 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Nanoscale drug carriers have been extensively developed to improve drug therapeutic efficiency. However, delivery of chemotherapeutic agents to tumor tissues and cells has not been favorably managed. In this study, we developed a novel “intelligent” nanoparticle, consisting of a gelatinase-cleavage peptide with poly(ethylene glycol) (PEG) and poly(ɛ-caprolactone) (PCL)-based structure for tumor-targeted docetaxel delivery (DOC-TNPs). The docetaxel-loaded PEG-PCL nanoparticles (DOC-NPs) that did not display gelatinase-stimuli behaviors were used as a control. We found clear evidence that the DOC-TNPs were transformed by gelatinases, allowing drug release and enhancing the cellular uptake of DOC (P < 0.01). In vivo biodistribution study demonstrated that targeted DOC-TNPs could accumulate and remain in the tumor regions, whereas non-targeted DOC-NPs rapidly eliminated from the tumor tissues. DOC-TNPs exhibited higher tumor growth suppression than commercialized Taxotere® (docetaxel; Jiangsu Hengrui Medicine Company, Jiangsu, China) and DOC-NPs on hepatic H22 tumor model via intravenous administration (P < 0.01). Both in vitro and in vivo experiments suggest that the gelatinase-mediated nanoscale delivery system is promising for improvement of antitumor efficacy in various overexpressed gelatinase cancers.
Introduction
Active targeting delivery systems have become a large area of focus in cancer therapeutics. Targeting ligands, such as aptamer,Citation1 RGD peptide,Citation2 antibody,Citation3 folate,Citation4 are peripherally conjugated to nanocarriers for binding to particular receptors overexpressed by tumor cells or vasculatures. However, much ligand-targeted therapeutics is only effective in certain types of cancer cases. For example, the anti-HER2 antibody delivery system only increased the therapeutic index in HER2 positive tumor.Citation5 Another new targeting strategy in development is employing stimuli-sensitive nanocarriers, such as thermo-sensitive micelles,Citation6 magnetic nanoparticles,Citation7 and photo-responsive polymers.Citation8 The structures of these nanocarriers could be transformed by external or internal triggering elements, facilitating drug release from nanoparticles or interacting with the tumor cells targetedly.Citation9 Although these reported nano-drugs are sensitive enough in laboratory research, limitations for clinical applications still exist. For instance, the thermo-responsive or magnetic nanocarriers need external heating or magnets to guide nanoparticles to the tumors, so the tumor locations should be clear and definite before treatment. As a result, treatment of disseminated tumors, which are an obstacle of clinical therapeutics, is a great challenge for these stimuli-responsive nanocarriers.Citation10 Therefore, exploration of more universal and targeted delivery systems is necessary for cancer therapy.
Proteases, which can recognize and hydrolyze peptide substrates, are one of the most promising triggers for stimuli-responsive drug delivery.Citation11 Considerable efforts have been made to develop protease-targeted drugs and have demonstrated promising results.Citation12 For example, in vitro experiments, the esterase and phospholipase A2 activated strategies have been developed to improve intercellular uptake efficacy.Citation13,Citation14 However, the environment in vivo is much more complex than that during in vitro experiments. So, it is not surprising that although some protease-stimuli strategies work effectively in vitro, the in vivo results are disappointing. In general, pervasiveness and selectivity of targeted proteases are two primary factors affecting the results of in vivo experiments.
Matrix metalloproteases (MMPs) have received great attention in cancer research due to their important role in cancer progression.Citation15 Notably, MMPs are a large family of enzymes with different groups of MMPs playing different roles in cancer progression. Some MMPs, such as MMP12, might help defy cancers.Citation16 Presently, many functions of MMPs are still unknown. Loss of selectivity for specific MMPs might induce side effects. Among various tumor-associated MMPs, gelatinases (MMP2/9, Collagenases IV) are known to be abundantly present in most tumors and their basic function is to degrade the extracellular matrix (ECM). Many studies have verified that gelatinases are ‘bad’ MMPs, playing an important role in numerous malignant tumor behaviors, such as angiogenesis, apoptosis, aggressiveness, metastasis, and poor survival.Citation17,Citation18 Therefore, gelatinases may be suitable candidates for stimuli-responsive targeted strategies, although MMP-stimuli strategies have been applied to prodrugs,Citation19 and liposomes.Citation20,Citation21 Nanoparticles, which have their own advantages on drug delivery, using such strategies are rare. Some gelatinase-mediated drug delivery strategies do not pay attention to the different roles of MMP subtypes, which are directly related to the therapeutic effects. Therefore, we developed the gelatinase-stimuli nanoparticle for the improvement of anticancer efficacy.
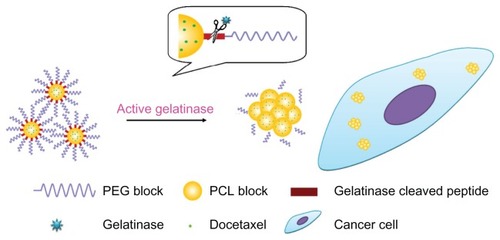
We focus on the enzyme-stimuli strategy also because PEGylated nanoparticles have some shortcomings. The PEGylated nanoparticles are primarily unable to interact with tumor cells thoroughly, leading to decreased efficacy and damage to normal cells. The gelatinase-stimuli nanoparticles can strike a balance between PEGylation and dePEGylation (). The PEG outer shell provides nanoparticles with stealth and increases their blood circulating time. Once the nanoparticles accumulate in the tumors, the peptides are degraded by gelatinases. The PEG blocks are cleaved, and the remaining PCL blocks aggregate, resulting in PEG-uncoated nanoparticles being retained in the tumor region, efficiently reaching more tumor cells, and presenting high antitumor activity. Additionally, the carriers can easily deform without entering cells, because active gelatinases are secreted exogenously.
Figure 1 The gelatinase-stimuli strategy enhanced nanoparticles interactions with cancer cells in the tumor tissues. After release from tumor capillaries, the PEG-Pep conjugates were cleaved by gelatinases, which were specifically secreted in the tumor microenvironment. The remained PCL blocks aggregated and the PEG-uncoated PCL nanoparticles interacted efficiently with cancer cells, resulting in fast drug release and effective therapeutics.
Abbreviations: PEG, poly(ethylene glycol); PCL, poly(ɛ-caprolactone); Pep, peptide.
In the present study, the poly(ɛ-caprolactone) (PCL) polymer was chosen because of its excellent biocompatibility and biodegradability properties. To emphasize the superiority of this novel nanocarrier, we used the well-recognized, effective carriers, PEG-PCL nanoparticles as a positive control. We confirmed the specific gelatinase-responsive profile from physicochemical characterization, in vitro cellular uptake, and in vivo real-time near-infrared fluorescent (NIRF) imaging. Further, we assessed the anticancer superiority of DOC-TNPs by tumor volume observation, historical study, positron emission tomography, and computed tomography (PET/CT) real-time scanning on murine hepatic H22 tumor-bearing mice as a model for comparison to Taxotere® and common DOC-NPs through intravenous (IV) administration.
Materials and methods
Materials
Methoxy-polyethyleneglycol-NHS (mPEG-NHS) was purchased from Beijing Jiankai Technology Co, (Beijing, China). The gelatinase-cleavable peptide (PVGLIG) was obtained from Shanghai HD Biosciences Co (Shanghai, China). Docetaxel (DOC) and Taxotere® (Taxotere® is the commercial name of DOC) were provided by Jiangsu Hengrui Medicine Company (Jiangsu, China). Coumarin-6 and gelatinases were purchased from Sigma-Aldrich® (St Louis, MO). ɛ-caprolactone (ɛ-CL) and dimethyl formamide (DMF) were purified by being dehydrated over CaH2 at room temperature and distillated under reduced pressure. All other chemicals were used as received without further treatment.
Synthesis of PEG-Pep-PCL copolymers and PEG-Peptide conjugates
The mPEG-NHS (200 mg) and peptides (26 mg) were dissolved in DMF (3 mL) containing 3% Et3N and stirred for 3 hours at room temperature to prepare PEG-Peptide conjugates. The unconjugated peptides were removed by dialysis in a 3500 Da MWCO membrane (Sigma-Aldrich) against deionized water for 24 hours.
Synthesis of PCL-NH2 polymers
A polymerization tube containing ɛ-CL and L-leucine was vacuated and sealed off. The polymerization was carried out at 160°C for 24 hours. Then the resulting complexes were dissolved in dichloromethane (DCM) and precipitated into a large amount of cold ethyl ether to produce PCLCOOH. The DMF dissolved PCL-COOH polymers were added with 4-dimethylamiopryidine (DMAP), 1-ethyl-3-[3-dimethylaminopropyl] carbodimide hydrochloride (EDC), ethylenediamine and N-hydroxysulfosuccinimide sodium salt (NHS), and stirred at 37°C for 18 hours. The mixture was precipitated in ethanol, filtered through medium speed filter paper, and dried under vacuum to prepare the PCL-NH2.
Synthesis PEG-Pep-PCL and PEG-PCL copolymers
The mixture of PCL-NH2 (0.002 mol), PEG-Peptide conjugates (0.002 mol), DMAP (0.003 mol), EDC (0.002 mol) and NHS (0.002 mol) in DMF was stirred for 24 hours at 32°C. The crude PEG-Peptide-PCL copolymers were purified by dialysis (MWCO 13 kDa) for 24 hours, lyophilized to powder, and stored at 4°C for further use. The PEG-PCL di-block copolymers were synthesized by a ring opening copolymerization as we previously described.Citation22
Preparation of DOC-loaded nanoparticles
DOC-loaded nanoparticles were prepared by a modified nano-precipitation method.Citation23 Briefly, 20 mg of copolymers and 10 mg of DOC were dissolved in 0.2 mL hot acetone by heating to 60°C. Subsequently, the mixture was added into 6 mL water quickly to form nanoparticles. The remaining acetone was removed by rotary vacuum evaporation. The resultant solution was filtered to remove non-incorporated DOC and the suspension was freeze-dried for further use. The empty nanoparticles were prepared similarly in the absence of DOC.
Particle size and morphology of the particles
The particle size and polydispersity of PEG-Pep-PCL and PEG-PCL nanoparticles were measured by dynamic light scattering (DLS) (Brookhaven Instruments Corporation, NY). The values were the average of triplicate measurements for a single sample.
PEG-Pep conjugates cleavage in response to gelatinases
We then investigated the cleavage of PEG-Pep conjugates in the presence of gelatinases. PEG-Pep-PCL nanoparticles (0.25 mM) were incubated in PBS solution with or without gelatinases at 37°C for 24 h. The morphology of the nanoparticles was determined by scanning electron microscopy (SEM) (S-4800, HITACHI, Tokyo, Japan).
In vitro DOC release from the nanoparticles
Twenty-five milligram lyophilized DOC-loaded nanoparticles were redispersed in 1 mL of phosphate buffer solution (PBS, 0.01 M, pH 7.4) with or without 1 mg/mL gelatinases and put into a dialysis bag (MWCO 12 kDa). The dialysis bag was immersed into 5 mL release medium (PBS containing 0.1% v/v Tween 80® [Sigma-Aldrich]) with gentle agitation at 37°C for 96 hours.Citation24 At different time points, the release medium was withdrawn for HPLC analysis, and equivalent release medium was added.
In vitro cell uptake assays
Human colon cancer carcinoma cell line LOVO, human Lewis lung carcinoma (LLC), and murine hepatic carcinoma cell line H22 were purchased from Shanghai Institute of Cell Biology (Shanghai, China). The tumor cells were routinely cultured in RPMI 1640 medium (Sigma-Aldrich), supplemented with 10% fetal bovine serum (FBS) at 37°C. The gelatinase-stimuli property and biocompatibility of nanoparticles were confirmed by in vitro cellular uptake study. We measured the gelatinases expressions of different adherent cancerous cells by gelatin zymography (see “Supplemental data”). The colon carcinoma cells LOVO and Lewis lung cancer cells were chosen for their different gelatinases levels. 105 LOVO and LLC cells were seeded in 30-mm Petri dishes and incubated with 500 μL medium containing coumarin-6 loaded nanoparticles (6.25 μg/mL calculated by coumarin-6) for 2 hours at 37°C. Then the media were replaced with fresh cell culture media and the fluorescent signals of cells were imaged using the laser scanning confocal microscopy (LSM710, Carl Zeiss MicroImaging GmbH, Berlin, Germany) with an excitation wavelength of 460 nm.
We also carried out the cellular uptake studies on suspended cells H22. The particles were labeled with Rhodamine B isothiocyanate, which was reacted with hydroxyl end group of PCL block in PEG-Pep-PCL copolymers. Briefly, 1 mg Rhodamine B isothiocyanate and 30 mg PEGPep-PCL copolymers were dissolved in 3 mL DMF, and the system was allowed to react for 10 hours. After which, the Rhodamine B-labeled PEG-Pep-PCL nanoparticles were purified by dialysis against deionized water for 48 hours. The Rhodamine B-labeled PEG-PCL nanoparticles were prepared in the same way. The H22 cell uptake studies were taken in the above mentioned way. The quantitative study was also carried out as we previously described.Citation25 Briefly, H22 cells were seeded in a 96-well black plate at a density 1 × 104 cells per well. Coumarin-6 loaded Rhodamine B-labeled PEG-Pep- PCL and PEG-PCL nanoparticles (6.25 μg/mL calculated by coumarin-6) were added into the cultured medium and incubated for 0.5, 1, 2, and 4 hours at 37°C. At different time points, the cells were collected by centrifugation, washed three times by PBS and lysed by 200 μL cell lysis buffer (50 mM Tris-HCl pH 7.4,150 mM NaCl, 1% Triton X-100). The fluorescence of whole cell lysate was determined by a fluorescence microplate reader (Safire, Tecan, Männedorf, Switzerland). The excitation and emission wavelength was 460 nm and 500 nm for coumarin-6, respectively. The cell uptake efficiency can be calculated by this equation:
The Isample, Ipositive and Inegative are the fluorescence intensities of the sample, positive control and negative control, respectively. In these tests, particle suspension was added to five wells as the positive control. In contrast, the negative control refers to the nanoparticles without coumarin-6 that were added in wells with cells.
Real-time NIRF imaging
In vivo distribution of nanoparticles was studied by NIR- 797 labeled nanoparticles. PEG-PCL or PEG-Pep-PCL nanoparticles were labeled with NIR-797-isothiocyanate (Sigma-Aldrich). Briefly, 2 mg NIR-797-isothiocyanate and 200 mg PEG-Pep-PCL were dissolved in 4 mL anhydrous DMF for 18 hours with agitation. After the reaction, unconjugated NIR-797 was removed by dialysis (MWCO 3000 Da) for 2 days. The NIR-797-labeled PEG-PCL nanoparticles were prepared in the same way and lyophilized for further use. 107 H22 tumor cells were inoculated subcutaneously into the right axilla of ICR mice (18–22 g, 4–5weeks, male) to establish the tumor model. The NIR-797 labeled PEG-PCL and PEG-Pep- PCL nanoparticles (equivalent to the dose DOC 10 mg/kg) were tail vein injected into H22 tumor bearing mice (the same tumor model as that in the antitumor study) on the seventh day after inoculation. The time-dependent biodistribution in mice was imaged by the Maestro™ in-vivo imaging system (CRi Inc, Woburn, MA) at 1, 2, 4, 12, 24, 48, and 72 hours after intravenous administration.
In vivo antitumor efficacy of DOC-loaded nanoparticles
All animal studies were performed in compliance with guidelines set by the Animal Care Committee at Drum Tower Hospital, Nanjing, China. ICR mice (18–22 g, 4–5 weeks, male) were inoculated with 107 H22 cells subcutaneously on the right axilla. Tumor dimensions were measured with vernier calipers and the volumes were calculated as follows: tumor volume (mm3) = width2 × length/2. When the tumors reached 100 mm3 (designated as day 1), the animals were randomized into the following treatment groups: saline (0.5 mL), empty NPs, empty TNPs, Taxotere® (10 mg/kg), DOC-NPs (10 mg/kg DOC equal), and DOC-TNPs (10 mg/kg DOC equal). For the treatment, 0.5~0.6 mL of different DOC formations and empty nanoparticles dissolved in saline were intravenously administered into the mice every 6 days, three times per day. On day 21 after the first treatment, the tumor tissues from each group were dissected and fixed in 10% neutral buffered formalin. The tumor paraffin slices with a thickness of 5 μm were stained with hematoxylin and eosin (H&E).
In vivo PET/CT imaging
A combined PET/CT scanner (Gemini GXL, Philips, Amsterdam, The Netherlands) was used to estimate the early antitumor efficacy on Day 7 after DOC administration. After anesthesia with pentobarbital sodium (40 mg/kg), the H22 bearing mice that received Taxotere®, DOC-NPs, and DOC-TNPs were tail vein injected with approximately 100 μCi/0.3 mL fluorin-18-2-fluoro-2-deoxy-d-glucose (18F-FDG) as radiotracer. PET and CT images were collected in the prone position 30 minutes after injection. The standard uptake values (SUVs) in regions of interests (ROIs) were calculated by the following equation:
Statistical analysis
The ANOVA and student’s t-test (two tailed) were used for statistical analyses. P-values less than 0.05 were considered statistically significant.
Results
Characterization of PEG-Pep-PCL nanoparticles
We synthesized the PEG-Pep-PCL copolymers via ring-opening copolymerization and amidation (Figure S1 in “Supplemental data”). Comparing the 1H NMR of PEGPep-PCL and PEG-PCL copolymers, the proton signal from methyl groups in the peptide (0.823–1.006 ppm) demonstrated that the specific substrates of gelatinases were successfully inserted into the intermediate of PEG and PCL blocks (Figure S2 in “Supplemental data”). The diameter of DOC-TNPs was about 85.7 nm measured by DLS and the polydispersity index was 0.131. The drug loading content was 20.3% and drug encapsulation efficiency was 80.7% with the initial feed amount of [DOC/nanoparticles] = 1:2 (Table S1 in “ Supplemental data”). DOC-NPs were prepared as a control to investigate the carriers’ property and intracellular uptake.
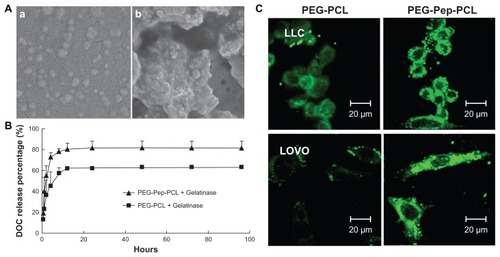
The SEM images verified the response of DOC-TNPs to gelatinases. As shown in , the DOC-TNP was a monolayer spherical particle with a diameter of approximately 60 nm, which was smaller than the size obtained by DLS due to the shrinking in dry state. Such size ranged nanoparticles appreciably accumulate in tumors due to the EPR effect. After co-incubation with gelatinases for 24 hours, the nanoparticles aggregated () and the diameters significantly increased. In addition, these reunited nanoparticles did not break up and still kept the spherical shape.
Figure 2 The gelatinase-stimuli characterization of PEG-Pep-PCL nanoparticles. (A) SEM images of PEG-Pep-PCL nanoparticles in response to gelatinases (a) PEG-Pep-PCL, (b) PEG-Pep-PCL co-incubation with gelatinases. (B) In vitro release of DOC from the nanoparticles in the presence of gelatinases. (C) The cellular uptake of coumarin-6-loaded PEG-PCL and PEG-Pep-PCL nanoparticles in two kinds of cancer cells, which express different gelatinases levels (LOVO cells expressed relatively high gelatinases compared with LLC cells).
Abbreviations: DOC, docetaxel; LLC, Lewis lung carcinoma; PCL, poly(ɛ-caprolactone); PEG, poly(ethylene glycol); Pep, peptide; SEM, scanning electron microscope.
In vitro DOC release from DOC-loaded nanoparticles
To evaluate the diffusion of DOC from the inner core of nanoparticles in the presence of gelatinases, DOC-TNPs and DOC-NPs were incubated in release medium with or without gelatinases. Both nanoparticles exhibited a steady continued-release pattern with a certain amount of initial burst release and a sustained release in the following time in the absence of gelatinases (Figure S3 in “Supplemental data”). The release of DOC from DOC-TNPs exhibited faster release than that of DOC-NPs in gelatinases solution (19% versus 14% at 1 hour, and 82% versus 64% at 96 hours []).
In vitro cellular uptake studies
We next examined whether or not the cellular uptake efficiency of PEG-Pep-PCL nanoparticles depended on the gelatinase levels. For this purpose, we first quantified the active gelatinases via gelatin zymography to screen suitable cancer cells (Figure S4 in “Supplemental data”). Colon carcinoma cells LOVO expressing high gelatinase levels were used as a positive control for comparison with the Lewis lung cancer cells, which express relatively low gelatinases compared to LOVO cells. The fluorescence intensity of PEG-PCL in LOVO cells is weaker than LLC, while the fluorescence intensity of PEG-Pep-PCL in LOVO cells is similar to LLC (). The enhanced cellular uptake of coumarin-6-loaded PEG-Pep-PCL nanoparticles to PEG-PCL nanoparticles was found in both LLC and LOVO cells, and the fluorescence difference was more distinguished in LOVO cells, which relatively express more active gelatinases.
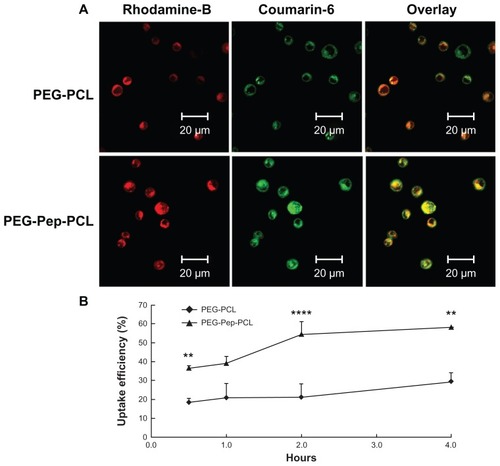
The intracellular uptake was also carried out on suspended cells H22, which highly expressed gelatinases (Figure S5 in “Supplemental data”). The nanoparticles were marked with Rhodamine B isothiocyanate to track the position of nanoparticles. Insoluble dye, coumarin-6, dispersed evenly in particles to imitate the actions of DOC. demonstrates that the green fluorescence from coumarin-6 and red color from Rhodamine B labeled nanoparticles were overlapped in the cytoplasm, suggesting that coumarin-6 entered cytosol together with nanoparticles. Besides the fluorescence densities of the PEG-Pep-PCL nanoparticles, treated groups were always higher than that of PEG-PCL nanoparticles, illustrating that PEG-Pep-PCL nanoparticles entered the cancer cells more efficiently. To confirm this supposition, the fluorescence microplate reader was used to quantify the cellular uptake efficiency, the ratio between the amounts of coumarin-6 loaded nanoparticles taken up by cells with the total amount. The uptake efficiency increased over the course of 4 hours, and a higher cell internalization of PEG-Pep-PCL nanoparticles was observed. Statistically, compared to PEG-PCL nanoparticles, the cellular uptake of PEG-Pep-PCL (0.5 hours, P < 0.01; 2 hours, P < 0.0001; 4 hours, P < 0.01) clearly showed this trend (). Especially at 2 hours where the cellular uptake of PEG-Pep- PCL was 159.55% higher than that of PEG-PCL.
Figure 3 The in vitro H22 cellular uptake studies of nanoparticles. (A) Confocal microscopy images of H22 cells after incubation with coumarin-6-loaded Rhodamine-B labeled PEG-PCL and Rhodamine-B labeled PEG-Pep-PCL nanoparticles. (B) The uptake efficiencies of coumarin-6-loaded PEG-PCL and PEG-Pep-PCL nanoparticles at different time points.
Notes: **P < 0.01; ****P < 0.0001.
Abbreviations: PCL, poly(ɛ-caprolactone); PEG, poly(ethylene glycol); Pep, peptide.
The real-time biodistribution in tumor bearing mice and in vivo antitumor effect of nanoparticles
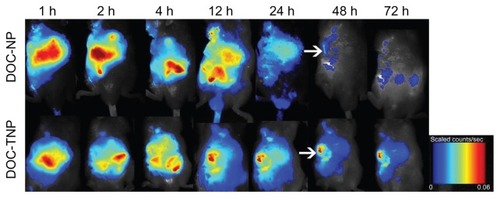
The real-time biodistribution of two nanoparticles in H22 tumor bearing mice was measured by a non-invasive near-infrared fluorescence imaging owing to dye-labeled PCL chain ends. depicts the NIRF dye NIR-797 labeled DOC-TNPs and DOC-NPs in vivo distribution during 72 hours. One hour after IV injection, the fluorescence signals of DOC-TNPs were found in the tumor regions, however, the same signals were mostly observed on the abdomen, indicating a fast hepatobiliary excretion tract. As time went on, the abdomen NIR fluorescence signals decreased, while the tumor signals became higher. Interestingly, the increasing NIR fluorescence densities of tumor zones were not observed in the DOC-NPs treated mice. Furthermore, at 48 hours post-injection, few fluorescence signals from PEG-PCL could be observed, suggesting a large amount of PEG-PCL nanoparticles were eliminated from the body. The fluorescence signals of PEG-Pep-PCL group in the tumor regions of DOC-TNPs were always stronger than the signals of the DOC-NPs group during the whole 72 hours, suggesting that DOC-TNPs preferred to concentrate and retain in the tumor regions. NIR fluorescence signals were clearly observed for NIR-797 labeled PEG-PCL nanoparticles, which were likely mediated by the enhanced permeability and retention (EPR) effect and the stealth effect afforded by PEGylation.
Figure 4 In vivo NIRF images of H22 tumor bearing mice following intravenous administration of NIR-797 labeled DOC-NPs and DOC-TNPs during 72 hours. The tumors were marked by arrows.
Abbreviations: DOC-NP, docetaxel-loaded nanoparticle; DOC-TNP, tumor-targeted docetaxel-loaded nanoparticle; NIR, near-infrared; NIRF, near-infrared fluorescent.
The in vivo antitumor studies of DOC-TNPs were performed on H22 tumor bearing mice, which highly expressed gelatinases. DOC-TNPs were given by intravenous administration every sixth day for 21 days. Saline, empty PEG-PCL nanoparticles, empty PEG-Pep-PCL nanoparticles, Taxotere®, and DOC-NPs were also given as controls for comprehensive comparison. On day 21 after the first treatment, the tumor inhibition rates in DOC-NPs and DOC-TNPs treated groups were 39.52% and 55.14%, respectively (). The DOC-TNPs treated group showed the highest antitumor efficiency and the smallest tumor volumes (P < 0.01 of 10 mg/kg Taxotere® equal; P < 0.01 of 10 mg/kg DOC-NPs equal).
Figure 5 In vivo antitumor effects of different groups. (A) The tumor growth curves of H22 tumor-bearing mice that received different treatments. The same DOC dose (10 mg/kg) was given by intravenous injection on days 1, 7 and 13 (marked by arrows). For Taxotere®, DOC-NPs and DOC-TNPs. **P < 0.01 versus Taxotere® treated group or DOC-NPs group. (B) H&E stained slices of tumors from mice on the day 21 at the end of treatment with saline (a), empty PEG-PCL nanoparticles (b), empty PEG-Pep- PCL nanoparticles (c), Taxotere® (d) (10 mg/kg), DOC-NPs (e) (10 mg/kg DOC equal), DOC-TNPs (f) (10 mg/kg DOC equal) (×40).
Abbreviations: DOC, docetaxel; DOC-NPs, docetaxel-loaded nanoparticles; DOC-TNPs, tumor-targeted docetaxel-loaded nanoparticles; H&E, hematoxylin and eosin; PCL, poly(ɛ-caprolactone); PEG, poly(ethylene glycol); Pep, peptide.
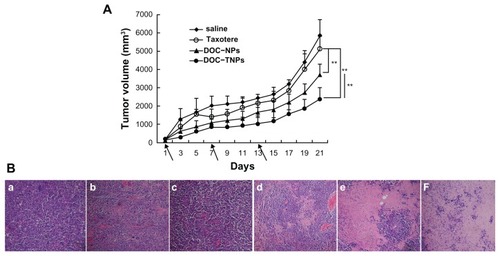
The pathology examination clearly demonstrated the significant therapeutic efficacy of DOC-TNPs. The tumors in each experimental group were obtained 21 days after treatment and the paraffin tumor slices stained by H&E were used to evaluate the necrosis levels of tumors. Small necrotic regions were observed in the saline, empty PEG-PCL and empty PEG-Pep-PCL treated groups (). The Taxotere® treated group displayed a larger necrotic region surrounded with numerously active cancer cells. A much larger necrotic level could be observed in the nanotherapeutic groups, especially in the DOC-TNPs treated group.
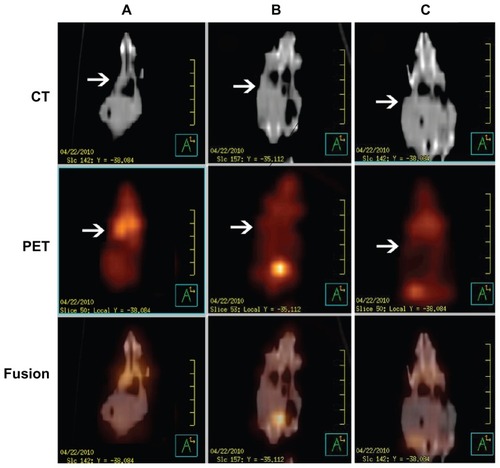
The real time therapeutic effects of DOC-TNPs, DOCNPs and Taxotere® on tumor activity were compared using positron emission tomography/computer tomography of deoxy-2-[18F]Fluoro-D-glucose (18FDG-PET/CT). The advantages of this technique include the visualization and quantification of disseminated tumors’ metabolism of tumor cells without the need of animal sacrifice for analysis. PET scanning images clearly revealed invisible metabolic activity within the transplanted tumors of the DOC-TNPs treated mice, and CT results confirmed the slowest tumor growth in this group. As displayed by , the mice treated with Taxotere® demonstrated the highest signal intensity, mice treated with DOC-NPs and DOC-TNPs presented gradually decreased signals. The mean SUVs of tumors in the mice that received Taxotere®, DOC-NPs and DOC-TNPs were 0.83 ± 0.07, 0.47 ± 0.06, 0.20 ± 0.01, respectively. As the CT imaging described, the tumor volume of Taxotere® treated mice was about 1000 mm3, while the tumors of DOC-NPs and DOC-TNPs treated groups were smaller, about 500 and 300 mm3, respectively.
Figure 6 PET/CT images of mice administered 100 μCi of 18F-FDG and scanned 30 minutes post injection. From up to down, CT, PET and fusion views. (A) Taxotere® (10 mg/kg DOC equal). (B) DOC-NPs (10 mg/kg DOC equal). (C) DOC-TNPs (10 mg/kg DOC equal).
Abbreviations: CT, computed tomography; DOC, docetaxel; DOC-NPs, docetaxel-loaded nanoparticles; DOC-TNPs, tumor-targeted docetaxel-loaded nanoparticles; PCL, poly(ɛ-caprolactone); PEG, poly(ethylene glycol); Pep, peptide; PET, positron emission tomography.
Discussion
PEG-Pep-PCL nanoparticles, which contained the active peptide substrates of gelatinases, were prepared to improve delivery of chemotherapeutic agents to tumor tissues and cells. We confirmed the gelatinase-responsive PEG-Pep-PCL nanoparticles were not only effectively targeted to the tumor tissues but also were taken up into tumor cells, and exhibited distinguished antitumor potency when they were used to load chemotherapeutic agent DOC. To emphasize the superiority of this nanoparticle, the PEG-PCL nanoparticles without targeting peptide were compared as the control. The PEGPep- PCL nanoparticles not only kept the passively targeted advantages of common nanocarriers, but also overcame some shortcomings of PEGylation. That is why PEG-TNPs showed high tumor growth suppression.
MMPs have received great attention in cancer research due to their important role in cancer progression. A variety of small-molecular MMPs inhibitors are under clinical trials or have been developed. However, few MMP inhibitors (MMPIs) are used clinically.Citation26,Citation27 The anticancer effects of MMPIs are mainly based on blocking either MMPs activity or secretion but the functions of MMPs are complex and variable. Some MMPs can accelerate tumor progression while others are protective. Specific MMPs even exhibit both positive and negative functions at different stages of the same disease. That’s why the use of MMPIs in multiple clinical trials has often been aborted due to severe side-effects. We need to rethink how to better use MMPs in cancer therapeutics. As drug-activators for tumor therapeutic strategies, gelatinases have their own advantages. Firstly, instead of MMPs inhibitors disturbing the proteases synthesis, activity and function, the enzyme-responsive strategy just takes advantage of the hydrolytic functions of MMPs. The gelatinase-stimuli strategy can avoid the bottleneck in the development of MMPs inhibitors. Secondly, among various MMPs, gelatinases are significantly up-regulated in various malignant tumors, while almost undetectable in non-pathological environments. MMPs-stimuli strategy will exhibit superior antitumor effects with limited side effects on a number of cancers. Thirdly, gelatinases are widely known as tumor associated malignant enzymes and they are susceptible to recognize and hydrolyze IV collagen, a major component of the extracellular matrix (ECM). It is known that the ECM is the first barrier for invasive and metastasis tumor cells to overcome. Therefore, the gelatinase-stimuli strategy may present its advantages on primary tumors as well as micrometastatic or disseminated tumors.
During the past couple of years, PEGylation has been widely used in the development of intravenously injected drug delivery system.Citation28 PEGylation prolongs blood circulation, increases tumor accumulation, reduces serum protein adherence and creates a stealth surface to avoid the uptake by the reticuloendothelial systems (RES). PEGylated nanoparticles are also preferable with good biocompatibility, biodegradability, low antigenicity, and immunogenicity. However, recent work reported that the introduction of PEG chains on the surface of copolymers decreased endocytosis and hindered incorporation of into cells.Citation23,Citation29,Citation30 In our cellular uptake studies, the gelatinase-stimuli strategy improved the low uptake of PEGylated nanoparticles in both attached and suspended cancer cells, and the added value depended on the gelatinases’ expression. This phenomenon may be ascribed to the dePEGylation caused by gelatinases. The gelatinases degraded the specific substrates, the linkers of hydrophilic PEG, and hydrophobic PCL blocks. The PEG conjugates were cleaved and the remaining PCL blocks gathered together for high hydrophobicity. The resulting PEG-uncoated nanoparticles had a higher affinity for cells than PEGylated nanoparticles, inducing more nanoparticles to enter tumor cells. Walker et al and Lee et al found such a trend in case of pH-triggering PEGylated-liposome and quantum dots (QD) with MMP-2 cleavage PEG conjugates.Citation31,Citation32 The enhanced cellular uptake of PEG-Pep-PCL nanoparticles is important for ideal therapeutic efficiency.
The gelatinases-stimuli DOC-TNPs, which could be transformed by both MMP-2 and MMP-9, showed distinguished tumor distribution dominance and antitumor efficiency over non-targeting DOC-NPs. Considering the synthesis feasibility and targeting specificity, we chose PVGLIG, a sequence with gelatinase selectivity, which has never been reported by other drug delivery studies,Citation33 as the bridge substrate of PEG and PCL blocks. Although, previous studies confirmed that MMP-2 cleavable liposomes could improve gene silencing effect.Citation34,Citation35 There remain many challenges in the protease-activatable area, including limited systemic evaluation and successful in vivo reports. The in vivo NIRF imaging clearly signified the gelatinases-stimuli profile altered the distribution of nanoparticles. The PEGPep- PCL nanoparticles can target and deliver to tumor tissues through EPR effect and PEGylation. Once arriving at the tumor tissues, the PEG conjugates were cleaved by tumor gelatinases and the PEG-uncoated particles aggregated. The gaps of tumor blood vessels are about 10–1000 nm,Citation36,Citation37 and larger particles are not easily extracted from tumor tissues. The penetration of nanoparticles is seriously impaired by some physiological barriers,Citation38,Citation39 especially the high interstitial fluid pressure. Small molecule drugs, such as DOC, diffused more easily in tumor tissues than nanoparticles. In vitro drug release studies showed gelatinase-stimuli profile induced DOC release quickly from PEG-Pep-PCL nanoparticles. This means dePEGylated nanoparticles not only induced drug accumulation in the tumors, but also delivered more drugs to tumor parenchyma and enhanced their efficacy. DOC-TNPs would enhance the distribution of the delivered drugs in the tumors and eradicate the tumors more completely. In the meantime, the nanoparticles that are accumulated in the liver and other tissues are able to clear, implying minimal side effects from these drugs.
Recent studies also found repeated injections of the conventional and noncleavable PEGylated nanoparticles could result in the “accelerated blood clearance (ABC)” phenomenon, which decreases antitumor effects and increase side effects.Citation40,Citation41 During the antitumor study, different DOC formations were given three times. As time went on, the difference of antitumor efficacy between DOC-TNPs and DOC-NPs became more obvious. Body weight variations, blood chemistry test and H&E-stained sections of main organs were also done to evaluate the adverse effects of different docetaxel formations (Figure S6 in the “Supplemental data”). The body weights in the group receiving DOC-NPs showed a remarkable loss compared with DOC-TNPs, especially on the last few days of the experiment. More recently, PEG-cleavage workCitation42,Citation43 showed that cleavable PEG derivatives on nanocarriers surface can weaken or eliminate the ABC phenomenon by reducing accumulation in liver and spleen. It is a clue for our design of the PEG-Pep-PCL nanoparticles, which should show similar effect on the ABC phenomenon. In fact, the data of the in vivo biodistirbution of fluorescent nanoparticles show that the PEG-cleaved DOC-TNPs had less accumulation both in liver and spleen compared with DOCNPs, indicating the gelatinases-specific strategy can improve pharmacokinetic and pharmacodynamic profiles. Thus, the gelatinase-cleavage of PEG could minimize or eliminate the shortages caused by ABC phenomenon.
This article is focused on the advantages of PEGPep- PCL nanoparticles in the tumor targeting and cellular uptake. Therefore, we concentrated on the gelatinase-stimuli property and the biodistribution of nanoparticles. As the current platform can be universally applied to almost any malignant tumors, and is adaptable to load a wide range of anticancer drugs, work on choosing the most suitable chemotherapy agents and animal models is currently underway and will likely demonstrate the techniques thoroughly. Consequently, the antitumor superiority of DOC-TNPs is associated with the following factors: (1) superior intracellular drug delivery; (2) maximal accumulation, delay and release in tumors; (3) reduced toxicity to other organs and tissues; (4) maintenance of the “passive targeting” advantages of nanoparticles.
Conclusion
In this paper, the gelatinase-stimuli PEG-Pep-PCL nanoparticles were prepared through inserting the enzyme-specific substrate between PEG and PCL blocks. Compared with PEG-PCL nanoparticles, the gelatinases strategy overcame some shortcomings of PEGylation, mediated specific, and sufficient interactions with cancer cells and maximum nanoparticles distribution. In tumors, the convertible PEGPep- PCL nanoparticles entered tumor cells more easily than PEG-PCL nanoparticles by the dePEGylation of gelatinases. In vivo NIRF imaging revealed that DOC-TNPs have prominent tumor targeting ability. The PEG-Pep-PCL nanoparticles kept high tumor concentration at 72 h pi, while PEG-PCL nanoparticles were almost completely excreted from the mice. More importantly, the DOC-TNPs showed higher antitumor efficacy and lower toxicity than Taxotere®, DOC-NPs by the tumor volumes observation, histological staining as well as PET/CT imaging. These results imply that the PEG-Pep-PCL drug delivery system will provide a promising strategy to improve clinical cancer therapy while minimizing side effects.
Results
Figure S1 Synthesis scheme of PEG-Pep-PCL copolymers.
Abbreviations: PCL, poly(ɛ-caprolactone); PEG, poly(ethylene glycol); Pep, peptide.
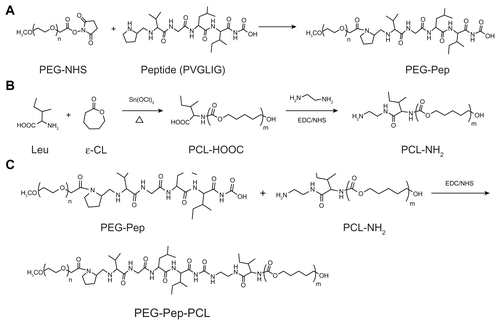
Figure S2 1H nuclear magnetic resonance spectra (300 MHz, 25°C) of polymers in CDCl3: (A) PEG-Pep-PCL copolymers; (B) PEG-PCL copolymers.
Note: The insert in (A) shows the proton signal from methyl groups in peptide (0.816–1.032 ppm), thus indicating portions of peptide were successfully conjugated into the copolymers.
Abbreviations: PCL, poly(ɛ-caprolactone); PEG, poly(ethylene glycol); Pep, peptide.
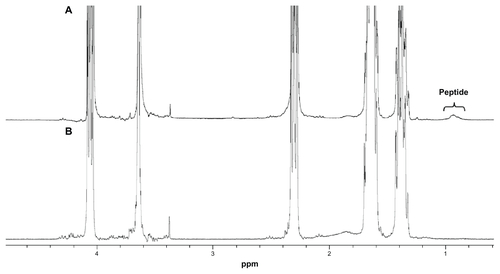
Table S1 Characterization of the DOC-TNPs and DOC-NPs
Figure S3 In vitro DOC release of DOC-TNPs and DOC-NPs in phosphate buffered saline.
Abbreviations: DOC, docetaxel; DOC-NPs, docetaxel-loaded nanoparticles; DOC-TNPs, tumor-targeted docetaxel-loaded nanoparticles; PCL, poly(ɛ-caprolactone); PEG, poly(ethylene glycol); Pep, peptide.
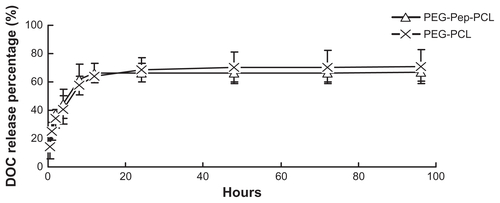
Figure S4 Detection of gelatinases by gelatin zymography, which can quantitatively measure the activity of gelatinases (MMP2/9). Following the Coomassie blue staining, gelatinases activity is detected as a white zone on black background and quantified by densitometry. (A) Scanning images of the gelatin zymography for LLC and LOVO cancer cells. (B) The expressions of LLC and LOVO cells for MMP-2 and MMP-9 by a semi-quantitative technique.
Abbreviations: LLC, Lewis lung carcinoma; MMP, matrix metalloproteases.
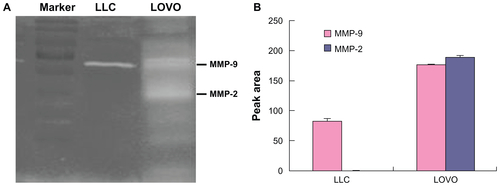
Figure S5 (A) Gelatin zymography of the gelatinases (MMP2/9) expression by in H22 cells. (B) Immunohistochemical analysis of the gelatinases (MMP2/9) expression in H22 tumours (×100).
Abbreviation: MMP, matrix metalloproteases.
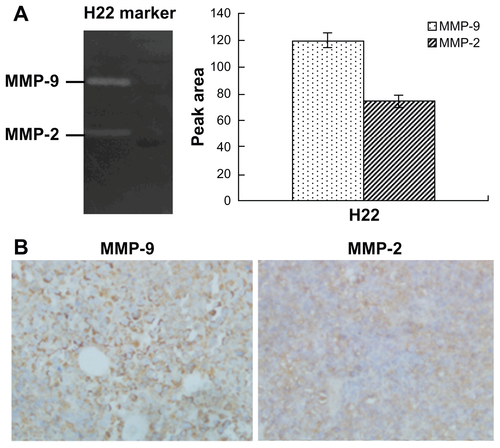
Figure S6 (A) Body weight curves of each group during the whole experiment. (B) Blood chemistry ALT and AST test was performed on Day 21 after initiation of the treatment. (C) Blood chemistry BUN and Cr test was performed on Day 21 after initiation of the treatment. (D) Abnormal damage was not observed in the H&E stained sections of main organs (×40). The result was consistent with the normal hepatic enzyme levels measured in the blood test (a) saline; (b) empty PEG-PCL nanoparticles; (c) empty PEG-Pep-PCL nanoparticles; (d) Taxotere®; (e) DOC-NPs; (f) Doc-TNPs.
Abbreviations: ALT, alamine aminotransferase; AST, aspartate aminotransferase; BUN, blood urea nitrogen; Cr, creatinine; DOC-NP, docetaxel-loaded nanoparticle; DOC-TNP, tumor-targeted docetaxel-loaded nanoparticle; H&E, hematoxylin and eosin; PCL, poly(ɛ-caprolactone); PEG, poly(ethylene glycol); Pep, peptide.
Acknowledgments
This work has been supported by the National Natural Science Foundation of China (Nos 30872471, 81071815, 81001408) and international cooperation plan of Nanjing Science and Technology Bureau (No 201001137). We also thank Mr Tom Morse for his helpful advice in editing the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
Electronic supplementary data
Materials and methods
Docetaxel and Taxotere® were kindly provided by Jiangsu Hengrui Medicine Company (Jiangsu, China). RPMI 1640 and MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) were purchased from Sigma Chemical Company (St Louis, MO). Murine hepatic carcinoma cell line H22 and human colon carcinoma cell line LOVO were purchased from Shanghai Institute of Cell Biology (Shanghai, China).
Particle size and morphology characterization
1H-NMR spectra were recorded on an AVANCE 300 MHz spectrometer (Bruker, Germany) using chloroform-d1 as solvent sparately in 5 mm NMR tubes. In all the spectra tetramethylsilane (TMS) was used as the internal reference.
The nanoparticles were suspended in distilled water to achieve an appropriate level of scattering. Particle size and polydispersity of PEG-Pep-PCL and PEG-PCL were measured by dynamic light scattering (DLS) using a Brookhaven BI-9000 AT system (Brookhaven Instruments Corporation, Long Island, NY). Each sample was diluted to filtered water and measured for three times.
Drug loading content and encapsulation efficiency
The lyophilized Doc-loaded nanoparticles were dissolved in methanol (HPLC grade, Merck Sharp and Dohme, Whitehouse Station, NJ) and sonicated for 15 minutes, centrifuged at 1200 g for 5 minutes. The supernatant fractions were prepared for HPLC analysis.
Chromatographic separation was achieved using a HC-C18 column (250 × 4.6 mm, 5 μm), (Agilent Technologies, Palo Alto, CA) at 35°C. The mobile phase consisted of 50/50 double-distilled water (Millipore, Milford, MA)/acetonitrile (HPLC grade, Merck Sharp and Dohme). The flow rate was set to 1.0 mL/minute, UV detection wavelength was 230 nm. The retention time of DOC was about 3.4 minutes. The drug loading content and encapsulation efficiency were calculated by Equationequations (1)(1) and Equation(2)
(2) , respectively.
Gelatin zymography
Gelatin zymography was used to quantify the active MMP-2 and MMP-9 extracellular levels. Briefly, 100 μL culture medium was subjected to SDS-PAGE in a gel containing 10 mg/mL of gelatin. The gels were then incubated in 2.5% Triton X-100 for 2 hours and rinsed in nanopure distilled H2O. Gels were further incubated at 37°C for 24 hours in 200 mM NaCl, 5 mM CaCl2, 0.02% Brij-35, 50 mM Tris-HCl buffer (pH 8.8), 1 μM ZnCl2, then stained with 0.2% Coomassie brilliant blue R-250, and destained in 10% acetic acid and 30% methanol in H2O. Gelatinolytic activity was detected as unstained bands on a blue background. Three independent experiments were performed.
Immunohistochemistry
Tumor samples fixed in 10% neutral buffered formalin were embedded in paraffin using automatic embedding equipment, after which 5-Am sections were prepared. Immunohistochemical analysis for MMP-9 and MMP-2 was done on paraffin-embedded H22 tumor sections of mice.
Side effects study
On Day 21 after treatment, the tissues including tumor, liver, spleen, lung, kidney and heart from the tested groups treated were dissected and fixed in 10% neutral buffered formalin. The tissues were processed routinely into paraffin, sectioned at a thickness of 5 μm, stained with H&E, and examined by optical microscope. The blood biochemistry and weights of all tested mice were also scrutinized.
References
- FarokhzadOCChengJTeplyBATargeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivoProc Natl Acad Sci U S A2006103166315632016606824
- LiuZCaiWHeLIn vivo biodistribution and highly efficient tumour targeting of carbon nanotubes in miceNat Nanotechnol200721475218654207
- LiLYWartchowCADanthiSNA novel antiangiogenesis therapy using an integrin antagonist or anti-FLK-1 antibody coated Y-90-labeled nanoparticlesInt J Radiat Oncol200458412151227
- WernerMEKarveSSukumarRFolate-targeted nanoparticle delivery of chemo- and radiotherapeutics for the treatment of ovarian cancer peritoneal metastasisBiomaterials201132338548855421843904
- TaiWYMahatoRChengKThe role of HER2 in cancer therapy and targeted drug deliveryJ Control Release2010146326427520385184
- DaiSRaviPTamKCThermo- and photo-responsive polymeric systemsSoft Matter200951325132533
- ChenBChengJWuYReversal of multidrug resistance by magnetic Fe3O4 nanoparticle copolymerizating daunorubicin and 5-bromotetrandrine in xenograft nude-miceInt J Nanomedicine20094737819421372
- KonnoTHasudaHIshiharaKItoYPhoto-immobilization of a phospholipid polymer for surface modificationBiomaterials200526121381138815482825
- DanhierFFeronOPreatVTo exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug deliveryJ Control Release2010148213514620797419
- SteegPSTheodorescuDMetastasis: a therapeutic target for cancerNature Clinical Practice Oncology200854206219
- LawBTungCHProteolysis: A biological process adapted in drug delivery, therapy, and imagingBioconjugate Chem200920916831695
- ZhuLXieJSwierczewskaMReal-time video imaging of protease expression in vivoTheranostics20111182721461134
- DavidsenJVermehrenCFrokjaerSMouritsenOGJorgensenKDrug delivery by phospholipase A(2) degradable liposomesInt J Pharm20012141–2676911282239
- AimettiAATibbittMWAnsethKSHuman neutrophil elastase responsive delivery from poly(ethylene glycol) hydrogelsBiomacromolecules20091061484148919408953
- KessenbrockKPlaksVWerbZMatrix Metalloproteinases: Regulators of the Tumor MicroenvironmentCell20101411526720371345
- Lopez-OtinCMatrisianLMTumour micro environment-opinion-emerging roles of proteases in tumour suppressionNat Rev Cancer200771080080817851543
- GuptaGPMassagueJCancer metastasis: building a frameworkCell2006127467969517110329
- RoyRYangJMosesMAMatrix metalloproteinases as novel biomarkers and potential therapeutic targets in human cancerJ Clin Oncol200927315287529719738110
- AlbrightCFGracianiNHanWMatrix metalloproteinase-activated doxorubicin prodrugs inhibit HT1080 xenograft growth better than doxorubicin with less toxicityMol Cancer Ther20054575176015897239
- TeradaTIwaiMKawakamiSNovel PEG-matrix metalloproteinase-2 cleavable peptide-lipid containing galactosylated liposomes for hepatocellular carcinoma-selective targetingJ Control Release2006111333334216488046
- ElegbedeAIBanerjeeJHansonAJMechanistic studies of the triggered release of liposomal contents by matrix metalloproteinase-9J Am Chem Soc200813032106331064218642903
- LiRTLiXLXieLPreparation and evaluation of PEG-PCL nanoparticles for local tetradrine deliveryInt J Pharm2009379115816619524653
- ZhuZSLiYLiXLPaclitaxel-loaded poly(N-vinylpyrrolidone)- b-poly(epsilon-caprolactone) nanoparticles: preparation and antitumor activity in vivoJ Control Release2010142343844619896997
- ChoYWLeeJLeeSCHuhKMParkKHydrotropic agents for study of in vitro paclitaxel release from polymeric micellesJ Control Release200497224925715196752
- HuYXieJTongYWWangCHEffect of PEG conformation and particle size on the cellular uptake efficiency of nanoparticles with the HepG2 cellsJ Control Release2007118171717241684
- FingletonBMMPs as therapeutic targets - Still a viable option?Semin Cell Dev Biol2008191616817693104
- OverallCMKleifeldOTowards third generation matrix metalloproteinase inhibitors for cancer therapyBrit J Cancer200694794194616538215
- JoralemonMJMcraeSEmrickTPEGylated polymers for medicine: from conjugation to self-assembled systemsChem Commun201046913771393
- WuWLiRTBianXCCovalently combining carbon nanotubes with anticancer agent: preparation and antitumor activityAcs Nano2009392740275019702292
- KarakotiASDasSThevuthasanSSealSPEGylated inorganic nanoparticlesAngew Chem Int Edit201150919801994
- WalkerGFFellaCPelisekJToward synthetic viruses: endosomal pH-triggered deshielding of targeted polyplexes greatly enhances gene transfer in vitro and in vivoMol Ther200511341842515727938
- LeeSRyuJHParkKPolymeric nanoparticle-based activatable near-infrared nanosensor for protease determination in vivoNano Lett20099124412441619842672
- TurkBEHuangLLPiroETCantleyLCDetermination of protease cleavage site motifs using mixture-based oriented peptide librariesNat Biotechnol200119766166711433279
- HatakeyamaHItoEAkitaHA pH-sensitive fusogenic peptide facilitates endosomal escape and greatly enhances the gene silencing of siRNA-containing nanoparticles in vitro and in vivoJ Control Release2009139212713219540888
- HatakeyamaHAkitaHKogureKDevelopment of a novel systemic gene delivery system for cancer therapy with a tumor-specific cleavable PEG-lipidGene Ther2007141687716915290
- PetrosRADesimoneJMStrategies in the design of nanoparticles for therapeutic applicationsNat Rev Drug Discov20109861562720616808
- RuoslahtiEBhatiaSNSailorMJTargeting of drugs and nanoparticles to tumorsJ Cell Biol2010188675976820231381
- NaganoSPerentesJYJainRKBoucherYCancer cell death enhances the penetration and efficacy of oncolytic herpes simplex virus in tumorsCancer Res200868103795380218483263
- GoodmanTTOlivePLPunSHIncreased nanoparticle penetration in collagenase-treated multicellular spheroidsInt J Nanomedicine20072226527417722554
- MizushimaTIshiharaTTakedaMAccelerated blood clearance phenomenon upon repeated injection of PEG-modified PLA-nanoparticlesPharmaceut Res2009261022702279
- ParkKTo PEGylate or not to PEGylate, that is not the questionJ Control Release2010142214714820096317
- ChenDLiuWShenYMuHZhangYLiangREffects of a novel pH-sensitive liposome with cleavable esterase-catalyzed and pH-responsive double smart mPEG lipid derivative on ABC phenomenonInt J Nanomedicine201162053206121976980
- XuHWangKQChenDWDengYHEffects of cleavable PEG-cholesterol derivatives on the accelerated blood clearance of PEGylated liposomesBiomaterials201031174757476320303164