Abstract
Background
Systemic delivery of small interfering RNA (siRNA) is limited by its poor stability and limited cell-penetrating properties. To overcome these limitations, we designed an efficient siRNA delivery system using polyethyleneimine-coated virus-like particles derived from adeno-associated virus type 2 (PEI-AAV2-VLPs).
Methods
AAV2-VLPs were produced in insect cells by infection with a baculovirus vector containing three AAV2 capsid genes. Using this method, we generated well dispersed AAV2-VLPs with an average diameter of 20 nm, similar to that of the wild-type AAV2 capsid. The nanoparticles were subsequently purified by chromatography and three viral capsid proteins were confirmed by Western blot. The negatively charged AAV2-VLPs were surface-coated with PEI to develop cationic nanoparticles, and the formulation was used for efficient siRNA delivery under optimized transfection conditions.
Results
PEI-AAV2-VLPs were able to condense siRNA and to protect it from degradation by nucleases, as confirmed by gel electrophoresis. siRNA delivery mediated by PEI-AAV2-VLPs resulted in a high transfection rate in MCF-7 breast cancer cells with no significant cytotoxicity. A cell death assay also confirmed the efficacy and functionality of this novel siRNA formulation towards MCF-7 cancer cells, in which more than 60% of cell death was induced within 72 hours of transfection.
Conclusion
The present study explores the potential of virus-like particles as a new approach for gene delivery and confirms its potential for breast cancer therapy.
Introduction
Small interfering RNA (siRNA) mediates sequence-specific binding and degradation of mRNA, and is one of the most important findings over the past decade, with great potential in the treatment of diseases caused by abnormal mRNA overexpression, such as cancer.Citation1 However, clinical application of siRNA is limited by its poor intracellular uptake, rapid enzymatic degradation, and inefficient systemic delivery.Citation2 Introduction of nanoparticles could overcome these limitations. Many nanoparticles have been investigated for gene delivery, including solid inorganic particles, such as gold,Citation3 silver,Citation4 and carbon nanotubes,Citation5–Citation7 or “soft” nanoparticles, such as liposomes,Citation8–Citation10 polymersomes,Citation11 and polyplexes. Use of nanoparticles enables targeting of tumor tissue through the enhanced permeability and retention effect. siRNA is a negatively charged molecule, which allows formation of electrostatic associations with positively charged coatings or complexes. One of the strategies to incorporate siRNA into nanoparticles is to coat these particles with positively charged polymers, such as polyethyleneimine (PEI), poly beta-amino ester, or poly L-lysine. Electrostatic coating can greatly enhance siRNA efficacy in systemic delivery due to its protective effects and improved cellular uptake.Citation12
Virus-like particles are hollow protein cages derived from viruses but do not contain viral genomes. They are an ideal nanoscale biomaterial due to their biocompatibility, regular structure, and homogenous size. Virus-like particles are noninfectious, but by nature they inherit the essential features of the viruses from which they are derived, including cell-penetrating ability, capacity to pack nucleic acids or structurally similar molecules, and specific tissue tropism. These features make virus-like particles excellent nanocarriers for delivery of therapeutics. In addition, extensive viral genomic studies allow genetic modification of viral surface proteins for bioconjugation of targeting molecules, and introduction of, eg, foreign targeting proteins, which are beneficial for site-specific delivery. Many biomedical applications, including vaccine development,Citation13 delivery of peptides, drugs, imaging reagents, have been approached using virus-like particles.Citation14
In recent years, adeno-associated virus (AAV) has been extensively investigated as gene therapy for many genetic disorders, including lysosomal storage disorders and hemophilia.Citation15 AAV is a small, nonenveloped parvovirus with a size of around 22 nm. The wild-type AAV genome contains about 4700 bases of linear, single-stranded DNA. The genome has two open frames with two genes, ie, rep and cap, which encode viral replication and capsid proteins respectively (). There are 11 known AAV serotypes to date, and AAV2 is the best studied among them. AAV2 virus-like particles (AAV2-VLPs) can be spontaneously assembled by capsid proteins. A single cap gene encodes three capsid proteins, ie, VP1, VP2, and VP3, with a molecular weight of 87, 73, and 62 kDa, respectively (). Strategies for expression of these three capsid proteins are involved in alternative splicing and an unusual translation mechanism. The cap gene can generate two transcripts, in which VP1 is expressed from the minor transcript mRNA, and VP2 and VP3 are expressed from the major transcript. Translation of VP2 is initiated from ACG, a nonconventional translation initiation codon; however, the expression rate of VP2 is less inefficient because ribosomes can easily bypass ACG to initiate expression of VP3 from ATG, the next inframe. The differences in translational initiation frequency and in the number of transcripts generated lead to a specific ratio of 1:1:10 in wild-type AAV2.Citation16 It has been shown that AAV2 is well tolerated in human clinical trials, infects both dividing and nondividing cells, and is able to target cancer cells without affecting healthy cells.Citation17 These features make AAV2-VLPs a potentially useful agent in biomedical applications.
Figure 1 Schematics of novel AAV2-VLPs siRNA delivery design strategy and their use in cancer therapy AAV2 cap gene was previously constructed into baculovirus vector (denoted as BAC-cap) under polyhedrin (polh) promoters for high amplification. The AAV2-VLPs were produced in bioreactor by infection of insect cells with BAC-cap. The generated crude AAV2-VLPs were purified by chromatography. For siRNA delivery, cationic polymer polyethylene imine (PEI) were applied to coat negative charged AAV-VLPs by electrostatic interactions. Delivery of cell death siRNA in breast cancer cells led to destruction of cancer cells.
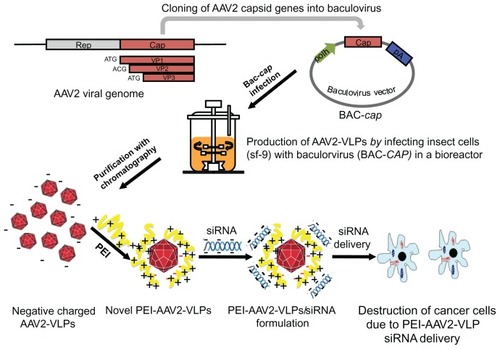
Several systems have been investigated for expression of recombinant AAV2, eg, cotransfection of plasmids containing separate AAV2 genes in HeLa or HEK293 cells, and infection of insect cells with AAV2 genes constructed from baculovirus.Citation18 The capsids of recombinant AAV2 generated by different expression methods have different stoichiometric ratios, ranging from 1:1:5 to 1:1:20.Citation19 In this study, we used an insect/baculovirus expression system to produce AAV2-VLPs. As shown in , the AAV2 cap gene was previously constructed into a baculovirus vector (denoted as BAC-cap) under a polyhedrin promoter for high amplification. The AAV2-VLPs were produced in a bioreactor by infection of insect cells with BAC-cap. For siRNA delivery, cationic PEI was used to coat negatively charged AAV-VLPs via electrostatic interactions. In this study, the production conditions for AAV-VLPs were optimized. We demonstrate that PEI-AAV2-VLPs could condense siRNA and protect it from enzymatic degradation; the PEI-AAV2-VLP complexes showed high siRNA transfer efficiency in vitro using MCF-7 breast cancer cells. Delivery of siRNA induced pronounced cell death in MCF-7 breast cancer cells, which holds promise for breast cancer therapy.
Materials and methods
Insect cell culture
Spodopterafrugiperda (Sf-9) cells were propagated in serum-free medium (Sf-900 II SFM Gibco cell culture, Invitrogen, Burlington, ON, Canada) at 27°C in shake flasks (Corning Glass Works, Corning, NY) with a working volume of 20% of the total volume and cultured in suspension in an orbital shaking incubator at 110 rpm. Cells were maintained in the exponential growth phase.
Expression of AAV2-VLPs by baculovirus infection in flask mode
The baculovirus construct, BAC-cap, containing the AAV2 capsid gene, was kindly provided by RM Kotin from the National Institutes of Health (Bethesda, MD). For AAV2-VLP expression, Sf-9 cells were inoculated at a density of 0.5 × 106 cells/mL and 20 mL of culture in 125 mL shake flasks. The cells were infected at a density of 2 × 106 cells/mL with BAC-cap in different multiplicities of infection (MOI) and maintained at 27°C and 110 rpm. Samples were taken every 24 hours post-infection. Cell density, viability, and diameter were measured using the Cedex Cell counting system (Innovatis, Bielefeld, Germany).
Production of AAV2-VLPs
Production of AAV2-VLPs was carried out in a 3.5 L Chemap bioreactor (Chemap AG, Mannedorf, Switzerland) equipped with a pitch blade impeller having a working volume of 2.8 L. Sf-9 cells were inoculated in the bioreactor at a density of 0.5 × 106 cells/mL in 2 L of culture medium. When the cell density reached 2 × 106 cells/mL, the cells were infected with BAC-cap at MOI 1. The dissolved oxygen concentration was controlled at 40% of air saturation. The O2 consumption, pH, and CO2 were monitored during the whole cell culture. Cell density and viability were examined by sampling every 12 hours and measured using the Cedex Cell counting system. The cells were harvested when viability was around 30%.
Purification of AAV2-VLPs
For purification of AAV2-VLPs, Sf-9 cells were firstly lysed to release virus-like particles from cells by adding triton-X100 at a final concentration of 0.1%, 5 U benzonase per million cells, and 2 mM MgCl2, then incubated at 37°C for one hour with shaking; MgSO4 was added to 37.5 mM, and incubated at 37°C for another 30 minutes with shaking. The cell lysates were centrifuged at 4000 g for 15 minutes, and the supernatant was collected and filtered through a 0.45 μm cellulose membrane (Amicon, Beachwood, OH) before loading onto purification columns.
AAV-VLPs were purified using two chromatography columns, ie, an ion exchange column and a hydrophobic interaction column, as described by Chahal et al.Citation20 For ion exchange chromatography, Fractogel SO3-, a cation exchange resin, was packed into an XK 50 column (GE Healthcare, Waukesha, WI) with a bed height of 9 cm. A step change of 340 mM NaCl was applied to elute the portion containing AAV2-VLPs. For the hydrophobic interaction chromatography, Butyl-650M (TosoHaas, Toyopearl) was packed into a XK50 column (GE Healthcare) with a bed height of 7.4 cm. The hydrophobic interaction chromatography column was eluted by applying a gradient from 1500 to 0 mM (NH4)2SO4. Fractions were collected and examined by Western blot for the presence of AAV2-VLPs.
SDS-PAGE and Western blot for AAV2 viral capsid proteins
Insect cell samples were lyzed by adding 0.1% triton-X100, after which 60 μL of lysates were mixed with 20 μL of dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) buffer, and boiled for 10 minutes at 70°C. After this, 10 μL of prepared samples were loaded into each well and resolved in NuPAGE 4%–12% Bis-Tris gel (Invitrogen). For Western blotting, the protein bands from the SDS-PAGE gel were transferred to a nitrocellulose membrane. A monoclonal antibody (dilution 1:1000) against the AAV2 capsid proteins, VP1, VP2, and VP3 (Maine Biotechnology Services Inc, Portland, ME) was used as the primary antibody and anti-mouse IgG (dilution 1:5000) as the secondary antibody for Western blotting.
ELISA for detection of AAV-VLPs
The amount of AAV-VLPs assembled was determined using an AAV2 titration ELISA kit (Progen, Heidelberg, Germany). An anti-AAV2 monoclonal antibody specific for the assembled capsid proteins was precoated with multititer strips and the AAV2 particles were detected with biotinylated AAV2 antibody and horseradish peroxidase-conjugated streptavidin. A standard curve was prepared using the standard AAV2 materials included in the kit. The ELISA procedure was performed according to the manufacturer’s instructions.
Transmission electron microscopy
The size and shape of the AAV2-VLPs were examined using transmission electron microscopy (Philips CM200, 200 kV), whereby 5 μL of purified AAV2-VLPs was deposited on a carbon-coated copper grid and allowed to dry for one hour, and then viewed under a transmission electron microscope.
Preparation of PEI-AAV2-VLPs
The purified AAV2-VLPs were first washed five times with phosphate-buffered solution (pH 7.4) using centrifugal filtration columns (Amicon) with a cutoff of 100 kDa to remove the (NH4)2SO4 present in the chromatograph purification elution. For each wash, three volumes of phosphate-buffered solution were added and centrifuged at 4000 g for 15 minutes. PEI (25 kDa, Sigma, St Louis, MO) was prepared as 1 mg/mL in phosphate-buffered solution. The AAV2-VLPs were mixed with varying amounts of PEI by vortexing for 5 seconds and then left to sit for 30 minutes at room temperature to form PEI-AAV2-VLP complexes. Excess PEI was removed by washing five times with phosphate-buffered solution using a centrifugal filtration column with a cutoff of 100 kDa.
Measurement of nanoparticle size and zeta potential
The PEI-AAV2-VLPs were diluted in phosphate-buffered solution to a final viral particle concentration of 1 × 107 VLPs/mL. The size and zeta potential of the particles were measured using a particle size and zeta potential analyzer (Brookhaven Instruments Corporation, Holtsville, NY). Five measurements were taken for each sample, and the results were expressed as the mean ± standard error of the mean.
Cell viability assay
The cytotoxic effect of PEI-AAV2-VLPs was evaluated in MCF-7 cells (American Type Culture Collection, Manassas, VA) by MTS assay using the Cell Titer 96® Aqueous non-radioactive cell proliferation MTS assay kit (Promega, Madison, WI). Briefly, triplicate samples of 1 × 104/well in 96-well plates were exposed to varying amounts of PEI-AAV2-VLPs at 37°C for 24 hours. The MTS assay was performed according to the manufacturer’s instructions. The 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) was reduced by mitochondrial enzyme dehydrogenase to form formazan in viable cells. The formazan concentration in the cell culture was measured at an absorbance of 490 nm using a 1420-040 Victor-3 multilabel counter (Perkin Elmer, Waltham, MA). The amount of formazan is proportional to the number of viable cells. Cell viability was calculated as the percentage of viable cells relative to that in the untreated control sample.
Gel electrophoresis assay for binding of siRNA to PEI-AAV2-VLPs
For gel electrophoresis, 4% agarose gel was used to examine the affinity of siRNA to PEI-AAV2-VLPs. siRNA (Allstar Cell Death, Qiagen, Quebec, Canada) was mixed with PEI-AAV2-VLPs by gently vortexing and incubation at room temperature for 30 minutes to allow formation of siRNA-VLP complexes. The mixture was loaded onto agarose gel containing ethidium bromide and run in Tris-acetate-EDTA buffer (40 mM Tris-HCl, 1% (v/v) acetic acid, 1 mM (ethylenediamine tetra-acetic acid) at 100 V. Migration of siRNA was viewed under ultraviolet light.
Transfection of siRNA in MCF-7 cells using PEI-AAV2-VLPs
MCF-7 breast cancer cells were maintained in Dulbecco’s modified Eagle’s medium (Invitrogen) with 10% fetal bovine serum (Invitrogen). The cells were cultured in a humidified incubator with 5% CO2 at 37°C. One day before transfection, the cells were seeded into a 96-well plate at a density of 1 × 104 cells/well in 200 μL medium. On the day of infection, the PEI-AAV2-VLPs were mixed with fluorescent siRNA (rhodamine, Qiagen) or Cell Death siRNA (Allstar, Qiagen) and left to sit for 30 minutes at room temperature, and then added to cells dropwise with gentle shaking. The transfection reagent (HiPerFect (Qiagen) was used as a positive control. For fluorescent siRNA transfection, the cells were washed once with phosphate-buffered solution to remove free particles in culture media and then viewed under a fluorescence microscope (TE2000-U, Nikon) after 4 hours of incubation. Transfection efficiency was calculated by the fluorescence intensity measured using a plate reader relative to that in the positive control sample. Transfection efficiency was further confirmed by counting the number of cells exhibiting fluorescence in five different fields of views under a fluorescent microscope.
Results
Expression, production, and purification of AAV2-VLPs
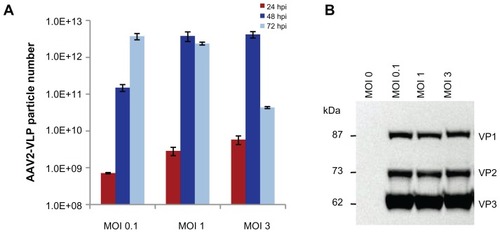
Two key factors could influence the yield of AAV2-VLPs in an insect cell/baculovirus expression system, ie, the baculovirus dosage used for infection and the timing of harvest. To obtain optimized conditions for production of AAV-VLPs in a bioreactor, production was first evaluated in flask culture with three baculovirus dosages, with a multiplicity of infection (MOI) of 0.1, 1, and 3. Insect cell growth was assessed by measuring cell density, diameter, and viability every 24 hours post-infection. The constitutive proteins (VP1–3) of the virus-like particles were examined by Western blot. The number of AAV2-VLPs assembled was measured using an ELISA kit. For all three infection conditions evaluated, cell viability dropped to less than 10% at 72 hours post-infection, indicating that the cell cultures had stopped by this point. The ELISA results showed that the yield of AAV2-VLPs assembled reached a peak at 48 hours post-infection for MOI 1 and 3; and at 72 hours post-infection for MOI 0.1 (). However, Western blotting showed that expression of the virus-like particle constitutive proteins (VP1–3) reached a similar level at 72 hours post-infection for all three different baculovirus infection dosages (). This suggests that the AAV2-VLPs assembled could dissociate with time in a cellular condition. Therefore, the timing of harvest is critical for recovery of intact virus-like particles.
Figure 2 Effect of baculovirus dosage on production of AAV2-VLPs in insect cell/BEV system (A) ELISA of assembled AAV2-VLPs with different baculovirus infection moi at different post infection time points (B) Western blot of AAV2-VLP constitution proteins at 72 hrs post infection. The sf-9 cells (at density of 2 × 106) were infected with baculovirus multiplicity of infection (moi) of 0.1, 1, 3 respectively, assembled VLPs numbers and the expressions of VLP constitution proteins were examined by ELISA and Western blot respectively. ELISA results showed the yield of assembled AAV2-VLPs peaked at 48 hpi for moi 1 and 3; and at 72 hpi for moi 0.1; Western blot showed that the VLP constitution proteins (VP1–3) expressions reached the similar level at end point (72 hpi) for all three different baculovirus infection dosages. The assembled AAV2-VLPs could disassemble with time, although the VLP constitution proteins were not degraded in cellular condition.
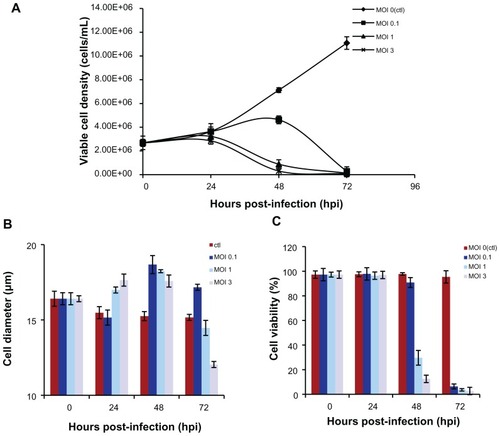
A change in cell diameter is an indication of virus infection in insect cells. shows that at MOI 1 and 3, the Sf-9 cells increased in size and stop doubling by 24 hours post-infection, but that cell enlargement and cessation of cell growth were delayed to 48 hours post-infection with baculovirus infection at MOI 0.1. Therefore, a baculovirus infection dosage of MOI 1 was adequate for synchronizing Sf-9 cell infection at 24 hours post-infection. For MOI 1 infection, Sf-9 cell viability was around 30% when the maximum yield of AAV2-VLPs assembled occurred (at 48 hours post-infection, ), so a cell viability of 30% was set as an endpoint for AAV2-VLP production. For large-scale AAV2-VLP production, 2.5 L of Sf-9 cell culture was used in the bioreactor. The Sf-9 cells were infected with baculovirus at MOI 1. Similar cell growth and infection patterns were observed in the bioreactor as in flask mode. Therefore, cell growth using the shake flask method was reproducible in the bioreactor. The cell culture was stopped when cell viability reached 30%.
Figure 3 Dynamics of insect cell (sf-9) growth in AAV2-VLPs production with different baculovirus dosages (A) viable cell density (B) cell viability (C) average cell diameters with different infection moi over time. The sf-9 cells (density of 2 × 106) were infected with baculovirus at moi 0.1, 1, 3 respectively, cell density, viability, cell diameter were examined by cell counting system Cedex. With moi 1 and 3, the sf-9 cells increased in size and stop doubling at 24 hpi, but cell enlargement and cease of cell growth were delayed with baculovirus infection of moi 0.1. So, baculovirus infection dosage of moi 1 was adequate for synchronizing the infection at 24 hpi.
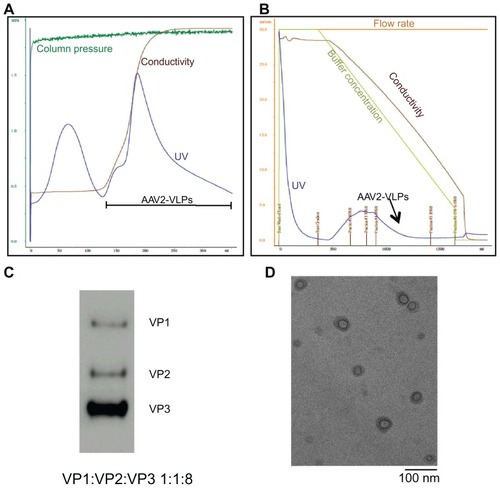
Purification of AAV2-VLPs was achieved using a two-step chromatography method developed earlier by Chahal et al.Citation20 show the eluted peaks containing AAV collected from the ion exchange and hydrophobic interaction chromatography columns. The fractions containing AAV2-VLPs were identified by Western blot. The concentration of pure AAV2-VLPs was 1.3 × 1012 VLPs/mL, as evaluated by ELISA. Band intensity analysis showed that pure AAV2-VLPs had a constitutive protein ratio of 1:1:8 (VP1–3, ). Under transmission electron microscopy, it was observed that the morphology of pure AAV2-VLPs included a round shape and a size of about 20 nm (), which is similar to that reported for wild-type AAV2 capsids.
Figure 4 Purification and Characterization of AAV2-VLPs (A) step 1: ion exchange chromatography (IEC) (B) step 2: hydrophobic interaction chromatography (HIC) (C) western blot of purified AAV2-VLPs (D) TEM image of purified AAV2-VLPs. The entire 2.5 L of cell lysates from bioreactor were subjected to equilibrated IEC column. During purification process, column pressure, flow rate, conductivity and UV of flow through were monitored. The column was washed with buffer containing 150 mM NaCl until a base line was reached. A step change of 340 mM NaCl was applied to elute AAV2-VLPs. There were 2 elution peaks detected by UV. AAV2-VLPs were eluted in the second elution peak. The fraction containing AAV2-VLPs was collected and adjusted to 1.5 M (NH4)2SO4 concentration and then loaded to HIC column. In HIC column, a concentration gradient from 1500 to 0 mM (NH4)2SO4 was applied to elute AAV2-VLPs. Pure AAV2-VLPs was detected in the fraction between 962 and 625 mM of (NH4)2SO4 (indicated with black arrow). Constitution proteins of purified AAV2-VLPs were examined by western blot, band density band intensity analysis displayed protein ratio of 1:1:8 (VP1–3). TEM analysis showed that the morphology of pure AAV2-VLPs was round in shape with size of around 20 nm, which was similar to that of reported wildtype AAV2 capsids.
PEI and AAV2-VLP interactions
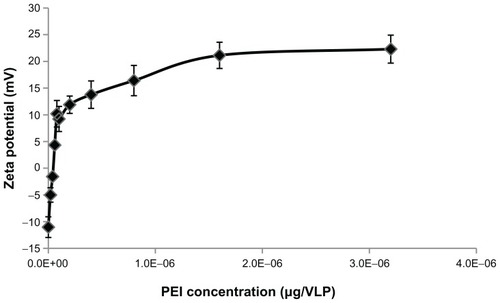
The AAV2 capsid surface has a zeta potential (a measure of net surface charge density) of −9.4 mV, according to the literature.Citation21 The AAV2-VLPs generated by our method displayed a zeta potential of −11 mV as measured by zeta potential analyzer, which is similar to that previously reported. Negatively charged AAV2-VLPs can be coated with a cationic polymer via electrostatic association. PEI, a cationic polymer, was used to coat the AAV2-VLPs for the purpose of siRNA delivery. Our experiments were designed to determine the optimal PEI quantity needed for coating AAV2-VLPs. The PEI coating amounts were evaluated in the range of 1 × 10−8 to 3.2 × 10−6 μg/VLP. A coating of PEI led to a sharp increase in the zeta potential from −11.0 mV to +11.9 mV with 5 × 10−8 μg/VLP of PEI added in the solution, and thereafter the extent of increase decreased significantly (). The amount of PEI at the transition point was 5 × 10−8 μg/VLP, which could be the minimum amount for forming a stable layer on the surface of the AAV2-VLP complex. Therefore, in the subsequent experiment, a PEI amount of 2 × 10−7 μg/VLP was used for coating, which was four times the minimum amount in order to compensate for inaccuracy of the viral particle number used. The unbound PEI was removed using a centrifugation column with a cutoff of 100 kDa.
Figure 5 Investigation of PEI and AAV2-VLPs interaction by zeta potential analysis. The PEI coating amount evaluated in the range of 1 × 10−8 to 3.2 × 10−6 μg/VLP. After coating, the excess PEI was removed by washing with 100 kDa cut-off centrifugation column. Zeta potential of PEI-AAV2-VLPs was measured in PBS buffer (pH 7.4). Total of five measurements were taken from each points. The results were plotted as means ± standard error. Coating of PEI led to a sharp increase of zeta potential of particles from −11 to +11.9 mV within 5 × 10−8 μg/VLP of PEI used. The amount of PEI in transition point (5 × 10−8 μg/VLP) was determined as the minimum amount for forming a stable layer on AAV2-VLP surface.
Toxicity of PEI-AAV2-VLPs
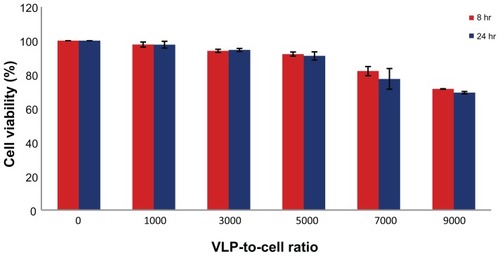
The toxicity of PEI-AAV2-VLPs was examined using MCF-7 cells. The cells were treated with varying amounts of PEI-AAV2-VLPs, with a VLP-to-cell ratio of 1000:1–9000:1, and cell viability was examined by MTS assay. Cell viability in the untreated group was deemed to be 100%, and the optical density from the MTS assay was used to calculate normalized cell viability in the other groups. The results showed that at a treatment dosage of less than 7000:1, toxicity was minor (cell viability >90%) for both 8-hour and 24-hour incubations; at the maximum dosage tested (9000:1), cell viability was around 70% ().
Figure 6 PEI-AAV2-VLPs Cytotoxicity evaluation MCF-7 cells were incubated with PEI-AAV2-VLPs at increased VLP-to-cell ratio for 8 and 24 hrs respectively. Cell viability was measured by MTS assay. Cell viability in untreated groups was assigned as 100% and its O.D from MTS assay was used to calculate cell viability in treatment groups. With treatment dosage of less than 7000:1 VLP-to-cell ratio, toxicity effects were minor (cell viability >90%) for both 8 hr and 24 hr incubations; at maximum dosage tested (VLP-to-cell ratio of 9000:1), cell viability was around 70%.
PEI-AAV2-VLPs and siRNA binding affinity/protection effect
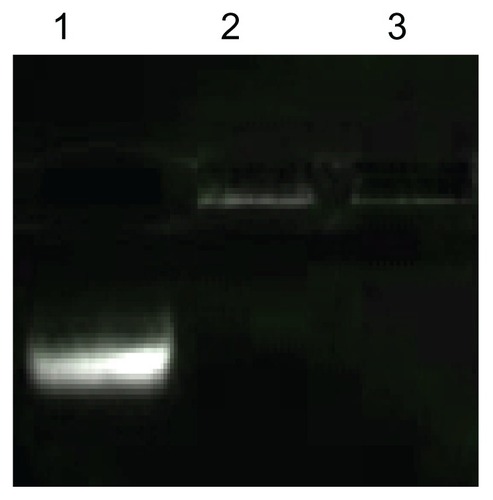
It was expected the negatively charged siRNA would form complexes with positively charged PEI-AAV2-VLPs via electrostatic interactions. Gel electrophoresis was applied to evaluate binding ability of virus-like particles to siRNA. 0.1 μg of siRNA was mixed with 0.5 × 109 PEI-AAV2 virus-like particles and incubated for 30 minutes at room temperature to form a VLP-siRNA complex. Experiments were designed to investigate the efficacy of siRNA protection afforded by PEI-AAV2-VLPs. For this, siRNA-VLP complexes were incubated with RNAse A for one hour at 37°C. shows that the migration of siRNA was fully arrested due to its association with PEI-AAV2-VLPs, because the particles were not able to pass through the gel due to their size (lane 2); with RNAase treatment, siRNA was not degraded (lane 3) and showed the same migration pattern as the sample not treated with RNAase (lane 2). This result suggested that PEI-AAV2-VLPs could associate tightly with siRNA and protect siRNA effectively from nuclease degradation.
Figure 7 siRNA binding affinity and siRNA protection effect of PEI-AAV2-VLPs. lane 1: free siRNA; lane 2: siRNA incorporated PEI-AAV2-VLPs; lane 3 cell siRNA incorporated PEI-AAV2-VLPs incubated with Rnase A for 1 hr at 37°C. 0.1 μg of siRNA was mixed with 0.5 × 109 PEI-AAV2-VLPs and incubated for 30 min at room temperature to form VLP-siRNA complex, and then incubated with or without RNAse A for 1 hr at 37°C. siRNA migration was assessed by gel electrophoresis using 4% agarose gel. The migration of siRNA was fully arrested due to association with PEIAAV2- VLPs; with RNAase treatment, siRNA was not degraded (lane 3) showing the same pattern as in RNAase untreated one (lane 2).
PEI-AAV2-VLP formulation for siRNA delivery
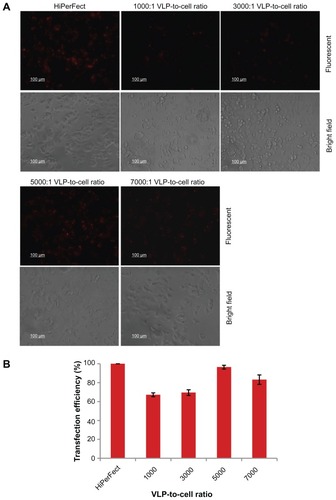
siRNA labeled with rhodamine, a fluorescent dye, was used to examine the delivery efficiency of PEI-AAV2-VLPs. Cell Death siRNA 40 ng was mixed with PEI-AAV2 virus-like particles of 1, 3, 5, and 7 × 107 VLPs, respectively, and left to sit for 30 minutes at room temperature to allow formation of nanoparticles. MCF-7 cells were plated at a density of 1 × 104 cells/well. siRNA incorporated into PEI-AAV2-VLPs was added to each well. After 4 hours of transfection, the cells were viewed under fluorescent microscopy and the fluorescence intensity was measured using a plate reader at excitation 560 nm and emission 630 nm. The siRNA delivery efficiency was evaluated by normalized fluorescence intensity. HiPerFect, a commercial transfection reagent, was used as the positive control. The siRNA delivery efficiency with HiPerFect was assigned to 100% and its fluorescent intensity was used to calculate siRNA delivery efficiency with PEI-AAV2-VLPs. Significant fluorescence was observed in MCF-7 cells (). The efficiency was 67%, 70%, 96%, and 83% for 1000:1, 3000:1, 5000:1, and 7000:1 VLP-to-cell ratios, respectively. The delivery efficiency was further confirmed by counting transfected cells under the microscope. The decreased transfection efficiency at a ratio of 7000:1 may have been caused by decreased cell numbers due to the cytotoxicity of a high amount of PEI-AAV2-VLPs.
Figure 8 siRNA delivery efficiency of PEI-AAV2-VLPs evaluated by fluorescent siRNA (A) fluorescent microscope images of siRNA transfected MCF-7 under fluorescent light (upper panel) and bright field (lower panel) (B) siRNA delivery efficiency evaluated by fluorescence intensity. The MCF-7 cells were plated at density 1 × 104 cells/well in 96-well plate. 40 ng of siRNA was mixed with PEI-AAV2-VLPs of 1, 3, 5 and 7 × 107 VLPs respectively. After 30 min incubation at room temperature, the preparations were added to each well. After 4 hr of transfection, the cells were viewed under fluorescent microscopy and fluorescence intensity was measured by plate reader. Commercial transfection reagent, HiPerFect, was used as a positive control. The siRNA delivery efficiency with HiPerFect was assigned to 100% and its fluorescent intensity was used to calculate siRNA delivery efficiency with PEI-AAV2-VLPs. Significant fluorescence was observed in MCF-7 cells. The siRNA delivery efficiency was 67%, 70%, 96% and 83% for 1000:1, 3000:1, 5000:1, 7000:1 VLP-to-cell ratio, respectively.
Efficacy of PEI-AAV2-VLPs siRNA delivery in breast cancer cell lines
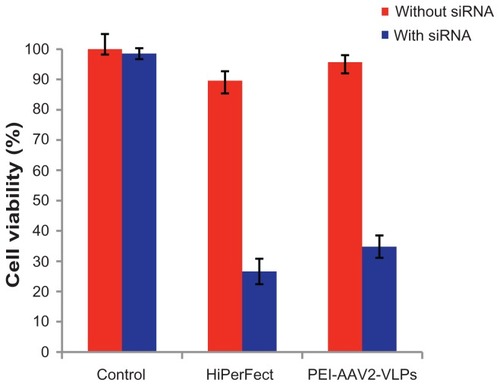
siRNA-induced cell death was used to evaluate the efficacy and functionality of siRNA delivery by PEI-AAV2-VLPs. MCF-7 breast cancer cells were preplated in 96-well plates at a density of 1 × 104 cells/well one night before. Each 40 ng of Cell Death siRNA was formulated with 5 × 107 PEI-AAV2-VLPs and added to 1 × 104 cells/well in 96-well plates (VLP-to-cell ratio 5000:1), which is the optimal transfection condition showing good transfection efficiency and a non-toxic VLP dosage. The sizes of PEI-AAV2-VLPs and PEI-AAV2-VLPs formulated with siRNA were 63.5 ± 0.5 nm and 114.7 ± 3.4 nm in diameter, respectively, as measured by particle size analyzer. The commercially available siRNA transfection reagent, HiPerFect, was used as a positive control. After 72 hours of transfection, cell viability was measured by MTS assay. Cell viability in the untreated group was assigned as 100% and its optical density by MTS assay was used to calculate normalized cell viability. siRNA delivery by PEI-AAV2-VLPs caused cell viability to change from 98% to 34.8%, which is comparable with HiPerFect (90% to 27% reduction in cell viability, ). Thus, PEI-AAV2-VLPs/siRNA exhibited a great gene silencing activity against cancer cells.
Figure 9 Cell death induced by PEI-AAV2-VLPs delivery of cell death siRNA in MCF-7 cells compared to commercially available siRNA transfection reagent HiPerFect MCF-7 cells were plated in 96-well plate at density of 1 × 104 cells/well. 40 ng of cell death siRNA was mixed with 5 × 107 PEI-AAV2-VLPs and then added to 1 × 104 cells in each well. After 72 hrs of transfection, the cell viability was measured by MTS assay. Cell viability in untreated group was assigned as 100% and its O.D from MTS assay was used to calculate normalized cell viability. Cell death siRNA delivery by HiPerFect caused cell viability to reduce from 90% to 27%; using PEI-AAV2-VLPs for siRNA delivery, cell viability reduced from 98% to 34.8%.
Discussion
Viruses typically consist of proteinaceous capsids and viral genomes, and have been traditionally thought of as pathogens because viral genomes are able to replicate inside the host and cause disease. However, by removing the viral genomes inside the capsids, the viruses become nonpathogenic. Nature has developed a wide range of capsids that vary in size, stability, and functionality, facilitating them to infect their hosts. Many of the essential features of viruses are derived from viral capsids, such as cell-penetrating capacity and specific tissue tropism. From the materials science viewpoint, viral capsids or virus-like particles are superior nanosized natural materials due to their good stability, well defined geometry, and homogeneity in particle size. One advantage of virus-like particles over other conventional materials is that they can be genetically manipulated for insertion of peptides and ligands, so that a variety of ligands can be displayed on the surface of virus-like particles. This level of control is not possible for nanomaterials made from inorganic or organic materials. As a new class of nanomaterials for biomedical application, virus-like particles are worthy of intensive investigation.
Some applications of virus-like particles for delivery of solid nanoparticles,Citation22 imaging reagents,Citation23 and small molecule drugsCitation24 have already been reported, but they have mainly focused on particles derived from plant viruses or bacteriophages, eg, Cowpea mosaic virus, Cowpea chorotic mottle virus, and bacteriophage MS2. Very few such applications of virus-like particles have been derived from human viruses due to the fact that human viruses may cause immune responses. However, virus-like particles derived from human viruses do have many advantages over other types of virus-like particles in terms of infectivity to specific human cells and tissues. The type of virus-like particles can be carefully selected so as to maximize its favorable functions. In this study, we used virus-like particles from AAV2, which has been widely used in clinical gene therapy trials and has been shown to induce a very mild immune response.Citation25 Therefore, use of virus-like particles of this type would definitely bring more benefits.
Another important factor for clinical application of virus-like particles is their ease of production, because a considerable amount of virus-like particles is needed for human trials. The insect cell/baculovirus expression system is able to produce sufficient quantities to meet the requirements of human clinical applications, and can be used as “a factory” for production of AAV2 and AAV2-VLPs.Citation26 Although it is known that the insect cell/baculovirus expression system is able to produce high levels of AAV2-VLPs, the optimal production conditions have not been determined. In this study, we optimized the production conditions, enabling 1012 functional baculoviruses per mL to be produced, which is sufficient for clinical scale application. It is known that the purification method used can influence the properties of the virus. For example, the classic cesium chloride density gradient centrifugation purification method causes over 50% loss of particle infectivity.Citation27 Other methods are limited by scalability, eg, iodixanol gradient purification, or by cost considerations, eg, immunoaffinity chromatography. Based on these considerations, we chose to use a scalable two-step chromatography method for purification of AAV2-VLPs. By applying scalable virus-like particle production and purification methods, further biomedical applications for the AAV2-VLPs generated will be easily amplified to human trial scale in future. Characterization of the virus-like particles by transmission electron microscopy (), Western blot (), and zeta potential analysis revealed that the AAV2-VLPs generated have a structure similar to that of wild-type AAV2. Therefore, it was expected that the AAV2-VLPs produced by the insect cells/baculovirus expression system could retain the infectivity and tissue tropism of wild-type AAV2.
Cationic groups are beneficial for electrostatic binding with negative charged siRNA. It has been shown that cationic polymers can be used to functionalize nanoparticles for efficient delivery of siRNA. Coating polymers on the surface of nanoparticles is a strategy to increase the solubility and stability of the particles. Cationic lipids and polymers have been shown to improve AAV2 transduction efficiency.Citation28,Citation29 In one study, AAV-mediated insulin gene therapy was combined with preinjection of PEI in mice to achieve increased infection efficiency.Citation21 However, to our knowledge, we have used electrostatic surface coating of AAV2-VLPs for siRNA delivery for the first time. Coating of virus-like particles with polymers could have other benefits, such as protection of virus-like particles from neutralization by the immune system and improved infection efficiency.Citation28–Citation30 It is important to know the mechanism of cell penetration and the mechanism of siRNA protection by the novel PEI-AAV2-VLPs. Investigation of these mechanisms in future studies will definitely provide more understanding of this new formulation.
Conclusion
In this study, a novel siRNA delivery formulation was developed using virus-like particles derived from adeno-associated virus, and the results are promising. The production and purification system for AAV2-VLPs is scalable for ease of possible future applications in clinical trials. The novel PEI-AAV2-VLPs are effective in siRNA delivery and are safe, and therefore suitable for biomedical applications. High siRNA delivery efficiency and a good silencing effect has been shown in cancer cells using PEI-AAV2-VLPs. In addition, PEI-AAV2-VLPs could protect siRNA from enzymatic degradation, which is beneficial for its in vivo delivery. Furthermore, the size of siRNA formulated PEI-AAV2-VLPs (diameter 113.7 nm) is within the optimal range of sub-150 nm for tumor targeting via the enhanced permeability and retention effect.Citation31 Our results suggest that this delivery system could be developed as a potential therapy for breast cancer.
Acknowledgments
This work was partially supported by a research grant to SP from the Canadian Institutes of Health Research (MOP 93641). WS acknowledges the Excellence Award from Biomedical Engineering Department, McGill University. AP acknowledges the Alexander Graham Bell Post Graduate Scholarship-Doctoral from Natural Sciences and Engineering Research Council of Canada. The authors are grateful for the assistance provided for transmission electron microscopy imaging by Xue-Dong Liu, Department of Physics, McGill University.
Disclosure
The authors report no conflicts of interest in this work.
References
- ShiQZhangXLDaiKRBenderdourMFernandesJCsiRNA therapy for cancer and non-lethal diseases such as arthritis and osteoporosisExpert Opin Biol Ther201111151621058934
- TokatlianTSeguraTsiRNA applications in nanomedicine WileyInterdiscip Rev Nanomed Nanobiotechnol201023305315
- SongWJDuJZSunTMZhangPZWangJGold nanoparticles capped with polyethyleneimine for enhanced siRNA deliverySmall20106223924619924738
- LiuJSonshineDAShervaniSHurtRHControlled release of biologically active silver from nanosilver surfaceACS Nano20104116903691320968290
- NunesAAmsharovNGuoCHybrid polymer-grafted multiwalled carbon nanotubes for in vitro gene deliverySmall20106202281229120878655
- KrajcikRJungAHirschANeuhuberWZolkOFunctionalization of carbon nanotubes enables non-covalent binding and intracellular delivery of small interfering RNA for efficient knock-down of genesBiochem Biophys Res Commun2008369259560218298946
- YangRYangXZhangZSingle-walled carbon nanotubes-mediated in vivo and in vitro delivery of siRNA into antigen-presenting cellsGene Ther200613241714172316838032
- EwertKKZidovskaAAhmadACationic liposome-nucleic acid complexes for gene delivery and silencing: pathways and mechanisms for plasmid DNA and siRNATop Curr Chem201029619122621504103
- HughesJYadavaPMesarosRLiposomal siRNA deliveryMethods Mol Biol201060544545920072900
- KimHKDavaaEMyungCSParkJSEnhanced siRNA delivery using cationic liposomes with new polyarginine-conjugated PEG-lipidInt J Pharm20103921–214114720347025
- TannerPBaumannPEneaROnacaOPalivanCMeierWPolymeric vesicles: from drug carriers to nanoreactors and artificial organellesAcc Chem Res201144101039104921608994
- SunXZhangNCationic polymer optimization for efficient gene deliveryMini Rev Med Chem201010210812520408796
- RoldaoAMelladoMCCastilhoLRCarrondoMJAlvesPMVirus-like particles in vaccine developmentExpert Rev Vaccines20109101149117620923267
- GarceaRLGissmannLVirus-like particles as vaccines and vessels for the delivery of small moleculesCurr Opin Biotechnol200415651351715560977
- Van VlietKMBlouinVBrumentNAgbandje-McKennaMSnyderROThe role of the adeno-associated virus capsid in gene transferMethods Mol Biol2008437519118369962
- MuralidharSBecerraSPRoseJASite-directed mutagenesis of adeno-associated virus type 2 structural protein initiation codons: effects on regulation of synthesis and biological activityJ Virol19946811701768254726
- AlamSSenEBrashearHMeyersCAdeno-associated virus type 2 increases proteosome-dependent degradation of p21 WAF1 in a human papillomavirus type 31b-positive cervical carcinoma lineJ Virol200680104927493916641284
- AucoinMGPerrierMKamenAACritical assessment of current adeno-associated viral vector production and quantification methodsBiotechnol Adv2008261738817964108
- AucoinMGPerrierMKamenAAProduction of adeno-associated viral vectors in insect cells using triple infection: optimization of baculovirus concentration ratiosBiotechnol Bioeng20069561081109216952153
- ChahalPSAucoinMGKamenAPrimary recovery and chromatographic purification of adeno-associated virus type 2 produced by baculovirus/insect cell systemJ Virol Methods20071391617017055590
- HsuPYYangYWEffect of polyethylenimine on recombinant adeno-associated virus mediated insulin gene therapyJ Gene Med20057101311132115906397
- ChenCDanielMCQuinkertZTNanoparticle-templated assembly of viral protein cagesNano Lett20066461161516608253
- DixitSKGoicocheaNLDanielMCQuantum dot encapsulation in viral capsidsNano Lett2006691993199916968014
- RenYWongSMLimLYFolic acid-conjugated protein cages of a plant virus: a novel delivery platform for doxorubicinBioconjug Chem200718383684317407258
- BankiewiczKSEberlingJLKohutnickaMConvection-enhanced delivery of AAV vector in parkinsonian monkeys; in vivo detection of gene expression and restoration of dopaminergic function using pro-drug approachExp Neurol2000164121410877910
- MeghrousJAucoinMGJacobDChahalPSArcandNKamenAAProduction of recombinant adeno-associated viral vectors using a baculovirus/insect cell suspension culture system: from shake flasks to a 20-L bioreactorBiotechnol Prog200521115416015903253
- AuricchioAHildingerMO’ConnorEGaoGPWilsonJMIsolation of highly infectious and pure adeno-associated virus type 2 vectors with a single-step gravity-flow columnHum Gene Ther2001121717611177544
- MoulayGBoutinSMasurierCSchermanDKichlerAPolymers for improving the in vivo transduction efficiency of AAV2 vectorsPLoS One2010512e1557621203395
- FeinDELimberisMPMaloneySFHeathJMWilsonJMDiamondSLCationic lipid formulations alter the in vivo tropism of AAV2/9 vector in lungMol Ther200917122078208719638960
- LeHTYuQCWilsonJMCroyleMAUtility of PEGylated recombinant adeno-associated viruses for gene transferJ Control Release2005108116117716125817
- NomuraTKoreedaNYamashitaFTakakuraYHashidaMEffect of particle size and charge on the disposition of lipid carriers after intratumoral injection into tissue-isolated tumorsPharm Res19981511281329487559