 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The aim of this research was to increase the oral bioavailability of daidzein by the formulations of poly(lactic-co-glycolic) acid (PLGA) nanoparticles loaded with daidzein. Amongst the various traditional and novel techniques of preparing daidzein-loaded PLGA nanoparticles, daidzein-loaded phospholipid complexes PLGA nanoparticles and daidzein-loaded cyclodextrin inclusion complexes PLGA nanoparticles were selected. The average drug entrapment efficiency, particle size, and zeta potential of daidzein-loaded phospholipid complexes PLGA nanoparticles and daidzein-loaded cyclodextrin inclusion complexes PLGA nanoparticles were 81.9% ± 5%, 309.2 ± 14.0 nm, −32.14 ± 2.53 mV and 83.2% ± 7.2%, 323.2 ± 4.8 nm, −18.73 ± 1.68 mV, respectively. The morphological characterization of nanoparticles was observed with scanning electron microscopy by stereological method and the physicochemical state of nanoparticles was valued by differential scanning calorimetry. The in vitro drug-release profile of both nanoparticle formulations fitted the Weibull dynamic equation. Pharmacokinetic studies demonstrated that after oral administration of daidzein-loaded phospholipid complexes PLGA nanoparticles and daidzein-loaded cyclodextrin inclusion complexes PLGA nanoparticles to rats at a dose of 10 mg/kg, relative bioavailability was enhanced about 5.57- and 8.85-fold, respectively, compared to daidzein suspension as control. These results describe an effective strategy for oral delivery of daidzein-loaded PLGA nanoparticles and might provide a fresh approach to enhancing the bioavailability of drugs with poor lipophilic and poor hydrophilic properties.
Introduction
Daidzein (4′,7-dihydroxylisoflavone) is a water-insoluble isoflavone,Citation1 isolated mainly from leguminous plants, which is used in the treatment of hypertension, coronary heart disease, cerebral thrombosis, and menopause syndrome. Recently, studies demonstrated that daidzein could inhibit the growth of cancer cells by activating a cell death pathway and prevent the onset of diabetes.Citation2–Citation4 Thus, daidzein is a promising multipurpose treatment. However, the oral bioavailability of daidzein is very poor, which limits its curative effect.Citation5–Citation8 Animal experiments also showed that the absolute bioavailability of daidzein suspension after oral administration to rats was only 6.1%.Citation6 There is a consensus that the low bioavailability of daidzein is related to the drug’s physicochemical properties which include low solubility, low partition coefficient of oil/water, and, in particular, strong metabolism that occurs in the intestine and liver.Citation9 Many microparticulate systems have been used in an attempt to address this issue, such as self-microemulsion and solid lipid nanoparticles drug delivery systems, and have demonstrated that bioavailability of daidzein increased by 2.5-, 2.9-, and 6.9-fold.Citation10–Citation13 An oral route of administration – attractive due to its convenience, patient acceptance, and cost-effectiveness, particularly in the case of chronic therapies – was developed.Citation14 In recent years, nano-based drug delivery systems, such as polymeric nanoparticles, have demonstrated obvious advantages in improving the oral bioavailability and pharmacological activities of poorly soluble drugs;Citation15 in particular, nanoparticles of synthetic polyester such as poly(lactic-co-glycolic) acid (PLGA) is often chosen due to its biocompatibility and versatility in encapsulating a variety of drugs.Citation16–Citation18 Oral delivery of PLGA nanoparticles has also been well studied.Citation19 Some of the commonly reported methods of preparing PLGA nanoparticles include emulsification-solvent evaporation and double-emulsification, which represent easy and reproducible techniques that have been widely used by several research groups to prepare nanoparticles.Citation20 However, the incorporation of daidzein into polymeric-based nanocarriers may be greatly restricted by these methods due to poor lipophilicity and poor hydrophilicity. To overcome these kinds of issues, efforts have been made to increase affinity for hydrophobic carrier materials of poor lipophilicity drugs.Citation11,Citation21 Additionally, cyclodextrins, well-known cyclic oligosaccharides with a lipophilic central cavity and a hydrophilic outer surface, are able to form inclusion complexes with insoluble molecules like camptothecin.Citation22 Therefore, by improving the liposolubility and solubility of daidzein with these techniques combined with the benefits of PLGA nanoparticles, it may be feasible to increase the oral bioavailability of daidzein.
Based on previous information, the aim of this work was, firstly, to separately prepare daidzein phospholipid complexes and daidzein cyclodextrin inclusion complexes and encapsulate into PLGA nanoparticles. Secondly, the physicochemical characteristics of nanoparticles were evaluated. Scanning electron microscopy (SEM) was used to characterize particle morphology, while the physical status of daidzein inside the nanoparticles was investigated by differential scanning calorimetry. Finally, an in vitro release test was used to analyze drug release behavior, and blood concentration was measured after oral administration to rats to measure the bioavailability of daidzein. This study concluded that PLGA combined with phospholipid or cyclodextrin as a carrier material could be applied to water soluble and poorly water soluble compounds, respectively, to control drug release. This is the first report which couples these two formulations with daidzein to improve its oral bioavailability and suggests that this innovative strategy could be effective in improving the oral absorption of poor hydrophilic and poor lipophilic drugs.
Materials
Daidzein (purity >98%) was obtained from Qingze Medical Technology Development Co Ltd (Nanjing, China). PLGA (lactic:glycolic 50:50, molecular weight [Mw] 15,000 Da), phosphatidylcholine, hydroxypropyl-beta-cyclodextrin (Mw 1400 Da), acetonitrile, methylene chloride, chloroform, and dimethyl sulfoxide were purchased from Sinopharm Chemical Reagent Co Ltd (Shanghai, China). Polyvinyl alcohol (PVA, viscosity 11–14 cp) was obtained from Kayon Biological Technology Co Ltd (Shanghai, China). Distilled water was produced by a Milli-Q® purification system (Millipore Corporation, Billerica, MA). The rest of the chemicals and reagents used were of analytical grade.
Methods
Solubility studies
Before the preparation of daidzein-loaded nanoparticles, the solubilization process of daidzein was studied. Briefly, an excess amount of daidzein was added into 1 mL of water, methylene chloride, chloroform, or 2% PVA aqueous solution in a centrifugal tube. Daidzein-phospholipid complexes were added into 1 mL of methylene chloride or chloroform and a certain amount of daidzein-cyclodextrin inclusion complexes were added into 1 mL of water or 2% PVA aqueous solution. These samples were shaken reciprocally at 25°C for 48 hours. This preexperiment showed that daidzein could reach solubility equilibrium and keep stability in 48 hours. After this period of time, the suspensions were filtered through a 0.45 μm membrane filter (Millipore) in order to remove the undissolved daidzein. The concentration of daidzein in the filtrate was measured by high performance liquid chromatography (HPLC) after appropriate dilution with methanol. The solubility experiments were conducted in triplicate and no interferences in the analysis of daidzein from the presence of both phosphatidylcholine and hydroxypropyl-beta-cyclodextrin were found.
Preparation of daidzein complexes
Daidzein-phospholipid complexes preparation
A schematic diagram of the formation of daidzein-loaded phospholipid complexes PLGA nanoparticles (D-PNPs) and daidzein-loaded cyclodextrin inclusion complexes PLGA nanoparticles (D-CNPs) is shown in . As described in , daidzein-phospholipid complexes were prepared according to a previous report in which insulin-phospholipid complexes were constructed.Citation23 In brief, daidzein (2 mg) was added to 5 mL ethanol, containing 20 mg phosphatidylcholine, in a water bath at 40°C. After stirring for 12 hours, ethanol was removed by nitrogen gas and the residues were collected to obtain the daidzein-phospholipid complexes.
Figure 1 Schematic diagram of the formations of daidzein-loaded phospholipid complexes poly(lactic-co-glycolic) acid nanoparticles and daidzein-loaded cyclodextrin inclusion complexes poly(lactic-co-glycolic) acid nanoparticles.
Abbreviations: HP-β cyclodextrin, hydroxypropyl-beta-cyclodextrin; PLGA, poly(lactic-co-glycolic) acid.
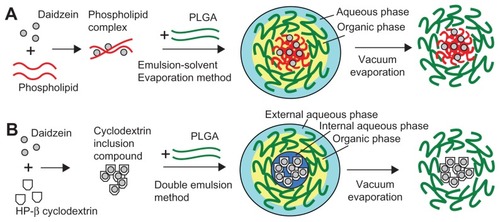
Daidzein-cyclodextrin inclusion complexes preparation
Daidzein-cyclodextrin inclusion complexes were prepared by a fluid inclusion method,Citation22 which is shown in . Briefly, daidzein was added to 5 mL methanol, containing 1:10 molar ratio of hydroxypropyl-beta-cyclodextrin, at 30°C. After stirring for 12 hours, methanol was volatilized by nitrogen gas and the residues were collected.
Preparation of nanoparticles
D-PNPs preparation
As shown in , D-PNPs were prepared by an emulsion-solvent evaporation method.Citation23 Briefly, the obtained daidzein-phospholipid complexes were dissolved in 2 mL of methylene chloride containing 60 mg PLGA previously dissolved. The organic phase was emulsified in 20 mL of aqueous phase containing PVA (1%, weight/volume [w/v]) by sonication over an ice-bath using a Scientz-IID ultrasonic probe (LifeScientz Bio-technology Co, Ltd, Ningbo, China) at an output of 450 W for 5 minutes to form an oil-in-water (o/w) emulsion. Then, the organic solvent was evaporated under vacuum at a temperature of 30°C in a rotary evaporator (RE52CS; Xian Depai Biotechnology Co, Ltd, Shaanxi, China). The residues were solidified by the addition of sodium chloride aqueous solution (1%, w/v) at 0°C. After the nanosuspensions were centrifuged and washed three times with ultra-pure water, the formulations were frozen and freeze-dried (ALPHA 1–2/LDplus; Martin Christ GmbH, Osterode am Harz, Germany) using trehalose (2%, weight/weight) as a cryoprotectant.
D-CNPs preparation
As shown in , D-CNPs were prepared by a double emulsion method.Citation20 Briefly, 1.0 mL of PVA aqueous solution (2%, w/v) containing daidzein-cyclodextrin inclusion complexes was emulsified in 2 mL of chloroform, containing 20 mg phosphatidylcholine and 60 mg PLGA, by high-speed homogenization at 10,000 rpm for 2 minutes. The primary emulsion was added to 20 mL of PVA solution (1%, w/v) and sonicated at 450 W for 5 minutes to form a double-emulsion. Organic solvent residues were removed and the following procedures were the same as described above.
HPLC analytical method
The amount of daidzein loaded into nanoparticles was evaluated by HPLC. Briefly, the chromatography system used was composed of a Shimadzu LC-20AT chromatographic system with an LC-20AT binary pump and an SPD-20A ultraviolet-visible detector (Shimadzu Corporation, Tokyo, Japan). Data processing was performed with LCsolution software (v1.0.0.1; Shimadzu). Analysis was carried out on a Dikma Dimonsil C18 column (200 mm × 4.6 mm, 5 μm; Dikma Technologies, Inc, Beijing China). The mobile phase was composed of methanol and deionized water (55:45 volume/volume),Citation9 and the flow rate was 1.0 mL/minute. Column temperature was maintained at 25°C, ultraviolet-visible detection wavelength was set at 249 nm, and injection volume was 20 μL.
Characterization of nanoparticles
Particle size and zeta potential of nanoparticles were determined by a particle size analyzer (90Plus; Brookhaven Instruments Corporation, Holtsville, NY). Nanoparticle suspension was analyzed by autocorrelation to determine both mean size and zeta potential. All measurements were performed in triplicate.
Morphological evaluation of the nanoparticles was performed using SEM (FEI™ Sirion 200/Inca Oxford; FEI Company, Hillsboro, OR). Briefly, a drop of the dilute nanoparticle suspension was placed onto a copper sheet and then dried under reduced pressure at 40°C. For SEM analysis, the surfaces of the corresponding membranes were covered with gold in a vacuum before viewing under the microscope.
Thermogram characteristics of selected batches of nanoparticles were determined by differential scanning calorimetry (Pyris 1; PerkinElmer, Waltham, MA). Samples (blank-PNPs, blank-CNPs, D-PNPs, D-CNPs, and pure daidzein as a control – calculated by drug loading which contained 2 mg daidzein) were placed separately in a series of sealed aluminum pans and detected. Samples were scanned at 200°C–400°C and heated at a rate of 10°C/minute.Citation24
Drug encapsulation efficiency and drug loading were analyzed by HPLC method. Briefly, 100 μL of evaporated nanoparticle suspension was sucked out and mixed with 900 μL dimethyl sulfoxide to destruct the nanoparticles. After being vortexed for 30 seconds, 100 μL of sample was withdrawn and 900 μL of methanol was added to precipitate insoluble polymers. After the mixture was vortexed again and centrifuged, the supernatant was filtered using a 0.45 μm membrane filter (Millipore) and the result was calculated as Wtotal. Similarly, the amount of daidzein entrapped in the nanoparticles was determined by the following procedure: evaporated nanoparticle suspension (100 μL) was sucked out and centrifuged (12,000 rpm, 30 minutes) in order to remove the nonentrapped daidzein. Then, 1 mL dimethyl sulfoxide was added to destruct the nanoparticles. After being vortexed for 30 seconds, 100 μL of sample was withdrawn and 900 μL of methanol was added to precipitate insoluble polymers. After the mixture was vortexed again and centrifuged, the supernatant was filtered using a 0.45 μm membrane filter (Millipore) and the yield was calculated as Wentrapped. Triplicate samples were prepared for each of the formulations. Drug encapsulation efficiency (%) and drug loading (%) were calculated by the following equations:
where Wtotal was the total amount of daidzein, Wentrapped was the amount of encapsulated drug, and WNPs was the weight of the freeze-dried nanoparticles.
In vitro release study
The in vitro release study was carried out using the dialysis bag diffusion technique. The release rate of daidzein from the nanoparticles was measured by HPLC and performed in a thermo shaker bath system (HZ-9212S Shaking Incubator; Taicang Science & Technology Equipment Inc, Taicang, China). which was maintained at 37°C and shaken horizontally at 100 rpm. Freeze-dried nanoparticles (calculated by drug loading which contained 1 mg daidzein and determined at the first time interval in the dissolution media by HPLC) or 1 mg pure daidzein was placed in a dialysis membrane bag with a molecular cut-off of 3500 Da (Shanghai Green-Bird Science & Technology Development Co Ltd, Shanghai, China), all groups were in sink condition with this amount of drug. Then, each group of samples was immersed in 30 mL of phosphate buffer solution (pH 7.4). After a particular time interval (0, 1, 2, 4, 6, 12, 24, 36, 48, 60, and 72 hours), 1 mL of the dissolution medium was withdrawn and the nanoparticles were resuspended in medium which was replaced with equal volume of fresh dissolution medium at the same temperature. The released daidzein in the dissolution media was determined by HPLC. Measurements were performed in triplicate for each batch.
Bioavailability studies
In vivo pharmacokinetics study was carried out using male Sprague Dawley rats (250 ± 10 g). The animal experimental protocols were performed according to the guidelines of the Experimental Animal Ethics Committee of Shanghai Jiao Tong University (Shanghai, China). These healthy rats were randomly divided into three treatment groups and fasted overnight with free access to water before drug administration. They were administered orally with D-PNPs, D-CNPs, or daidzein suspension (daidzein dispersed in 0.5% sodium carboxymethyl cellulose solution) at a dose of 10 mg/kg. After administration, 300 μL blood samples were collected from the retroorbital plexus at predetermined time points of 0.5, 1, 1.5, 2, 4, 8, 12, 24, 36, and 48 hours and placed into heparinized microcentrifuge tubes (100 IU/mL blood). The blood samples were centrifuged at 5000 rpm for 5 minutes and the separated plasma samples were stored at −80°C until analysis.
Daidzein concentration in plasma, with 0.5 μg/mL ethylparaben as the internal standard, was determined by HPLC method. Plasma samples were prepared in the following steps: 100 μL plasma was firstly mixed with 10 μL internal standard and then 900 μL acetonitrile was added to the plasma. Vortexed for 5 minutes, the plasma sample was centrifuged at 12,000 rpm for 10 minutes. After centrifuging, the supernatant was taken out, dried by nitrogen gas, and the residue was reconstituted in 100 μL methanol. Following further centrifugation at 12,000 rpm for 10 minutes, 20 μL of sample solution was injected into the HPLC system. Peak concentration and time to reach peak concentration were determined directly from the plasma concentration–time curves. Area under the curve was calculated by the trapezoidal method from zero to the final sampling time.
Statistics
All experiments in this study were performed at least three times and results expressed as mean ± standard deviation. Statistical significance was assessed by Student’s t-test or Dunnett’s test for multiple comparisons with P < 0.05 as the minimal level of significance. Pharmacokinetic analysis was performed using Kinetica software program (v4.4; Thermo Fisher Scientific, Waltham, MA).
Results and discussion
Solubility study
In order to study the role of complexes in promoting the solubility of daidzein, a solubility experiment was designed. As shown in , daidzein is not only poorly soluble in water (3.84 μg/mL) and 2% PVA aqueous solution (9.75 μg/mL), but also insoluble in the majority of organic solvents that are commonly used in preparation of nanoparticles, eg, methylene chloride (1.49 μg/mL) and chloroform (0.44 μg/mL). Interestingly, solubility of daidzein-phospholipid complexes in dichloromethane increased significantly, ie, 96 times (828.74 μg/mL) higher than its prototype. Similarly, solubility of daidzein-cyclodextrin inclusion complexes increased 95-fold to 757.92 μg/mL in 2% PVA compared to its prototype.
Table 1 Solubility of pure daidzein and daidzein in each complex form in different solvents
Technically, for a drug to be encapsulated in PLGA nanoparticles, it must be either lipophilic and combine with PLGA molecular chains by a single o/w method or hydrophilic and be encapsulated in an internal aqueous phase of nanoparticles by a double water-in-oil-in-water (w/o/w) emulsification method. Therefore, this must be the main factor limiting daidzein formulation development, which strongly suggests that it is possible to encapsulate daidzein in PLGA nanoparticles if the solubility is changed.
In the present study, to increase lipid or aqueous solubility of daidzein, two different kinds of complexes using phospholipid or cyclodextrin were prepared. Then, nanoparticle formulations of these complexes were prepared by different methods due to their different solubility characteristics. Daidzein-phospholipid complexes as lipophilic substances were easily encapsulated into PLGA nanoparticles by a single o/w method and daidzein-cyclodextrin inclusion complexes as hydrophilic substances were encapsulated in an internal aqueous phase of PLGA nanoparticles by a double w/o/w method.
Optimization of the preparation method
As shown in , the most important step was modifying the drug before it was encapsulated into the nanoparticles. In the schematic diagram of D-PNPs (), daidzein was first combined with phospholipid to form daidzein-phospholipid complexes and then totally codissolved in dichloromethane with PLGA. In the next ultrasound step, the daidzein-phospholipid complexes were fixed inside the PLGA lipophilic chain side while the oil phase formed droplets in water. This was the key step in the preparation of D-PNPs formation. Dichloromethane evaporation maintained the shape of the D-PNPs and created particles that could not fuse to one another.
Compared to D-PNPs, the schematic for preparation of D-CNPs () is more complicated. After the preparation of the daidzein-cyclodextrin inclusion complexes, a single emulsion was formed by high-speed mixing of the internal aqueous phase containing daidzein-cyclodextrin inclusion complexes and the organic phase containing PLGA and phosphatidylcholine. A stable w/o single emulsion was the key step in forming D-CNPs as the structure kept daidzein inside the PLGA layer. A w/o/w double emulsion was prepared after the single emulsion was obtained, and chloroform was evaporated to yield D-CNPs.
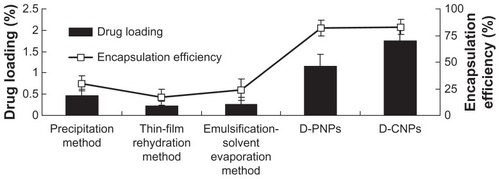
As a safe drug delivery system, PLGA nanoparticles have been used for hundreds of drugs in modern pharmaceutical research, but never for daidzein. Many traditional nanotechnical methods have been used previously in the authors’ lab for encapsulating daidzein directly into PLGA nanoparticles, including the precipitation method,Citation25,Citation26 thin-film rehydration method,Citation27,Citation28 and emulsification-solvent evaporation method.Citation29,Citation30 The nanoprecipitation technique for preparation of nanoparticles is based on the interfacial deposition of a polymer following displacement of a semipolar solvent miscible with water from a lipophilic solution. However, this method suffers the drawback of poor incorporation efficiency of poor lipophilic drugs due to rapid migration and, therefore, loss of drug into the aqueous phase. Compared to the nanoprecipitation method, thin film hydration technique for preparation of nanoparticles is suitable for lipid-soluble drugs, which poses a significant challenge to encapsulating poor lipophilic drugs directly into water-insoluble polymers efficiently. The emulsification-solvent evaporation method also is another commonly used method for preparation of nanoparticles.Citation30 However, one of the disadvantages of the traditional emulsification-solvent evaporation method is low encapsulation efficiencies of poor lipophilic drugs (for o/w emulsification method) or low encapsulation efficiencies of poor hydrophilic drugs (for w/o/w emulsification method). For o/w emulsification method, the drug would diffuse out or partition from the dispersed oil phase into the aqueous continuous phase and microcrystalline fragments of the hydrophilic drugs get deposited on the nanoparticle surface. This would result in low trapping of poor lipophilic drugs. The o/w emulsification process is therefore widely used to encapsulate lipid-soluble drugs. For w/o/w emulsification method, poor hydrophilic drugs would diffuse out or partition from the dispersed internal aqueous phase into the oil phase and this would result in low trapping of poor hydrophilic drugs. These traditional methods were compared to the ones created in this study, and the results are shown in . Unfortunately, the results show that encapsulation efficiency and drug loading of nanoparticles obtained from traditional methods were very low (encapsulation efficiency <25% and drug loading <0.5%). Surprisingly, both D-PNPs and D-CNPs exhibited better encapsulation efficiency and drug loading (encapsulation efficiency >80% and drug loading >1% for both; D-CNPs drug loading was much higher than D-PNPs).
Figure 2 Drug loading and encapsulation efficiency of daidzein-loaded poly(lactic-co-glycolic) acid nanoparticles obtained from traditional methods versus daidzein-loaded phospholipid complexes poly(lactic-co-glycolic) acid nanoparticles and daidzein-loaded cyclodextrin inclusion complexes poly(lactic-co-glycolic) acid nanoparticles.
Abbreviations: D-PNPs, daidzein-loaded phospholipid complexes poly(lactic-co-glycolic) acid nanoparticles; D-CNPs, daidzein-loaded cyclodextrin inclusion complexes poly(lactic-co-glycolic) acid nanoparticles.
These results confirm the assumption that increased affinity of daidzein to the organic phase or aqueous phase increases the amount of drug in PLGA or reduces the loss of drug in the external aqueous phase during the emulsification step. In the traditional methods mentioned above, the majority of daidzein cannot encapsulate into PLGA nanoparticles directly because daidzein cannot coexist in the organic phase (dichloromethane) with PLGA or in the internal aqueous phase with 2% PVA. Thus, daidzein did not appear to be suitable for encapsulation in PLGA nanoparticles directly using traditional methods. After formation of phospholipid complexes or cyclodextrin inclusion complexes, daidzein accumulated in either the organic phase or internal aqueous phase to substantially improve drug loading and encapsulation efficiency.
Overall, daidzein PLGA nanoparticles made by any other method were inferior to D-PNPs and D-CNPs described here (D-PNPs appear inferior to D-CNPs), and these improvements are due to the increased solubility of daidzein following complex formations.
Characterization of D-PNPs and D-CNPs
Preliminary studies were undertaken to determine the range of conditions (particle size and encapsulation efficiency) suitable for the formation of nanoparticles. As a first step, the present work more intensively investigated the possibility of forming nanoparticles with different Mw and concentration of PLGA in the organic phase. PLGA Mw and concentration had a great impact on size and encapsulation efficiency of nanoparticles. When PLGA Mw increased from 5000 Da to 15,000 Da, size and encapsulation efficiency of particles both increased significantly (data not shown). Similarly, the size and encapsulation efficiency of nanoparticles increased as the concentration of PLGA increased (from 20 mg/mL to 40 mg/mL). When concentration of PLGA in the organic phase increased from 30 mg/mL to 40 mg/mL, the size of nanoparticles increased significantly and there were no significant differences in encapsulation efficiency (data not shown). This finding is in agreement with other published studies.Citation31,Citation32 Taking particle size and drug encapsulation both as evaluation standards, PLGA with Mw of 15,000 Da and concentration of 30 mg/mL were selected.
Another aspect worth noting is the function of PVA during the preparation process. It was found that nanoparticles containing PVA exhibited higher encapsulation efficiency and small size compared to others. Some studies have also indicated that it was used as an emulsifier in the preparation of nanoparticles.Citation33 The present research proposes PVA at 1% (w/v) in the aqueous phase as an emulsifier during the preparation process of D-PNPs and D-CNPs, as it was easier to collect nanoparticles at this concentration than at 2% (w/v). The reason for PVA’s superior performance may be that it was easier to form a thin film on the particle surfaces, which could control particle size and prevent daidzein leakage. In addition, as described in , the stability of w/o single emulsion stability is a prerequisite of successful nanoparticle preparation. It was found that dissolved daidzein-cyclodextrin inclusion complexes in the high viscosity of PVA solution at 2% (w/v) as the internal aqueous phase increased encapsulation efficiency compared to the group without PVA. Regarding the effect of phosphatidylcholine in the D-CNPs preparation process, it was found that a stable single emulsion could be more easily formed to increase encapsulation efficiency of daidzein in nanoparticles in the presence of an emulsifier (phosphatidylcholine at 10% of PLGA weight).
After the nanoparticles were isolated, a series of detection experiments were conducted to demonstrate whether the structure and function of nanoparticles were consistent with expectations. Nanoparticle morphology, particle size, surface charge, and physical state of encapsulated drug were studied.
D-PNPs and D-CNPs yielded particles 323.2 ± 4.8 nm and 309.2 ± 14 nm in size (mean diameter), respectively, and zeta potential of D-PNPs and D-CNPs was −32.14 ± 2.53 mV and −18.73 ± 1.68 mV, respectively. Zeta potential is a function of the surface charge of particle dispersions which is commonly used to predict and control dispersion stability. Higher negative values were obtained for D-PNPs (−32.14 ± 2.53 mV) and a marked decrease in the surface charge for D-CNPs occurred (−18.73 ± 1.68 mV). It was speculated that the zeta potential of nanoparticles may be affected by the presence of cyclodextrins, but this theory was not explored in depth.
Encapsulation efficiency of daidzein in D-PNPs was 81.9% ± 5% and 83.2% ± 7.2% in D-CNPs. Drug loading of D-PNPs and D-CNPs was 1.27% ± 0.33% and 1.75% ± 0.24%, respectively, indicating that D-CNPs were more efficient for drug loading than D-PNPs.
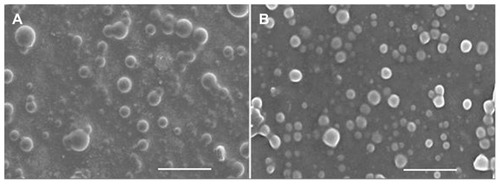
The morphological characterization of the optimized D-PNPs and D-CNPs was examined by SEM. SEM is expected to provide information on nanoparticle morphology and size. Examination of SEM photographs of the nanoparticles revealed that the surfaces were smooth and spherical ().
Figure 3 Scanning electron microscopic photographs of daidzein-loaded phospholipid complexes poly(lactic-co-glycolic) acid nanoparticles (A) and daidzein-loaded cyclodextrin inclusion complexes poly(lactic-co-glycolic) acid nanoparticles (B).
Note: Bar represents 1 μm (80,000× magnification).
The physicochemical state of nanoparticles was valued by differential scanning calorimetry. shows the differential scanning calorimetric thermograms of daidzein, blank-CNPs, blank-PNPs, D-CNPs, and D-PNPs. The melting peak of daidzein appeared at 331°C (). The curves of D-CNPs and D-PNPs showed the same endothermic trend as blank-CNPs and blank-PNPs (), but did not show the daidzein melting peak (). This result indicates that daidzein was not in a crystalline state but in an amorphous state in nanoparticles.
Figure 4 Differential scanning calorimetry curves of daidzein (A), blank phospholipid complexes poly(lactic-co-glycolic) acid nanoparticles (B), blank cyclodextrin inclusion complexes poly(lactic-co-glycolic) acid nanoparticles (C), daidzein-loaded phospholipid complexes poly(lactic-co-glycolic) acid nanoparticles (D), and daidzein-loaded cyclodextrin inclusion complexes poly(lactic-co-glycolic) acid nanoparticles (E).
Abbreviation: DSC, differential scanning calorimetry.

Taking into account that the primary goal of this work was to design a nanoparticulate drug delivery system intended for oral administration, a critical study of these particles would be the evaluation of their stability in the different media present in the gastrointestinal tract. PLGA nanoparticle stability in simulated gastrointestinal conditions can be reviewed in the literature. Previous research reported that PLGA nanoparticles were stable in gastric fluid (pH 1.2) and intestinal fluid (pH 7.5).Citation34,Citation35 Therefore, it was speculated that both D-CNPs and D-PNPs were stable in simulated gastrointestinal conditions.
In vitro release
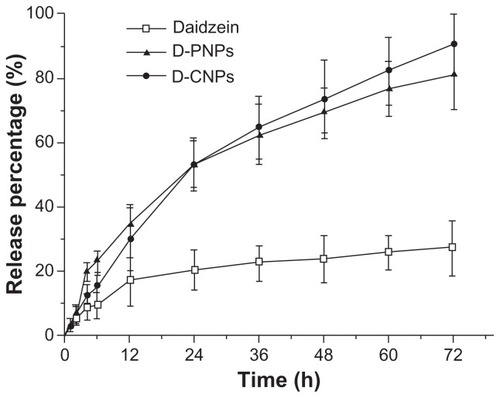
The in vitro release behavior of the nanoparticles is summarized in the cumulative percentage release shown in . The accumulative pure daidzein release rate was only 26.94% while D-PNPs and D-CNPs achieved 80.85% and 90.12%, respectively, in 72 hours. It was demonstrated that pure daidzein cannot be released effectively from the dialysis bag, even when the release solution was an efficient sink. This may be because most of the daidzein was attached to the dialysis bag wall or sank to the bag bottom. This explains why the bioavailability of prototype daidzein is low. A possible reason for this was that 1 mg pure daidzein was suspended in 2 mL of release solution in the dialysis bag, and small clumps of daidzein could have blocked the dialysis bag pores and kept other daidzein molecules from passing through. On the contrary, D-PNPs and D-CNPs were released completely. Each release pattern fitted the Weibull equation:
Figure 5 In vitro cumulate release of pure daidzein and daidzein from daidzein-loaded phospholipid complexes poly(lactic-co-glycolic) acid nanoparticles and daidzein-loaded cyclodextrin inclusion complexes poly(lactic-co-glycolic) acid nanoparticles in phosphate buffer solution (pH 7.4) at a temperature of 37°C and a rate of 100 rpm.
Abbreviations: D-PNPs, daidzein-loaded phospholipid complexes poly(lactic-co- glycolic) acid nanoparticles; D-CNPs, daidzein-loaded cyclodextrin inclusion complexes poly(lactic-co-glycolic) acid nanoparticles; h, hours.
There was no significant burst effect in the initial release phase, possibly because the washing process removed all daidzein adhered on the nanoparticle surface. The initial burst release is one of the major problems in the development of controlled release formulations, including drug-loaded microparticles and nanoparticles, especially with low Mw drugs. In the present experiment, both D-PNPs and D-CNPs released stably from the beginning and didn’t have a burst release, which is shown by their fit to the Weibull equation and illustrates the advantageous structure of D-PNPs and D-CNPs. In both D-PNPs and D-CNPs, daidzein was encapsulated within PLGA. PVA outside the D-PNPs is an important hydrophilic layer to keep daidzein away from the particle surface, while the three-tier w/o/w double emulsion structure in D-CNPs fixed daidzein in the inner core of the particle.Citation36 When nanoparticles were put in a release solution, PVA was dissolved and the carriers (PLGA or phosphatidylcholine) were dissolved gradually to form holes or disintegrated to release the drugs.Citation37 This steady release rate from nanoparticles will allow better drug control in vivo.
Bioavailability studies
D-PNPs, D-CNPs, and daidzein suspension were orally administrated to male Sprague Dawley rats. Plasma concentration– time curves are shown in . A remarkable increase in plasma concentrations and a much slower release of daidzein were observed when nanoparticle groups were studied compared to control group. This result clearly shows that there was a great difference in bioavailability between the two kinds of nanoparticles and daidzein alone. The profile of the D-CNPs curve can be divided in two parts: in the first part, D-CNPs had a high sharp absorption peak at 2 hours; in the second part D-CNPs had a low absorption peak at 12 hours.
Figure 6 Mean plasma concentration–time profiles of daidzein in rats after oral administration of daidzein-loaded phospholipid complexes poly(lactic-co-glycolic) acid nanoparticles, daidzein-loaded cyclodextrin inclusion complexes poly(lactic-co-glycolic) acid nanoparticles, and daidzein suspension at a dose of 10 mg/kg (n = 3).
Abbreviations: conc, concentration; D-PNPs, daidzein-loaded phospholipid complexes poly(lactic-co-glycolic) acid nanoparticles; D-CNPs, daidzein-loaded cyclodextrin inclusion complexes poly(lactic-co-glycolic) acid nanoparticles; h, hours.

shows the pharmacokinetic parameters (peak concentration, time to reach peak concentration, area under the curve0–48, half-life, and relative bioavailability) of daidzein after oral administration. As shown in , peak concentration was found to be significantly higher for D-PNPs (1.92 ± 0.69 μg/mL) and D-CNPs (2.44 ± 0.74 μg/mL) than for daidzein suspension group (1.35 ± 0.47 μg/mL). Similarly, half-life was faster for D-PNPs (8.62 ± 2.69 hours) and D-CNPs (3.38 ± 1.24 hours) compared to daidzein suspension group (1.15 ± 0.55 hours) and time to reach peak concentration was also faster for D-PNPs (5.97 ± 2.17 hours) and D-CNPs (2.04 ± 0.91 hours) compared to daidzein suspension group (0.76 ± 0.21 hours). In the same way, area under the curve0–48 for D-PNPs (10.63 ± 3.77 h μg/mL) and D-CNPs (16.90 ± 6.93 h μg/mL) was five- and eight-fold higher than that of daidzein suspension (1.91 ± 0.81 h μg/mL). Finally, relative bioavailability of D-PNPs and D-CNPs was calculated to be 556.98% and 885.62%, respectively, compared to daidzein suspension group. These results verify that both D-PNPs and D-CNPs were effective in improving the oral bioavailability of daidzein.
Table 2 Pharmacokinetic parameters of daidzein after oral administration of daidzein-loaded phospholipid complexes poly(lactic-co-glycolic) acid nanoparticles, daidzein-loaded cyclodextrin inclusion complexes poly(lactic-co-glycolic) acid nanoparticles, and daidzein suspension at the dose of 10 mg/kg
The pharmacokinetic parameters clearly demonstrate that the two kinds of nanoparticles studied greatly improved bioavailability for daidzein. The reasons are many, but among the most salient is the nano-based drug delivery system. On the one hand, particles that are nanoscale in size are easily absorbed into the folds of the intestinal wall, while large particle surface area to volume lends itself to rapid drug dissolution.Citation38–Citation40 On the other hand, particulate systems for oral delivery can protect the drug at low pH levels, facilitate absorption into the intestinal tract, and bypass unwanted metabolic degradation, and the nature of degradation of the carrier maintains a sustained release.Citation41,Citation42 Amorphous dispersion of daidzein in nanoparticles might be another reason for better absorption.Citation43,Citation44 Apart from these reasons, more importantly, daidzein complexes may play an important role in absorption. Some studies have demonstrated that phospholipid-drug complexes and cyclodextrin-drug complexes could increase oral bioavailability by improving the liposolubility and solubility of drugs.Citation21,Citation22 Furthermore, studies have also pointed out that daidzein as a hydrophilic compound could transport across the intestinal barrier via the paracellular pathway and passive transport is the only mechanism of daidzein absorption in the small intestine of rats, at least within the concentrations tested.Citation45 In addition, phospholipids have been proven to have a high affinity for flavonoids and accelerates daidzein transport across the cell membrane.Citation46–Citation48 Cyclodextrin has been proven as a drug absorption enhancer as well.Citation49,Citation50 Since daidzein is a typical poor hydrophilic and poor lipophilic drug with very low oral bioavailability,Citation7 the greatly improved liposolubility and solubility of daidzein by D-PNPs and D-CNPs should be favorable to its absorption in the gastrointestinal tract. Daidzein complex intermediated not only the increased drug loading of nanoparticles, but also promoted drug absorption in vivo.
Somewhat surprisingly, the in vivo bioavailability study of D-PNPs and D-CNPs in the present study showed that the absorption curves were different shapes. D-PNPs had an absorption peak at about 6 hours, while D-CNPs had a high sharp absorption peak at 2 hours and a low absorption peak at 12 hours. It is generally believed that the ileum and colon are the main absorption sites for nanoparticles; as daidzein-phospholipid complexes in D-PNPs are lipophilic substances, and because of their small size, they would be easily taken up by epithelial cells in the ileum and colon as whole nanoparticles or as drug released from PLGA nanoparticles.Citation51,Citation52 The absorption process may continue for 8–10 hours with a coincident absorption peak. However, a completely different situation may exist for D-CNPs. As far as is known, daidzein-cyclodextrin inclusion complexes are not likely to be absorbed directly by intestinal epithelial cells, but as hydrophilic compounds they will easily leak out of the nanoparticles into gastric juice. By using cyclodextrin as an absorption enhancer, daidzein would be absorbed easily in the small intestine. The first absorption peak may be due to this process. The second peak that appeared after 10 hours was lower and wider, suggesting that daidzein was absorbed into the blood slowly. Based on previous reports and the concentration–time curve of pure daidzein, the second peak was not caused by entry into hepatic circulation as this did not happen in the prototype daidzein absorption curve. As cyclodextrin cannot pass through intestinal epithelial cells, the most likely explanation was that D-CNPs or daidzein-cyclodextrin inclusion complexes were taken up by lymphocytes in the small intestine and daidzein eventually entered the blood after being transported in microfold cells through the ileum.Citation14,Citation53,Citation54
Conclusion
In the present study, an effective strategy for enhancing the oral absorption of daidzein, which has poor hydrophilicity and poor lipophilicity, was developed. Daidzein was incorporated into PLGA nanoparticles by two improved methods. Both of them used daidzein complexes as intermediates that increased the solubility of daidzein to either aqueous or organic solvents. The soluble complexes were successfully used to form PLGA nanoparticles under optimal preparation conditions. In this experiment, D-PNPs were prepared by an emulsion evaporation method using daidzein-phospholipid complexes as an intermediate, while D-CNPs were prepared by a double emulsion method using daidzein-cyclodextrin inclusion complexes as an intermediate. Detection of nanoparticle properties showed that both structures were consistent with theoretical assumptions. More importantly, the pharmacokinetic results showed that both of them improved oral bioavailability of daidzein in rats. These formulations overcome the problem of poor daidzein encapsulation in the lipophilic polymer carrier PLGA. Two distinct compound-nanoparticles were produced from the same drug by producing and using two different complexes as intermediates. This is a new approach for the formulation of drugs with both poor lipophilic and poor hydrophilic properties.
Acknowledgments
The work was supported by the National Natural Science Foundation of China (Grant No 30973644), the National Basic Research Program of China (973 Program; Grant No 2007CB936004), and Shanghai Municipal Committee of Science and Technology (Grant No 08DZ1971304).
Disclosure
The authors report no conflicts of interest in this work.
References
- GutierrezRMBaezEGCardioactive agents from plantsMini Rev Med Chem20099787889919519512
- LoFHMakNKLeungKNStudies on the anti-tumor activities of the soy isoflavone daidzein on murine neuroblastoma cellsBiomed Pharmacother200761959159517905565
- MollerFJDielPZierauOHertrampfTMaassJVollmerGLong-term dietary isoflavone exposure enhances estrogen sensitivity of rat uterine responsiveness mediated through estrogen receptor alphaToxicol Lett2010196314215320381596
- AllredCDTwaddleNCAllredKFSoy processing affects metabolism and disposition of dietary isoflavones in ovariectomized BALB/c miceJ Agric Food Chem200553228542855016248551
- JanningPSchuhmacherUSUpmeierAToxicokinetics of the phytoestrogen daidzein in female DA/Han ratsArch Toxicol200074842143011097378
- QiuFChenXYSongBZhongFLiuCXInfluence of dosage forms on pharmacokinetics of daidzein and its main metabolite daidzein-7-O-glucuronide in ratsActa Pharmacol Sin20052691145115216115384
- ZhengYLeeSOVerbruggenMAMurphyPAHendrichSThe apparent absorptions of isoflavone glucosides and aglucons are similar in women and are increased by rapid gut transit time and low fecal isoflavone degradationJ Nutr2004134102534253915465743
- LiXShenQYuanDMaAJiaWDetermination of daidzein in rat plasma by LCChromatographia2008683–4201205
- KullingSEHonigDMMetzlerMOxidative metabolism of the soy isoflavones daidzein and genistein in humans in vitro and in vivoJ Agric Food Chem20014963024303311410004
- ShenQLiXYuanDJiaWEnhanced oral bioavailability of daidzein by self-microemulsifying drug delivery systemChem Pharm Bull (Tokyo)201058563964320460789
- ZhangZHuangYGaoFBuHGuaWLiYDaidzein-phospholipid complex loaded lipid nanocarriers improved oral absorption: in vitro characteristics and in vivo behavior in ratsNanoscale2011341780178721350765
- ZhangZHuangYGaoFA self-assembled nanodelivery system enhances the oral bioavailability of daidzein: in vitro characteristics and in vivo performanceNanomedicine (Lond)2011681365137922026378
- GaoYGuWChenLXuZLiYThe role of daidzein-loaded sterically stabilized solid lipid nanoparticles in therapy for cardio-cerebrovascular diseasesBiomaterials200829304129413618667234
- LavelleECSharifSThomasNWHollandJDavisSSThe importance of gastrointestinal uptake of particles in the design of oral delivery systemsAdv Drug Deliv Rev1995181522
- AgüerosMZabaletaVEspuelasSCampaneroMAIracheJMIncreased oral bioavailability of paclitaxel by its encapsulation through complex formation with cyclodextrins in poly(anhydride) nanoparticlesJ Control Release201014512820347897
- JainRAThe manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devicesBiomaterials200021232475249011055295
- PanyamJLabhasetwarVBiodegradable nanoparticles for drug and gene delivery to cells and tissueAdv Drug Deliv Rev200355332934712628320
- ShiveMSAndersonJMBiodegradation and biocompatibility of PLA and PLGA microspheresAdv Drug Deliv Rev199728152410837562
- DamgeCAprahamianMMarchaisHBenoitJPPingetMIntestinal absorption of PLAGA microspheres in the ratJ Anat1996189Pt 34915018982822
- TewesFMunnierEAntoonBComparative study of doxorubicin- loaded poly(lactide-co-glycolide) nanoparticles prepared by single and double emulsion methodsEur J Pharm Biopharm200766348849217433641
- CuiFShiKZhangLTaoAKawashimaYBiodegradable nanoparticles loaded with insulin-phospholipid complex for oral delivery: preparation, in vitro characterization and in vivo evaluationJ Control Release2006114224225016859800
- CirpanliYBilensoyELale DoganAÇalisSComparative evaluation of polymeric and amphiphilic cyclodextrin nanoparticles for effective camptothecin deliveryEur J Pharm Biopharm2009731828919442723
- JefferyHDavisSSO’HaganDTThe preparation and characterisation of poly(lactide-co-glycolide) microparticles. I: oil-in-water emulsion solvent evaporationInt J Pharm1991772–3169175
- MuLFengSSA novel controlled release formulation for the anticancer drug paclitaxel (Taxol): PLGA nanoparticles containing vitamin E TPGSJ Control Release2003861334812490371
- FessiHPuisieuxFDevissaguetJPAmmouryNBenitaSNanocapsule formation by interfacial polymer deposition following solvent displacementInt J Pharm198955R14
- MolpeceresJGuzmanMAberturasMRChaconMBergesLApplication of central composite designs to the preparation of polycaprolactone nanoparticles by solvent displacementJ Pharm Sci19968522062138683450
- AdityaNPPatankarSMadhusudhanBMurthyRSSoutoEBArthemeter-loaded lipid nanoparticles produced by modified thin-film hydration: pharmacokinetics, toxicological and in vivo anti-malarial activityEur J Pharm Sci201040544845520493255
- AquilanoDCavalliRGascoMRSolid lipospheres obtained from hot microemulsions in the presence of different concentrations of cosurfactant: the crystallization of stearic acid polymorphsThermochim Acta19932302937
- HariharanSBhardwajVBalaISitterbergJBakowaskyURavi KumarMNDesign of estradiol loaded PLGA nanopaticles formulations: a potential oral delivery system for hormone therapyPharm Res200623118419516267632
- ArshadyRPreparation of biodegradable microspheres and microcapsules: 2. Polylactides and related polyestersJ Control Release1991171122
- LiXDengXYuanMInvestigation on process parameters involved in preparation of poly-DL-lactide–poly(ethylene glycol) microspheres containing Leptospira Interrogans antigensInt J Pharm1999178224525510205644
- ZhuKJJiangHLDuXYWangJXuWXLiuSFPreparation and characterization of hCG-loaded polylactide or poly(lactide-co-glycolide) microspheres using a modified water-in-oil-in-water (w/o/w) emulsion solvent evaporation techniqueJ Microencapsul200118224726011253941
- SahooSKPanyamJPrabhaSLabhasetwarVResidual polyvinyl alcohol associated with poly(D,L-lactide-co-glycolide) nanoparticles affects their physical properties and cellular uptakeJ Control Release200282110511412106981
- AhlinPKristlJKristlAVrecerFInvestigation of polymeric nanoparticles as carriers of enalaprilat for oral administrationInt J Pharm20022391–211312012052696
- YinYChenDQiaoMWeiXHuHLectin-conjugated PLGA nanoparticles loaded with thymopentin: ex vivo bioadhesion and in vivo biodistributionJ Control Release20071231273817728000
- HasanASSochaMLamprechtAEffect of the microencapsulation of nanoparticles on the reduction of burst releaseInt J Pharm20073441–2536117643878
- HurteauxREdwards-LevyFLaurent-MaquinDLevyMCCoating alginate microspheres with a serum albumin-alginate membrane: application to the encapsulation of a peptideEur J Pharm Sci2005242–318719715661490
- RatzingerGFillaferCKerletaVWirthMGaborFThe role of surface functionalization in the design of PLGA micro- and nanoparticiesCrit Rev Ther Drug Carrier Syst2010271183
- DelieFEvaluation of nano- and microparticle uptake by the gastrointestinal tractAdv Drug Deliv Rev1998342–322123310837679
- McCleanSProsserEMeehanEBinding and uptake of biodegradable poly-DL-lactide micro- and nanoparticles in intestinal epitheliaEur J Pharm Sci1998621531639795038
- DongYFengSSPoly(D,L-lactide-co-glycolide)/montmorillonite nanoparticles for oral delivery of anticancer drugsBiomaterials200526306068607615894372
- O’HaganDTJefferyHRobertsMJMcGeeJPDavisSSControlled release microparticles for vaccine developmentVaccine19919107687711759495
- HancockBCCarlsonGTLadipoDDLangdonBAMullarneyMPComparison of the mechanical properties of the crystalline and amorphous forms of a drug substanceInt J Pharm20022411738512086723
- CorriganDOHealyAMCorriganOIThe effect of spray drying solutions of bendroflumethiazide/polyethylene glycol on the physicochemical properties of the resultant materialsInt J Pharm20032621–212513712927394
- FotiPErbaDSpadafrancaACiappellanoSBrescianiJTestolinGDaidzein is absorbed by passive transport in isolated small intestine of ratsNutr Res2006266284288
- FrickerGKrompTWendelAPhospholipids and lipid-based formulations in oral drug deliveryPharm Res20102781469148620411409
- LeighMvan HoogevestPTiemessenHOptimising the oral bioavailability of the poorly water-soluble drug cyclosporin A using membrane lipid technologyDrug Deliv Syst2001137377
- PorterCJTrevaskisNLCharmanWNLipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugsNat Rev Drug Discov20076323124817330072
- HamadaHIshiharaKMasuokaNMikuniKNakajimaNEnhancement of water-solubility and bioactivity of paclitaxel using modified cyclodextrinsJ Biosci Bioeng2006102436937117116587
- MadyFMAbou-TalebAEKhaledKAEvaluation of carboxymethyl-beta-cyclodextrin with acid function: improvement of chemical stability, oral bioavailability and bitter taste of famotidineInt J Pharm20103971–21820600734
- MaitiKMukherjeeKGantaitASahaBPMukherjeePKCurcumin-phospholipid complex: preparation, therapeutic evaluation and pharmacokinetic study in ratsInt J Pharm20073301–215516317112692
- Sachs-BarrableKLeeSDWasanEKThorntonSJWasanKMEnhancing drug absorption using lipids: a case study presenting the development and pharmacological evaluation of a novel lipid-based oral amphotericin B formulation for the treatment of systemic fungal infectionsAdv Drug Deliv Rev200860669270118053611
- WinKYFengSSEffects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugsBiomaterials200526152713272215585275
- FlorenceATHilleryAMHusainNJaniPUNanoparticles as carriers for oral peptide absorption: studies on particle uptake and fateJ Control Release1995361–23946