustained release of lower amounts of DFO would be more appropriate to antagonize de novo iron accumulation.
The data suggest that DFO encapsulated within the PPAd-block-PCL copolymer core-shell nanoparticles may be suitable for pharmacological applications involving intravenous administration of the drug. Nevertheless, further studies with cultured cells and animal models are required to uncover and characterize the biological responses elicited by these novel DFO-nanoparticles.
Disclosure
The authors report no conflicts of interest in this work.
References
- HollandSJTigheBLGouldPLPolymers for biodegradable medical devices. 1. The potential of polyesters as controlled macromolecular release systemsJ Control Release198643155180
- RatnerBDNew ideas in biomaterials science – a path to engineered biomaterialsJ Biomed Mater Res19932778378508360211
- LiSLiuLGarreauHVertMLipase-catalyzed biodegradation of poly(epsilon-caprolactone) blended with various polylactide-based polymersBiomacromolecules20034237237712625734
- WangLMaWGrossRAMcCarthySPReactive compatibilization of biodegradable blends of poly(lactic acid) and poly(ɛ-caprolactone)Polym Degrad Stab1998591–3161168
- YuanMWangYLiXXiongCDengXPolymerization of lactides and lactones. 10. Synthesis, characterization, and application of amino-terminated poly(ethylene glycol)-co-poly(ɛ-caprolactone) block copolymerMacromolecules200033516131617
- BikiarisDNPapageorgiouGZGiliopoulosDJStergiouCACorrelation between chemical and solid state structures and enzymatic hydrolysis in novel biodegradable polyesters. The case of poly(propylene alkanedicarboxylate)sMacromol Biosci20088872874018615455
- ZorbaTChrissafisKParaskevopoulosKMBikiarisDNSynthesis, characterization and thermal degradation mechanism of three poly(alkylene adipate)s: comparative studyPolym Degrad Stab2007922222230
- WuXMansourHMNanopharmaceuticals II application of nanoparticles and nanocarrier systems in pharmaceutics and nanomedicineInt J Nanotechnol201181–2115145
- ShahMNaseerMIChoiMHKimMOYoonSCAmphiphilic PHA-mPEG copolymeric nanocontainers for drug delivery: preparation, characterization and in vitro evaluationInt J Pharm20104001–216517520713137
- ChenCCaiGZhangHJiangHWangLChitosan-poly (ɛ-caprolactone)-poly(ethylene glycol) graft copolymers: synthesis, self-assembly, and drug release behaviorJ Biomed Mater Res A201196111612421105159
- WangJFengSSWangSChenZYEvaluation of cationic nanoparticles of biodegradable copolymers as siRNA delivery system for hepatitis B treatmentInt J Pharm20104001–219420020801205
- LiDPingYXuFConstruction of a star-shaped copolymer as a vector for FGF receptor-mediated gene delivery in vitro and in vivoBiomacromolecules20101192221222920704346
- GaoZGJinZHYinXZTianLMultifunctional nanoparticles for targeting cancer therapyJ Nanosci Nanotechnol201010117743774621138023
- MiaoQXuDWangZAmphiphilic hyper-branched copolymer nanoparticles for the controlled delivery of anti-tumor agentsBiomaterials201031287364737520599267
- NeilandsJBMicrobial iron compoundsAnnu Rev Biochem1981507157316455965
- SebastianiGPantopoulosKDisorders associated with systemic or local iron overload: from pathophysiology to clinical practiceMetallomics201131097198621901209
- ToliyatTKhorasaniradZDevelopment of a Depofoam technology for the sustained delivery of desferrioxamine mesylateDaru20031138894
- ToliyatTJorjaniMKhorasaniradZAn extended-release formulation of desferrioxamine for subcutaneous administrationDrug Deliv200916741642119640256
- TabakAHofferETaitelmanUEvaluation of a liposome system for the delivery of desferrioxamine to lungs in ratsJ Pharm Pharmacol199446107897967699564
- SchlicherEJAMPostmaNSZuidemaJTalsmaHHenninkWEPreparation and characterization of poly(D,L-lactic-co-glycolic acid) microspheres containing desferrioxamineInt J Pharm19971532235245
- RossiNAMustafaIJacksonJKIn vitro chelating, cytotoxicity, and blood compatibility of degradable poly(ethylene glycol)-based macromolecular iron chelatorsBiomaterials200930463864818977029
- LiuGMenPHarrisPLRolstonRKPerryGSmithMANanoparticle iron chelators: a new therapeutic approach in Alzheimer disease and other neurologic disorders associated with trace metal imbalanceNeurosci Lett2006406318919316919875
- PapadimitriouSBikiarisDNNovel self-assembled core-shell nanoparticles based on crystalline amorphous moieties of aliphatic copolyesters for efficient controlled drug releaseJ Control Release2009138217718419446585
- MosmannTRapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assaysJ Immunol Methods1983651–255636606682
- NanakiSGKanazeFIBikiarisDNDevelopment of a simple validated isocratic HPLC method for the assaying of desferrioxamine in nanoparticles based on poly(ɛ-caprolactone)-block-poly(propylene adipate) copolymersGlobal J Anal Chem322011 [Epub ahead of print.]
- PapadimitriouSBikiarisDNChrissafisKParaskevopoulosKMMourtasSSynthesis, characterization, and thermal degradation mechanism of fast biodegradable PPSu/PCL copolymersJ Polym Sci A Polym Chem2007452250765090
- CayuelaJBounor-LegaréVCassagnauPMichelARing-opening polymerization of ɛ-caprolactone initiated with titanium n-propoxide or titanium phenoxideMacromolecules200639413381346
- ZhouSDengXYangHBiodegradable poly(epsilon-caprolactone)-poly(ethylene glycol) block copolymers: characterization and their use as drug carriers for a controlled delivery systemBiomaterials200324203563357012809785
- ChenCHPengJSChenMLuHYTsaiCJYangCSSynthesis and characterization of poly(butylene succinate) and its copolyesters containing minor amounts of propylene succinateColloid Polym Sci20102887731738
- WeiZLiuLYuFWangPQiMSynthesis and characterization of poly(ɛ-caprolactone)-b-poly(ethylene glycol)-b-poly(ɛ-caprolactone) triblock copolymers with dibutylmagnesium as catalystJ Appl Polym Sci20091111429436
- WangYBhattacharyaMManoJFThermal analysis of the multiple melting behavior of poly(butylene succinate-co-adipate)J Polym Sci B Polym Phys2005432130773082
- PapageorgiouGZBikiarisDNSynthesis and properties of novel biodegradable/biocompatible poly(propylene-co-ethylene succinate) random copolyestersMacromol Chem Phys20092101714081421
- BikiarisDNKaravelidisVKaravasENovel biodegradable polyesters. Synthesis and application as drug carriers for the preparation of raloxifene HCl loaded nanoparticlesMolecules20091472410243019633613
- AthanasiouKANiederauerGGAgrawalCMSterilization, toxicity, biocompatibility and clinical applications of polylactic acid/polyglycolic acid copolymersBiomaterials1996172931028624401
- PapadimitriouSBikiarisDNDissolution rate enhancement of the poorly water-soluble drug Tibolone using PVP, SiO2, and their nanocomposites as appropriate drug carriersDrug Dev Ind Pharm20093591128113819555245
- KaravelidisVGiliopoulosDKaravasEBikiarisDNNanoencapsulation of a water soluble drug in biocompatible polyesters. Effect of polyesters melting point and glass transition temperature on drug release behaviorEur J Pharm Sci201041563664320863892