Abstract
Background
Multidrug resistance (MDR) mediated by the overexpression of adenosine triphosphate (ATP)-binding cassette (ABC) transporters, such as P-glycoprotein (P-gp), remains one of the major obstacles to effective cancer chemotherapy. In this study, lipid/particle assemblies named LipoParticles (LNPs), consisting of a dimethyldidodecylammonium bromide (DMAB)-modified poly(lactic-co-glycolic acid) (PLGA) nanoparticle core surrounded by a 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) shell, were specially designed for anticancer drugs to bypass MDR in human breast cancer cells that overexpress P-gp.
Methods
Doxorubicin (DOX), a chemotherapy drug that is a P-gp substrate, was conjugated to PLGA and encapsulated in the self-assembled LNP structure. Physiochemical properties of the DOX-loaded LNPs were characterized in vitro. Cellular uptake, intracellular accumulation, and cytotoxicity were compared in parental Michigan Cancer Foundation (MCF)-7 cells and P-gp-overexpressing, resistant MCF-7/adriamycin (MCF-7/ADR) cells.
Results
This study found that the DOX formulated in LNPs showed a significantly increased accumulation in the nuclei of drug-resistant cells relative to the free drug, indicating that LNPs could alter intracellular traffic and bypass drug efflux. The cytotoxicity of DOX loaded-LNPs had a 30-fold lower half maximal inhibitory concentration (IC50) value than free DOX in MCF-7/ADR, measured by the colorimetric cell viability (MTT) assay, correlated with the strong nuclear retention of the drug.
Conclusion
The results show that this core-shell lipid/particle structure could be a promising strategy to bypass MDR.
Introduction
The development of multidrug resistance (MDR) is a major obstacle to effective cancer chemotherapy. After a long period of chemotherapy, many patients suffer from MDR, which can reduce therapy efficiency and lead to treatment failure.Citation1 The high level of resistance is usually caused by complex MDR mechanisms. Among them, overexpression of the adenosine triphosphate (ATP)-binding cassette transporters (ABC), such as P-glycoprotein (P-gp), is one of the most prevalent mechanisms.Citation2,Citation3 Tumor cells possess highly ordered internal resistance. This P-gp encoded by the MDR-1 gene acts as a drug efflux pump that exports a wide range of chemotherapeutic drugs and will reduce the accumulation of functional drugs in MDR cancer cells, resulting in low cancer chemotherapeutic efficacy.Citation4 Currently, only a few tissue-derived advanced cancers can be cured by chemotherapeutic drugs. Furthermore, these tumors may also recur and become drug resistant.Citation5 Therefore, overcoming the drug resistance of tumors is a serious challenge for successful chemotherapy.
With the development of nanotechnology, nanoformulations have been widely used to circumvent MDR.Citation6,Citation7 Previous studies have shown that these nano-sized particles, such as lipids, micelles, and inorganic hybrid particles, can bypass the P-gp efflux pumps and alter the intracellular accumulation of chemotherapeutic drugs.Citation8,Citation9 In a recent study, biodegradable polymer capsules offered a promising means to circumvent P-gp-mediated MDR.Citation10 The capsules were internalized by MDR cells via endocytosis, and were designed for the subsequent intracellular release of the drugs by enzymatic degradation in lysosomes. According to another strategy, low molecular weight polyethylenimine (PEI) conjugated to doxorubicin (DOX)-loaded particles could escape endosomal retention, and increase the intracellular DOX accumulation, thereby circumventing MDR-mediated drug efflux.Citation11 These synthetic biomaterials are able to escape the endosomal/lysosomal compartment and change intracellular trafficking to increase the drug concentration in MDR cancer cells. These are common mechanisms whereby MDR activity can be reversed. Thus, various endosomal escape pathways provide an opportunity for encapsulating drugs to overcome MDR in tumors.Citation12
Several synthetic agents that have been introduced, such as PEI and poly(amidoamine)s (PAAs), have the proton sponge effect, a property that can contribute to efficient endosomal escape.Citation13,Citation14 In this study, dimethyldidodecylammonium bromide (DMAB), which is a cationic emulsifier with double hydrocarbon chains, was used to escape the endosomal pathway. Early nanotechnology application of DMAB focused on oral drug delivery or inhibition of vascular restenosis because of its low critical micelle concentration and high zeta potential.Citation15,Citation16 Recent studies have shown that DMAB-modified nanoparticles are an ideal vehicle for nuclear targeting that could significantly improve the transfection rate in gene delivery.Citation17 The results demonstrated clearly that DMAB-modified nanoparticles could evade the arrest of the endosomal/lysosomal lumen and destabilize the membranes. This is similar to the effects elicited by the lysosomal disrupting agent L-Leucine-L-leucine methyl ester (LLME).Citation18 The carrier then escapes from the endosomes/lysosomes and is targeted to the cell nucleus. These and other studies with drug- or gene-loaded DMAB nanoparticles have demonstrated their advantage as efficient drug delivery vehicles with small particles sizes, enhanced cell uptake, and endosomal escape.Citation19,Citation20 However, no studies have been carried out to improve the biocompatibility of DMAB particles for in vivo applications, or to examine whether drug-loaded DMAB particles are able to circumvent MDR by efficiently escaping endosomes and lysosomes.
In recent years, the core-shell supramolecular structures, the lipid/particle assemblies – termed LipoParticles (LNPs) – have received considerable attention for their potential application in biomedicine and biotechnology.Citation21,Citation22 These LipoParticles assemblies, based on electrostatic interaction, were first reported by Carmona-Ribeiro et al.Citation23,Citation24 Later, Troutier et al reported an in-depth characterization on the basis of zeta potential measurements and microscopic morphology.Citation25,Citation26 Subsequent studies demonstrated that these unique hybrid assemblies, consisting of a solid polymer sphere surrounded by a lipid shell, could combine the advantages of lipid shells with those of solid support cores.Citation27 The lipid shell provides excellent biocompatibility, as well as the ability to encapsulate particles for in vitro and in vivo bioapplications for protein, peptide, and DNA (lipidic liposome/DNA complexes) delivery.Citation28 Lipid/particle assemblies, based on cationic polysaccharide nanoparticles as oligonucleotide carriers, appear to be endocytosed and accumulate intracellularly in the cytoplasm, further indicating additional structural advantages.Citation29
The current study designed a novel, core-shell structure lipid/particle assembly, LNPs, based on the cationic DMAB-modified poly(lactic-co-glycolic acid) (PLGA) nanoparticles to overcome MDR. The PLGA nanoparticle core was wrapped by 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) via hydrophilic and electrostatic interactions and forming a stable, cell-like structure.Citation30 Moreover, the phospholipid surface was expected to improve biocompatibility and enhance the penetration of barriers. DOX, a cell cycle toxic drug, was conjugated to PLGA, as reported previously.Citation31 In this manner, DOX was loaded in a stable manner, and the drug release could also be controlled. The core-shell LNP utilizes DMAB-modified DOX-PLGA nanoparticles as a strategy to escape the endosomal/lysosomal pathway by enzymatic degradation to overcome MDR in cancer cells.Citation10 The mechanism for reversing MDR was confirmed by intracellular accumulation observations.
Materials and methods
Materials
PLGA block copolymer (50:50, molecular weight: 8000) was purchased from Shandong Institute of Medical Instruments (Jinan, China). DPPC was purchased from Avanti Polar Lipids Inc (Alabaster, AL). DOX was purchased from Haikou Manfangyuan Chemical Company (Haikou, China). DMAB, Trinton® X-100, and thiazolyl blue tetrazolium bromide was obtained from Sigma-Aldrich® Chemical Co, (St Louis, MI). Ethyl acetate (EA, analytical grade) and acetonitrile (high-performance liquid chromatography (HPLC) grade) was from Merck (Darmstadt, Germany). Distilled water, Milli-Q® (18 MΩ; Millipore, Billerica, MA), was used in the synthesis. All other materials were analytical grade. Fetal bovine serum (FBS), RPMI-1640 medium (RPMI), penicillin-streptomycin (PS, 10000 U/mL), trypsin-EDTA solution (0.5% trypsin, 5.3 Mm EDTA tetrasodium), and Hank’s balanced salt solution (HBSS) were obtained from Gibco® BRL (Gaithersberg, MD).
Preparation of LNPs
DOX-PLGA conjugate was synthesized as reported, and was used to prepare nanoparticles. Nanoparticles were prepared by the emulsification-diffusion method. In detail, 200 mg PLGA-DOX conjugate was dissolved in 10 mL ethyl acetate. Then, the oil phase was slowly added into 20 mL of an aqueous solution containing 0.5% DMAB, and emulsified using a high speed homogenizer (FLUKO, Shanghai, China) twice at 10,000 r/min for 10 minutes, 5 minutes each time. The resulting oil-in-water emulsion solution was slowly poured into 100 mL water under the moderate magnetic stirring. The organic solvents were removed through rotary-evaporation. The formed nanoparticles were collected by centrifugation (J2-MC; Beckman Coulter, Fullerton, CA; 51,000 g, 30 minutes, 4°C) and washed with distilled water for three times. The nanoparticle pellet was resuspended in solution for further study. Blank nanoparticles were also prepared by the same method. DPPC was dissolved in chloroform; the solution was incubated at 40°C for 4 hours with backflow device; then chloroform was removed by rotary evaporation. A suspension of DMAB-modified nanoparticles was added to the DPPC lipid membranes. After stirring and 30 minutes incubation at 45°C, the suspension was homogeneous with Nuclepore™ 100 nm (Whatman®, Brentford, UK). The suspension was centrifuged (16,000 g, 30 minutes, 4°C) and washed with distilled water three times. The pellet was resuspended in solution or freeze-dried for further study.
Nanoparticle characterization
Particle size and size distribution
Mean hydrodynamic diameters and mean size distributions were determined at 25°C by means of dynamic-light scattering technique with particle size analyzer (Zetasizer Nano S90; Malvern Instruments, Malvern, UK). The measurement angle was 90°. The nanoparticle samples were freeze-dried and dispersed in a 1 mmol · L−1 NaCl solution or serum-supplemented RPMI-1640, and the results was given by the average of three measurements. The polydispersity index (PDI) was a dimensionless measure of size distribution, and was also recorded at this stage.
Surface charge
Zeta potential is an indicator of surface charge, which determines particle stability in dispersion. Zeta potential was performed on a zeta potential analyzer (90Plus; Brookhaven Instrument Corp, Huntsville, NY) by the same treatment as size measurement.
Transmission electron microscopy
Transmission electron microscopy (TEM) images were performed on a JEM-1230 electron microscope (JEOL, Tokyo, Japan) at an acceleration voltage of 80 kV. To prepare sample sections of bare particles or polymeric nanoparticles, the samples were diluted with phosphate-buffered saline (PBS) (NaOH 29 mM, KH2PO4, 50 mM, pH 7.0) and the final particle concentration was 1 mg/mL. The resulting sample was dropped on to a copper grid. Excess aqueous solution was blotted with filter paper and evaporated at room temperature for 2 hours before observation.
Scanning electron microscopy
Morphology of the nanoparticles was performed on a field emission-scanning electron microscopy (FE-SEM) (S-4800 UHR; Hitachi High Technologies, Tokyo, Japan). The sample was freeze-dried and fixed on metallic studs with double-sided conductive tape. It was then precoated with platinum before SEM observation.
Loading efficiency of nanoparticles
Untrapped drugs were removed by centrifugation. The nanoparticle samples were freeze-dried. Then 3 or 4 mL of nanoparticles or polymeric nanoparticles were dissolved in 10 mL dimethylformamide (DMF) solution. The drug concentration was determined by HPLC (Agilent 1100; Hewlett Packard HP/Agilent Technologies, Santa Clara, CA). The drug loading content (LC) was calculated.
Drug release
DOX-loaded polymeric nanoparticles 5 mL were diluted into 1 mL 1/15 M PBS (pH 5.5, 6.5, and 7.4), in a dialysis bag of MW 3500 (Boya Biotechnology, Shanghai, China). The dialysis bags containing samples were placed into 20 mL PBS, 37°C incubator shaker at 150 r/min. At appropriate intervals, 5.0 mL solution was removed for determination by HPLC, and fresh release medium was replaced immediately. Percent drug released from polymeric nanoparticles was calculated.
P-gp expression
MCF-7 cells and MCF-7/ADR cells were allowed to grow in 25 mL cell bottles separately for 72 hours. One million cells were collected for each sample and were centrifuged at 4000 r/min for 3–4 minutes. After washing each sample three times by bovine serum albumin (BSA) (0.1 mg/mL), P-gp specific primary antibody (Abcam, Cambridge, UK) were added and incubated for 1 hour. Then cells were washed another three times by BSA (0.1 mg/mL) and fluorescein isothiocyanate-labeled secondary antibody (MultiSciences Biotech Co, Ltd, Beijing, China) was added. Again 1 hour later, cells were washed twice by PBS (pH 7.4) and analyzed using flow cytometry (FACS) for the green fluorescence intensity. Untreated cells were taken as controls for both cells.
Cellular uptake assay
MCF-7 parental cells and resistant MCF-7/ADR cells were obtained from the Institute of Biochemistry and Cell Biology of Chinese Academy of Sciences (IBCB, Shanghai, China). Cells were cultured in RPMI-1640 medium with 1% penicillin-streptomycin solution, supplemented with 10% FBS at 37°C, 5% CO2, and 95% humidity. In the present study, 5 × 104 cells were seeded on 24 well black plates (Costar, Acton, MA), which were incubated until the cells reached 80% confluence. The medium was changed with Hank’s HBSS (pH 7.4) and incubated for 30 minutes. After equilibration, the cells were incubated with nanoparticle suspension (8 μg/mL free DOX or a concentration of LNPs-DOX equivalent to 8 μg/mL in HBSS). The samples were incubated with the cells for 0.50–4 hours at 37°C. Then, the cells were centrifuged and washed using PBS (pH 7.4) to eliminate free particles that were not preyed by the cells. After washing three times, FACS analysis was used to determine DOX intensity. Negative control was also determined for regulating the results.
Confocal laser scanning microscopy
Cells were seeded on glass-bottom culture dishes and incubated at 37°C, 5% CO2, and 95% relative humidity. After 80% confluent, the medium was removed and replaced by HBSS. Thirty minutes later, the solution was replaced with free DOX, or LNPs-DOX with the same DOX concentration. The cells were incubated with particles at 37°C for 4 hours. Then the cells were washed three times with PBS (pH 7.4) solution to eliminate free particles that were not removed by cell uptake. Cells were then fixed with 4% paraformaldehyde in PBS for 30 minutes, and nuclei were stained by Hoechst 33342 (Beyotime Biotechnology, Nanjing, China). The samples were examined with the confocal laser scanning microscope (LSM 510; Zeiss, Dresden, Germany). The excitation wavelengths were 405 nm and 488 nm (Hoechst, blue; DOX, red), respectively.
Dose-response experiments
MCF-7 parental cells and resistant MCF-7/ADR cells were seeded in 96 well plates at a density of 1 × 104 cells per well and incubated for 24 hours. After that, the medium was replaced with the specified samples. The cells were then treated with free DOX or polymeric nanoparticles at a certain concentration of DOX in 150 μL RPMI-1640 medium. Cell viability was determined after 24-hour incubation. Thiazolyl blue tetrazolium bromide solution was added into each well. Without any additions, the cells were incubated at 37°C and 5% CO2 for 4 hours. Subsequently, the culture medium in each well was removed, and 200 μL dimethyl sulfoxide (DMSO) was used to dissolve the formed formazan crystals. The absorbance was measured at 570 nm after shaking using an enzyme-linked immunosorbent assay (ELISA) plate reader (Thermo Scientific Multiskan® Spectrum; Thermo Scientific, Waltham, MA). Cells incubated with fresh RPMI-1640 medium were used as a control.
Statistical analysis
Statistical analysis was performed using t-test. Differences between groups were designated as P < 0.05(*).
Results
Preparation and characterization of LNPs
In this study, the LNPs were designed for reversing MDR with DMAB-modified PLGA nanoparticle (NP) core coated with DPPC (). DOX and PLGA were connected via an amide bond, as reported (supporting information, S1).Citation31 The DOX conjugation efficiency was 46% measured by nuclear magnetic resonance. This kind of DOX-PLGA conjugate could facilitate zero-order release of DOX at the tumor site for a long period. PLGA NPs prepared by DMAB dissolved in ethyl acetate had a uniform size distribution of about 150 nm. The LNPs were self-assembled from the cationic DMAB-modified PLGA NP core. The PLGA NPs were wrapped in DPPC via electrostatic forces, and the size of the formed LNPs increased to approximately 200 m. The DOX loading content of the PLGA NPs and LNPs was 0.71% ± 0.06% and 0.52% ± 0.10%, respectively, as measured by HPLC ().
Table 1 Physicochemical properties of different formulations (n = 3)
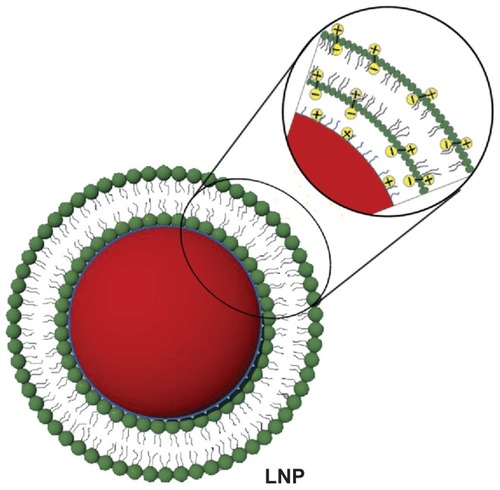
Figure 1 Schematic diagram showing the structure of lipid/polymer particle assemblies. The negative phosphate charges on the hydrophilic segment of DPPC (green) can be attracted to the oppositely charged ions of the ammonium bromide ammonia headgroup of DMAB (blue), which modify the surface of DOX-loaded PLGA nanoparticles (red).
Abbreviations: DMAB, dimethyldidodecylammonium bromide; DOX, doxorubicin; DPPC, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine; LNP, lipid/polymer particle assembly; PLGA, poly(lactic-co-glycolic) acid.
The surface morphology of LNPs was studied by means of TEM and SEM (). Based on TEM images (), the LNPs showed relatively uniform sizes and were surrounded by a layer of petal-like vesicles with a bilayer thickness of about 20 nm. SEM images () showed that the surface structure of the PLGA NPs modified by DMAB was fairly rigid, and did not show the decomposition under the effect of electron-beam bombardment. The surface of the LNPs showed evidence of degradation and cracking under the same voltage conditions. These results demonstrate that the nanoparticles were covered by a layer of DPPC.Citation32 PLGA NPs had a high positive zeta potential of 57 mV owing to the cationic DMAB. The surface properties of the PLGA core should be changed with the DPPC coating, and the zeta potential of LNPs was reduced from 57 mV to −1.8 mV (). This negative zeta potential may improve the biocompatibility and facilitate further application in vivo.Citation33 The colloidal stability of PLGA NPs and LNPs was determined in serum-supplemented RPMI-1640 by dynamic-light scattering. Compared to PLGA NPs, the LNPs showed excellent stability in this biomimetic medium over a 7-day period.
Figure 2 TEM and SEM images of LNPs formed with a core-shell supramolecular structure. A TEM image shows relatively uniform sized LNP particles (A), and a LipoParticle revealing its lipid shell thickness (B). A SEM image shows the fairly rigid surface morphology of PLGA particles by the electron-beam bombardment effect (C), and another SEM image displays the degradation and cracking of LNPs with a DPPC shell (D).
Abbreviations: DPPC, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine; LNPs, lipid/polymer particle assemblies; SEM, scanning electron microscopy; TEM, transmission electron microscopy.

In vitro drug release
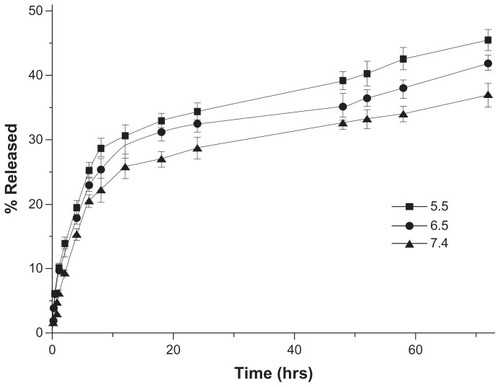
Drug release in vitro was determined in PBS of different pH (5.5, 6.5, and 7.4) to estimate the potential drug release characteristics of the nanoparticles under different physiological conditions. The release profiles revealed that the release of DOX from LNPs-DOX (with lipid shell, LC 0.52%) was accelerated at low pH. As shown in , the LNPs-DOX released its drug more readily at pH 5.5, 6.5, and 7.4. The LNPs with a lipid shell exhibited less than 20% DOX release after the first 6 hours at pH 7.4 (blood pH level), and approximately 32% slow leakage within 72 hours. This controlled release from LNPs-DOX was similar to that from NPs-DOX (without a lipid shell; supporting information S2), and indicates that aggregation proceeds independent of the spatial structure of the particle core and the lipid shell, and that the DOX release was affected more by the DOX-PLGA conjugation. In the more acidic tumor microenvironment (pH 6.5), and in endosomes/lysosomes (pH 5.5), DOX release from these assemblies was more rapid, suggesting that DOX can be released sharply during intracellular trafficking, which would be helpful to bypass drug efflux.
Figure 3 In vitro drug release profiles of DOX-loaded LNPs at different pH (5.5, 6.5, and 7.4) and 37°C.
Abbreviations: DOX, doxorubicin; LNPs, lipid/polymer particle assemblies.
Determination of MDR on MCF-7/ADR cells
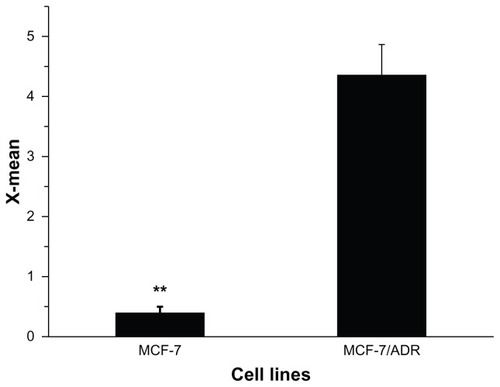
Parental MCF-7 and resistant MCF-7/ADR cells were purchased from ICBC, and maintained at 37°C in 5% CO2 in RPMI-1640 medium with a 1% penicillin-streptomycin solution. Preliminary experiments and the MTT assays here showed that MCF-7/ADR had significant MDR. MCF-7 and MCF-7/ADR cells were exposed to increasing doses of DOX for 24 hours. Cell viability and 50% viability (IC50) were determined (supporting information, S3). The IC50 of MCF-7/ADR (47.53 ± 2.53 μg/mL) was 228-fold greater than that in MCF-7 cells (0.221 ± 0.045 μg/mL). To further understand the drug resistance in MCF-7/ADR, P-gp protein expression was determined by flow cytometry. The results revealed that the greater drug resistance of MCF-7/ADR was accompanied by the higher expression of P-gp protein (), suggesting that reversal of resistance to DOX in MCF-7/ADR should escape this protein-mediated efflux.
Figure 4 P-gp expression in MCF-7 and MCF-7/ADR cells, as shown by flow cytometry analysis.
Note: **P < 0.01.
Abbreviations: MCF-7, Michigan Cancer Foundation-7; MCF-7/ADR, MCF-7/adriamycin; P-gp, P-glycoprotein.
Intracellular accumulation of doxorubicin
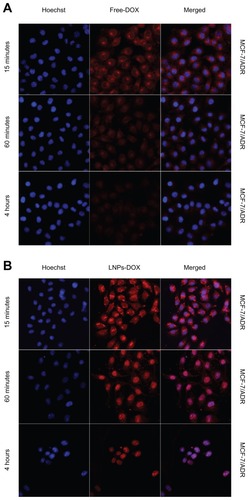
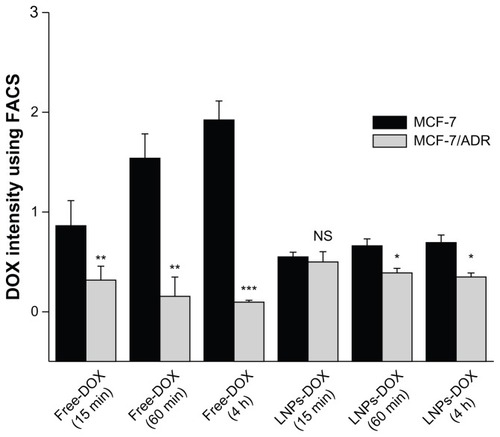
DOX is known to interact with DNA by intercalation and inhibition of macromolecular biosynthesis. One potential strategy to reverse DOX resistance in cells is to change the intracellular localization of the drug through endocytosis using carrier particles.Citation34 When drug-loaded nanocarriers enter cells via endocytosis, the particle-associated drugs can escape endosomal/lysosomal effect (eg, the proton sponge effect elicited by PEI) and increase the localization in the cytosol or nucleus of cells.Citation35 Here, to determine if LNPs- DOX could be accumulated efficiently in the nucleus, the intracellular localization of free DOX and LNPs-DOX was characterized in both parental MCF-7 and MCF-7/ADR cells. LNPs-DOX distribution in MCF-7 and MCF-7/ADR cells was visualized after 15 minutes, 1 hour, and 4-hour incubation (). Following treatment in MCF-7/ADR cells, little free DOX could diffuse into the nucleus, and most of the drug accumulated in the cytoplasm (15 minutes–1 hour). Then, the drug uptake and accumulation in MCF-7/ADR cells was reduced significantly, which should be the result of the efflux of DOX caused by the overexpression of the ATP-binding cassette (ABC) transporters (). By contrast, the intracellular localization of LNPs-DOX in MCF-7/ADR cells was changed. After a 1-hour exposure, significant DOX was accumulated in the cytoplasm of the parental MCF-7 cells, whereas substantially increased LNPs- DOX accumulated in the nuclei of the drug-resistant MCF-7/ADR cells (). This observation suggests that LNPs, as a drug carrier, could change the sub-cellular localization of DOX and bypass MDR. The results at 4 hours showed the long-term accumulation of LNPs-DOX in the nuclei.
Figure 5 Confocal images of the intracellular accumulation of free DOX and LNPs- DOX in MCF-7 (A) and MCF-7/ADR (B) cells. Cells were treated with 8 μg/mL free DOX or an equivalent concentration of LNPs-DOX for the different time periods as indicated. Nuclear (blue) uptake of DOX was increased dramatically by LNPs.
Abbreviations: DOX, doxorubicin; free DOX, free doxorubicin; LNPs-DOX, doxorubicin loaded in lipid/polymer particle assemblies; MCF-7, Michigan Cancer Foundation-7; MCF-7/ADR, MCF-7/adriamycin.
To further characterize the intracellular accumulation of DOX, the study determined the DOX concentration in MCF-7 and MCF-7/ADR cells (). After treatment with free DOX (15 minutes drug exposure), the amount of drug that accumulated in MCF-7 cells was 2.6-fold greater than that in MCF-7/ADR cells. Following a 1-hour exposure to free DOX, the drug concentration in MCF-7/cells was about tenfold greater than that in MCF-7/ADR cells, the DOX within which were reduced due to active drugs efflux. In contrast to free DOX, the accumulation of LNPs- DOX in MCF-7 and MCF-7/ADR cells were similar after 15 minutes drug exposure. After a 4-hour exposure, the LNPs-DOX concentration indicated no significant reduction in both cells. The phenomenon may demonstrate that the LNPs-DOX have successfully bypassed the enzymatic degradation and efflux pumps effect. More importantly, based on confocal laser scanning microscopy (), most of the DOX was located in the nuclei of the cancer cells at this time point. Since DOX is a drug that is toxic to the cell cycle by inhibiting the activity of topoisomerase II, the cytotoxicity of LNPs-DOX would be increased.
Figure 6 Accumulation of free DOX or LNPs-DOX in MCF-7 and MCF-7/ADR cells. Cells were treated with 8 μg/mL DOX or an equivalent concentration of LNPs-DOX for 15 minutes and 1 hour. The intracellular DOX was then determined by flow cytometry analysis.
Notes: *P < 0.05; **P < 0.01; ***P < 0.001.
Abbreviations: DOX, doxorubicin; free DOX, free doxorubicin; h, hours; LNPs- DOX, doxorubicin loaded in lipid/polymer particle assemblies; MCF-7, Michigan Cancer Foundation-7; MCF-7/ADR, MCF-7/adriamycin; NS, no significance.
Reversal of MDR in vitro by LNPs
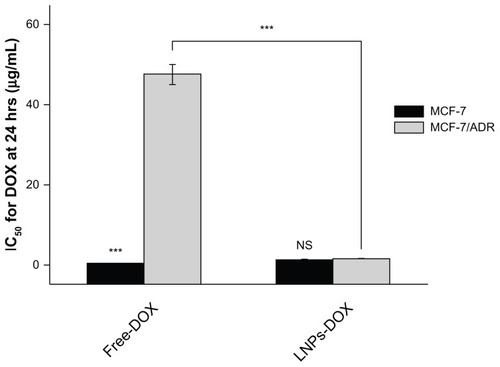
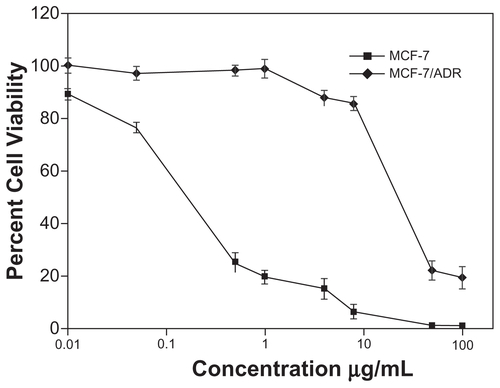
Following the analysis of the intracellular accumulation of LNPs-DOX, the ability of LNPs to overcome MDR was investigated. The study evaluated the cytotoxicity in vitro of DOX-loaded LNPs, blank LNPs, and free DOX to parental and resistant MCF-7 cells, using the MTT assay. The blank LNPs showed no cytotoxicity to either cell line; the viability values of MCF-7 and MCF-7/ADR cells were 98% and 97%, respectively, at the blank LNPs concentration of 50 mg/mL (). The LNPs-DOX and free DOX both exhibited significant cell growth inhibition of the parental cell line. The DOX-loaded LNPs exhibited a dose-dependent cytotoxicity against MCF-7/ADR cells (IC50:1.61 ± 0.13μg/ml). That is, DOX-loaded LNPs had a 30-fold lower IC50 value than free DOX in MCF-7/ADR, suggesting that LNPs can circumvent drug efflux-mediated MDR.
Figure 7 IC50 for free DOX or LNPs-DOX in MCF-7 and MCF-7/ADR cells. Cells were treated with various concentrations of DOX or LNPs-DOX at the same dose for 24 hours.
Note: ***P < 0.001.
Abbreviations: Free DOX, free doxorubicin; IC50, half maximal inhibitory concentration; h, hours; LNPs-DOX, doxorubicin loaded in lipid/polymer particle assemblies; MCF-7, Michigan Cancer Foundation-7; MCF-7/ADR, MCF-7/adriamycin; NS, no significance.
Discussion
Adsorption of lipids onto oppositely charged surfaces or particles has been studied in detail by analytical and simulation methods.Citation36 The study employed lipid/particle assemblies composed of positively charged PLGA nanoparticles (150 nm in diameter), covered by a bilayer of DPPC (200 nm for LNPs). In this context, DMAB consists of a bromide headgroup with two hydrophobic tails connecting them together; that is, one hydrophobic chain of DMAB is adsorbed onto the surface of PLGA nanoparticles; and the other acts as electrostatic glue that enhances the stability of the self-assembled particles by molecular electronegativity interaction with the oppositely charged phosphocholine headgroups.Citation25 Once these cell-like structures were formed, the adsorption of a lipid bilayer on the PLGA core could be seen by TEM. Based on SEM, the surface of the particles was covered with DPPC, which constituted a biocompatible membrane environment useful for in vivo biomedicine (eg, in the delivery of drugs, proteins, and peptides).
LipoParticles are self-assemblies with a particle core surrounded by a lipid shell. PLGA particles have certain cell cytotoxicity by generating transient holes on cell membranes with a positively charged ammonium bromide headgroup of the DMAB molecule;Citation37 however, the negative charges on the hydrophilic segment of DPPC could be attracted to the oppositely charged ions of the ammonium bromide ammonia headgroup.Citation38 This charge neutralization would then form the lipid/particle assemblies, and improve the biocompatibility of the particles, with a reduction of the zeta potential from 57 mV to −1.8 mV. The MTT assays also confirmed that the blank LNPs at 50 mg/mL were not cytotoxic, and cell membrane damage caused by DMAB was not found. Thus, the modification of the cationic polymer substance with a lipid shell should be a useful means to reduce the toxicity of positively charged biomaterials, and there is potential to expand the application of these materials in biomedicine.
The MDR of MCF-7/ADR was accompanied with overexpression of P-gp transporter. Bypassing these drug effluxes is a rational strategy to circumvent the MDR.Citation39 In the current study, the significant cytotoxicity of LNPs-DOX in the MCF-7/ADR has been validated. Overcoming the MDR was related to the enhanced intracellular accumulation and nuclear targeting of the drug mediated by LNPs. To elaborate the details of the MDR reversal, the intracellular accumulation and localization of free DOX and LNPs-DOX was investigated in the drug-resistant MCF-7/ADR cells. Since the intracellular trafficking pathways of drug-loaded carriers, such as polymer capsules, were previously reported,Citation10 the cellular uptake and fluorescence imaging studies were performed to determine how the newly designed lipid/particle assemblies were able to bypass the resistance to DOX. The results showed that LNPs-DOX could effectively increase the DOX concentration in MCF-7/ADR cells, and most of the LNPs-DOX located in the nuclei of the drug-resistant cells after about 1 hour of incubation. This observation demonstrated that the DMAB-modified particle core had the ability to escape from endosomes/lysosomes and localize in the nucleus. In summary, when the drug delivery system entered cells via endocytosis, after enzymatic degradation, the encapsulation of DMAB-modified particle core may be exposed to the endosomal/lysosomal lumen, and lead to the rupture of the particles.Citation17 Then, the positively charged core would deliver the encapsulated DOX to the nucleus, which could increase DOX localization in the nucleus, and thereby bypass the drug efflux mechanism mediated by transporter proteins. The lipid/particle assemblies developed here could produce intracellular targeting to the nucleus and overcome the resistance of the cancer cells to DOX.
Conclusion
In conclusion, it can be reported that the lipid/particle assemblies consisting in DMAB-modified PLGA NPs coated with DPPC lipid shell have been successfully developed for MDR reversal. The results demonstrate that these LNPs, with biomimetic properties of lipids, can improve the biocompatibility of cationic particles via charge neutralization. Importantly, the LNPs can reverse the transporter-mediated drug resistance by increasing the intracellular accumulation and improving the targeting to the nucleus. This drug delivery system has the potential for overcoming MDR in biomedicine.
Acknowledgments
Support from the National Natural Science Foundation of China (NSFC: 30973647) and National Basic Research Program of China (973 Program, 2009 CB930300) are acknowledged.
Disclosure
The authors report no conflicts of interest in this work.
Supplementary Data
Figure S1 Gel Permeation chromatogram of DOX-PLGA conjugate (solid line) and unconjugated PLGA (dotted line)
Abbreviations: DOX, doxorubicin; PLGA, poly(lactic-co-glycolic) acid.
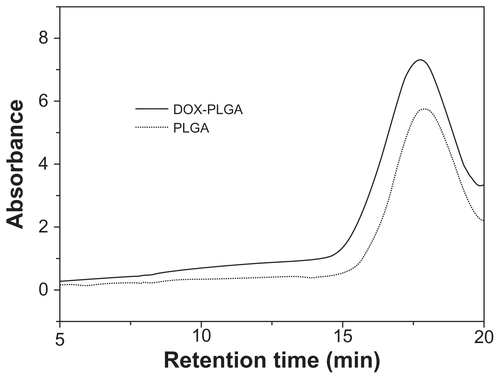
Figure S2 In vitro drug release profiles of DOX-loaded PLGA nanoparticles (without DPPC shell) in different pH (5.5, 6.5, and 7.4) at 37°C. Error bars indicate mean ± SD.
Abbreviations: DOX, doxorubicin; DPPC, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine; PLGA, poly(lactic-co-glycolic) acid; SD, standard deviation.
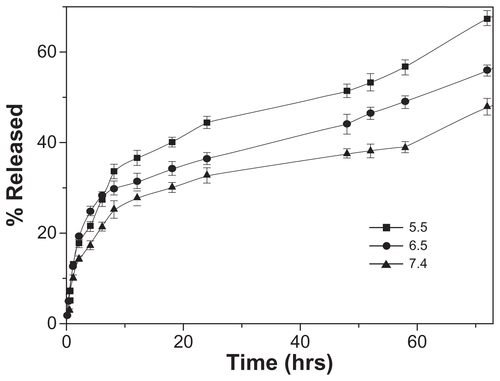
Figure S3 Cytotoxicity of Free-DOX against MCF-7 and MCF-7/ADR cells. Error bars indicate mean ± SD.
Abbreviations: DOX, doxorubicin; MCF-7, Michigan Cancer Foundation-7; MCF-7/ADR, MCF-7/adriamycin; SD, standard deviation.
References
- SzakácsGPatersonJKLudwigJATargeting multidrug resistance in cancerNat Rev Drug Discov20065321923416518375
- FojoATUedaKSlamonDJExpression of a multidrug-resistance gene in human tumors and tissuesProc Natl Acad Sci U S A19878412652692432605
- KohnoKSatoSTakanoHThe direct activation of human multidrug resistance gene (MDR1) by anticancer agentsBiochem Biophys Res Commun19891653141514212575381
- AmbudkarSVKimchi-SarfatyCSaunaZEP-glycoprotein: from genomics to mechanismOncogene200322477468748514576852
- FletcherJIHaberMHendersonMJABC transporters in cancer: more than just drug efflux pumpsNat Rev Cancer201010214715620075923
- RenFChenRWangYPaclitaxel-loaded poly(n-butylcyanoacrylate) nanoparticle delivery system to overcome multidrug resistance in ovarian cancerPharm Res201128489790621184150
- SharmaAKZhangLLiSPrevention of MDR development in leukemia cells by micelle-forming polymeric surfactantJ Control Release2008131322022718722489
- LeeESNaKBaeYHDoxorubicin loaded pH-sensitive polymeric micelles for reversal of resistant MCF-7 tumorJ Control Release2005103240541815763623
- PatelNRRathiAMongaytDReversal of multidrug resistance by co-delivery of tariquidar (XR9576) and paclitaxel using long-circulating liposomesInt J Pharm2011416129629921703341
- YanYOchsCJSuchGKBypassing multidrug resistance in cancer cells with biodegradable polymer capsulesAdv Mater201022475398540320976679
- KievitFMWangFYFangCDoxorubicin loaded iron oxide nanoparticles overcome multidrug resistance in cancer in vitroJ Control Release20111521768321277920
- VarkouhiAKScholteMStormGEndosomal escape pathways for delivery of biologicalsJ Control Release2011151322022821078351
- BoussifOLezoualchFZantaMAA versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimineProc Natl Acad Sci U S A19959216729773017638184
- LinCZhongZLokMCNovel bioreducible poly(amido amine)s for highly efficient gene deliveryBioconjug Chem200718113814517226966
- HariharanSBhardwajVBalaIDesign of estradiol loaded PLGA nanoparticulate formulations: a potential oral delivery system for hormone therapyPharm Res200623118419516267632
- SonajeKItaliaJLSharmaGDevelopment of biodegradable nanoparticles for oral delivery of ellagic acid and evaluation of their antioxidant efficacy against cyclosporine A-induced nephrotoxicity in ratsPharm Res200724589990817377747
- FayFQuinnDJGilmoreBFGene delivery using dimethyldido-decylammonium bromide-coated PLGA nanoparticlesBiomaterials201031144214422220185174
- CirmanTOresićKMazovecGDSelective disruption of lysosomes in HeLa cells triggers apoptosis mediated by cleavage of Bid by multiple papain-like lysosomal cathepsinsJ Bio Chem200427953578358714581476
- MeiLSunHSongCLocal delivery of modified paclitaxel-loaded poly(epsilon-caprolactone)/pluronic F68 nanoparticles for long-term inhibition of hyperplasiaJ Pharm Sci20099862040205018855915
- SahanaDKMittalGBhardwajVPLGA nanoparticles for oral delivery of hydrophobic drugs: influence of organic solvent on nanoparticle formation and release behavior in vitro and in vivo using estradiol as a model drugJ Pharm Sci20089741530154217722098
- TroutierALLadavièreCAn overview of lipid membrane supported by colloidal particlesAdv Colloid Interface Sci2007133112117397791
- SenguptaSEavaroneDCapilaITemporal targeting of tumour cells and neovasculature with a nanoscale delivery systemNature2005436705056857216049491
- Carmona-RibeiroAMInteractions between cationic liposomes and drugs or biomoleculesAn Acad Bras Cienc2000721394310932102
- CampanhãMTMamizukaEMCarmona-RibeiroAMInteractions between cationic liposomes and bacteria: the physical-chemistry of the bactericidal actionJ Lipid Res19994081495150010428986
- TroutierALVéronLDelairTNew insights into self-organization of a model lipid mixture and quantification of its adsorption on spherical polymer particlesLangmuir200521229901991016229507
- TroutierALDelairTPichotCPhysicochemical and interfacial investigation of lipid/polymer particle assembliesLangmuir20052141305131315697275
- ZuzziSCamettiCOnoriGPolyion-induced aggregation of lipidic-coated solid polystyrene spheres: the many facets of complex formation in low-density colloidal suspensionsLangmuir200824126044604918484758
- DebinAKravtzoffRSantiagoJVIntranasal immunization with recombinant antigens associated with new cationic particles induces strong mucosal as well as systemic antibody and CTL responsesVaccine20022021–222752276312034102
- BertonMSixouSKravtzoffRImproved oligonucleotide uptake and stability by a new drug carrier, the SupraMolecular Bio Vector (SMBV)Biochim Biophys Acta1997135517199030197
- ThevenotJTroutierALPutauxJLEffect of the polymer nature on the structural organization of lipid/polymer particle assembliesJ Phys Chem B200811244138121382218844402
- YooHSOhJELeeKHBiodegradable nanoparticles containing doxorubicin-PLGA conjugate for sustained releasePharma Res199916711141118
- JunYJParkMKJadhavVBTripodal amphiphiles tunable for self-assembly to polymersomesJ Control Release2010142113213719822179
- HePUrbanMWPhospholipid-stabilized Au-nanoparticlesBiomacromolecules2005631224122515877336
- Jabr-MilaneLSvan VlerkenLEYadavSMulti-functional nanocarriers to overcome tumor drug resistanceCancer Treat Rev200834759260218538481
- ShiehMJHsuCYHuangLYReversal of doxorubicin-resistance by multifunctional nanoparticles in MCF-7/ADR cellsJ Control Release2011152341842521435362
- SalvatiARistoriSOberdisseJSmall angle scattering and zeta potential of liposomes loaded with octa(carboranyl)porphyrazineJ Phys Chem B200711135103571036417696389
- VermaAUzunOHuYSurface-structure-regulated cell-membrane penetration by monolayer-protected nanoparticlesNat Mater20087758859518500347
- WangBZhangLBaeSCNanoparticle-induced surface reconstruction of phospholipid membranesProc Natl Acad Sci U S A200810547181711817519011086
- KopeckaJCampiaIOliveroPA LDL-masked liposomal-doxorubicin reverses drug resistance in human cancer cellsJ Control Release2011149219620520946921