Abstract
Background
Titanium dioxide (TiO2) has been widely used in many areas, including biomedicine, cosmetics, and environmental engineering. Recently, it has become evident that some TiO2 particles have a considerable cytotoxic effect in normal human cells. However, the molecular basis for the cytotoxicity of TiO2 has yet to be defined.
Methods and results
In this study, we demonstrated that combined treatment with TiO2 nanoparticles sized less than 100 nm and ultraviolet A irradiation induces apoptotic cell death through reactive oxygen species-dependent upregulation of Fas and conformational activation of Bax in normal human cells. Treatment with P25 TiO2 nanoparticles with a hydrodynamic size distribution centered around 70 nm (TiO2P25–70) together with ultraviolet A irradiation-induced caspase-dependent apoptotic cell death, accompanied by transcriptional upregulation of the death receptor, Fas, and conformational activation of Bax. In line with these results, knockdown of either Fas or Bax with specific siRNA significantly inhibited TiO2-induced apoptotic cell death. Moreover, inhibition of reactive oxygen species with an antioxidant, N-acetyl-L-cysteine, clearly suppressed upregulation of Fas, conformational activation of Bax, and subsequent apoptotic cell death in response to combination treatment using TiO2P25–70 and ultraviolet A irradiation.
Conclusion
These results indicate that sub-100 nm sized TiO2 treatment under ultraviolet A irradiation induces apoptotic cell death through reactive oxygen species-mediated upregulation of the death receptor, Fas, and activation of the preapoptotic protein, Bax. Elucidating the molecular mechanisms by which nanosized particles induce activation of cell death signaling pathways would be critical for the development of prevention strategies to minimize the cytotoxicity of nanomaterials.
Introduction
In recent years, advances in nanotechnology have been attributed to an increase in the production of nanomaterials. Nanomaterials have physical and chemical properties very different from those of bulk materials, even though they have the same chemical composition. The most important property of nanomaterials is their size, ranging from individual atoms and molecules to microscaled materials.Citation1,Citation2 The size and surface area of materials are crucial characteristics from a toxicological perspective.Citation3–Citation5 Because nanomaterials have an increased surface area compared with bulk materials, nanomaterials are expected to be more reactive and have more interaction with living organisms.
One of the widely used nanomaterials is titanium dioxide (TiO2) that is utilized as a white pigment for making paints, as a food colorant, and in plastics in its bulk form. As the demand for nanomaterials increases, nanosized TiO2 is used increasingly in cosmetics, especially for sunscreens, and as a photocatalyst in many industrial fields. Recently, it has become evident that some nanomaterials, including TiO2, have considerable biological activity and cytotoxic effects in normal cells. Previous studies have shown that nanosized TiO2 induces oxidative stress-mediated toxicity in many cell types. However, the molecular mechanism for the cytotoxic effects of nanosized TiO2 has yet to be defined. Several cellular stressors, including anticancer drugs and chemo preventive agents, disturb the natural oxidation and reduction equilibrium in cells via various mechanisms, which involve complex redox (reduction-oxidation) reactions with endogenous oxidants and cellular antioxidant systems. These stressors lead to an increase in reactive oxygen species by a variety of redox enzymes, including flavoprotein-dependent super oxide-producing enzymes such as NADPH oxidase,Citation6,Citation7 or by mitochondrial electron transport chain dysfunction.Citation8 In recent years, it has become apparent that reactive oxygen species play an important role during induction of apoptotic cell death.Citation9 Several studies have provided evidence that intracellular production of reactive oxygen species can lead directly to activation of mitochondrial permeability transition, loss of mitochondrial membrane potential, and cytochrome c release from mitochondria into the cytoplasm, followed by activation of the caspase cascade, and, ultimately, apoptotic cell death.Citation10
Further, according to the recent literature, various physicochemical properties of nanoparticles contribute to the cytotoxic mechanisms of nanoparticles. Because commonly used chemicals in aqueous media are unchanged during the exposure time and their intrinsic properties are well understood, treatment conditions, such as concentration and exposure time, provide sufficient information to produce reproducible and reliable dose-response relationships for the corresponding compound.Citation11–Citation13 In contrast, in the case of nanoparticles, various physicochemical characteristics (eg, hydrodynamic size distribution, surface charge, and dispersibility in cell culture media) can be drastically changed during the exposure time, making it difficult to generate a reproducible and reliable toxicity response and mechanism for nanoparticles. For instance, due to the differences in their dispersibility in cell culture media, nanoparticles can be highly agglomerated and sedimented, thereby causing a higher dose to adherent cells via gravitational settling as well as diffusion processes. However, when nanoparticles are well dispersed in cell culture media, they are transported to adherent cells only via a diffusion process and thereby provide a less effective dose to cells.Citation14 Additionally, due to the significant differences in their hydrodynamic sizes, the highly agglomerated nanoparticles are sedimented more on top of the adherent cells and may have completely different cellular uptake and cytotoxicity profiles, compared with those of well suspended nanoparticles.Citation15–Citation17 Thus, to have a more clear understanding of the cytotoxic mechanisms of nanoparticles, it is necessary to investigate the toxicity mechanisms in well characterized nanoparticles as well, together with reasonable control of those key physicochemical properties.Citation11–Citation14
Although TiO2 is widely used to make cosmetics and in sunscreens, TiO2 is known to be excited by ultraviolet light and to generate reactive oxygen species in aqueous solution.Citation18 In this study, we found that TiO2P25–70 nanoparticles with a hydrodynamic size distribution centered around 70 nm are stable without agglomeration and sedimentation for more than a week, compared with bigger sizes of nanoparticles, ie, the TiO2P25–300 and TiO2P25–130 fractions. Therefore, we examined the molecular basis for the biological activity and cytotoxicity of nanosized P25 TiO2 nanoparticles with a hydrodynamic size distribution centered around 70 nm (TiO2P25–70) under ultraviolet A irradiation. We found that treatment of normal human cells with TiO2P25–70 under ultraviolet A irradiation induced apoptotic cell death via reactive oxygen species-dependent upregulation of the death receptor, Fas, and activation of the preapoptotic protein, Bax, in human normal cells. Elucidating the molecular mechanisms by which nanosized particles induce activation of cell death signaling pathways is critical for the development of prevention strategies to protect against cytotoxicity when using nanomaterials.
Materials and methods
Preparation of TiO2 nanoparticle dispersions
P25 TiO2 powder (Aeroxide TiO2, Evonik Degussa GmbH, Germany) was dispersed to 10 g/L in deionized water and was then sonicated for 10 minutes using a probe sonicator (420 W, 20 kHz, Sonosmasher, Ulsso Hitech, South Korea).
The pH of the dispersed TiO2 stock suspension was approximately 3.6 at an initial concentration of 10 g/L. The TiO2 suspension was distributed to various sizes by differential speed of centrifugation; TiO2 stock suspension (20 mL) was centrifuged at 0 or 4000 (6570 × g) or 6000 (9870 × g) rpm for 20 minutes using a high-speed centrifuge (Mega 17R, Hanil Science Industrial, Gyeyang-gu Incheon, Korea), and the supernatant (15 mL) was carefully taken and designated as TiO2P25–300, TiO2P25–130, and TiO2P25–70, respectively.Citation19
Characterization of TiO2 nanoparticles
The morphology and size distribution of the TiO2 nanoarticles were analyzed using images of the nanoparticles and their aggregates taken by scanning electron microscopy (SU-70, Hitachi, Tokyo, Japan). The titanium concentration in the suspension was determined by inductively coupled plasma mass spectrometry (Elan 6100, Perkin-Elmer Sciex, Waltham, MA) after acid digestion procedure with mixtures of HNO3, HCl, and HF. The hydrodynamic size of TiO2 nanoparticles in aqueous media was measured using a dynamic light scattering instrument (Scattero scope I, Qudix Inc, Seoul, South Korea), while the zeta potential was measured by a zetasizer (Nano-ZS, Malvern Instruments, Worcestershire, UK). Relative amounts of the anatase and rutile phases were estimated by their powder x-ray diffraction patterns, as measured using a Rigaku x-ray diffraction instrument (D/max-2000, Rigaku Corporation, Tokyo, Japan). The surface area of the TiO2 powder was determined by multipoint Brunauer-Emmett-Teller analysis (nano porosity, Mirae Ultrasonic Tech Co, Bucheon Si, South Korea).
Cell culture, antibodies, and transfection of siRNA
Chang (normal human liver), MCF10A (normal breast epithelial), and WI38 (normal lung fibroblast) cell lines were obtained from the American Type Culture Collection (Manassas, VA). The cells were grown in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% fetal bovine serum, 100 units/mL penicillin, and 100 μg/mL streptomycin at 37°C in 5% CO2.
Polyclonal antibodies specif ic for Bcl-2, Bcl-xL, apoptosis- inducing factor, cytochrome c, caspase-8, caspase-9, caspase-3, α-tubulin, HSP60, DR4, and DR5, as well as monoclonal antibodies specific for Bax, Fas, and tumor necrosis factor receptor were purchased from Santa Cruz Biotechnology Inc (Santa Cruz, CA). β-actin was from Sigma (St Louis, MO). Monoclonal anti-Bak, anti-Bax, and anti-PARP antibodies were from BD Pharmingen (Franklin Lakes, NJ). All small interfering RNA duplexes were purchased from Samchully Pharmaceutical Co, Ltd (Seoul, Korea). Specific siRNA targeting of Bax (5′-CCGAGGCAGCUGACAUGTT-3′), Fas (5′-AAGUGCAAGUGCAAACCAGAC-3′), or green fluorescent protein (5′-CCACTACCTGAGCACCCAG-3′) was introduced to Chang cells using the transfection reagent, Lipofectamine® (Invitrogen, Carlsbad, CA), following the manufacturer’s recommendations. siRNA targeted to green fluorescent protein was used as the negative control.
Ultraviolet A irradiation of TiO2-treated cells
For treatment of cells, TiO2 nanoparticles were added to the cell culture medium at a concentration of 150 ppm. Cells were then irradiated with ultraviolet A (0.75 J/hour) using a Sankyo Denki lamp (FL20SBL, Nagano, Japan). After culture for 24 hours, the cells were harvested for further analysis.
Quantification of cell death
Cell death was analyzed using both propidium iodide staining and an Annexin V labeling kit according to the manufacturer’s recommendations (Sigma). For assessment of cell death, the cells were plated onto 60 mm dishes at a cell density of 2 × 105 cells per dish and treated with TiO2 the following day. At indicated time points, the cells were harvested and washed in phosphate buffer solution. Annexin V-positive early apoptotic cells or propidium iodide-positive cells were then quantified using a FACScan flow cytometer fitted with Cell Quest software (BD Biosciences, Franklin Lakes, NJ).
Measurement of reactive oxygen species generation
Briefly, the cells were incubated in 40 nM 3,3-dihexyloxa-carboxyanine iodide [DiOC6(3)] or 10 nM 2,7-dichlorodi-hydrofluorescein diacetate (DCFH-DA, Molecular Probes, Eugene, OR) at 37°C for 15 minutes and washed with cold phosphate buffer solution three times. The retained DiOC6(3) and DCFH-DA was analyzed using a flow cytometer fitted with Cell Quest Pro software (Becton Dickinson, Franklin Lakes, NJ). For MitoSox Red staining, the cells were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100 in phosphate buffer solution. Following fixation, the cells were incubated with MitoSox Red in phosphate buffer solution. The stained cells were then visualized using a fluorescence microscope (Olympus IX71) and quantified by flow cytometry.
Flow cytometric analysis of Bax and Bak activation
After fixation in 0.25% paraformaldehyde for 5 minutes and washing with phosphate buffer solution, the cells were incubated for 30 minutes in the presence of digitonin 100 μg/mL with antibodies recognizing the N-terminal epitopes of Bax (clone 6A7, BD Pharmingen) or Bak (AM03TC100, Oncogene Research Products). After incubation with a fluorescein isothiocyanate-conjugated anti mouse antibody for 30 minutes, the cells (10,000 per sample) were analyzed by flow cytometry.
Western blot analysis
Cell lysates were prepared by extracting proteins with lysis buffer (40 mM Tris-HCl [pH 8.0], 120 mM NaCl, 0.1% Nonidet-P40) supplemented with protease inhibitors. Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane (Amersham Biosciences, Arlington Heights, IL). The membrane was blocked with 5% nonfat dry milk in Tris-buffered saline and incubated with primary antibodies overnight at 4°C. Blots were developed using a peroxidase-conjugated secondary antibody, and the proteins were visualized by enhanced chemiluminescence (Amersham Biosciences), using the manufacturer’s protocol.
Preparation of cytosolic and mitochondrial fractions
Cells were washed with ice-cold phosphate buffer solution, left on ice for 10 minutes, and then resuspended in isotonic homogenization buffer (250 mmol/L sucrose, 10 mmol/L KCl, 1.5 mmol/L MgCl2, 1 mmol/L Na-EDTA, 1 mmol/L Na-EGTA, 1 mmol/L dithiothreitol, 0.1 mmol/L phenylmethylsulfonylfluoride, and 10 mmol/L Tris-HCl [pH 7.4]) containing a proteinase inhibitor mixture (Roche, Basel, Switzerland). Cells were then disrupted by a Dounce homogenizer at 80 strokes. The unbroken cells were excluded by spinning down at 30 g for 5 minutes. The mitochondrial fractions were then obtained by serial centrifugation of the supernatant at 750 g for 10 minutes and 14,000 g for 20 minutes, taking the supernatant at each step. For cytosolic fractionation, the cells were disrupted by a loose homogenizer at 10 strokes, and serially centrifuged at 750 g for 10 minutes and 14,000 g for 20 minutes, taking the supernatant at each step.
Cross-linking of Bax and Bak proteins
Cells were permeabilized at room temperature with 0.015%–0.02% digitonin for 1–2 minutes in an isotonic buffer (10 mM HEPES, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, pH 7.4) containing protease inhibitors. The permeabilized cells were then scraped at 4°C and incubated with cross-linker (dissuccinimidyl suberate, with linker lengths of 11.4 Å) on a head-to-head rocker for 30 minutes at room temperature. The unreacted cross-linkers were quenched with a 1/10 volume of 2 M Tris-HCl (pH 7.4) for another 30 minutes at room temperature, with rocking. The permeabilized and cross-linker-treated cells were then centrifuged at 15,000 g for 10 minutes at 4°C. The supernatant was taken for cytosol proteins and the pellet was further lysed with ice-cold lysis buffer, ie, 2% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate containing protease inhibitors, for 60 minutes at 4°C to determine the membrane fraction. For this, a nondenaturing loading buffer was added before running sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
Statistical analysis
Data were analyzed using the unpaired two-tailed Student’s t-test. Data were expressed as the mean ± standard error of the mean derived from at least three independent experiments. Differences were considered statistically significant at P < 0.005.
Results and discussion
Physicochemical characteristics of TiO2 nanoparticles and dispersions
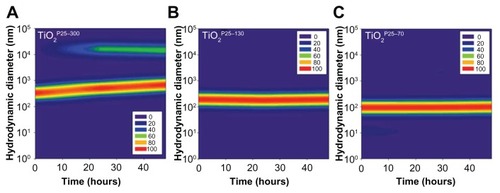
Based on our scanning electron microscopy and x-ray diffraction measurements, the P25 TiO2 powder had a primary particle size of approximately 21 nm and was composed mainly of anatase (87%) and rutile (13%) crystalline phase. The Brunauer-Emmett-Teller specific surface area was 57 m2/g and the point-of-zero charge was estimated to be in the vicinity of pH 5.8. These basic characteristics agree well with those provided by the manufacturer and by previous studies.Citation20–Citation22 In this study, we divided the P25 TiO2 suspension into three fractions with different hydrodynamic size distributions. To this end, the primary P25 TiO2 suspension was centrifuged using different speeds (0, 4000, 6000 rpm). TiO2 nanoparticles with different hydrodynamic size distributions were then obtained by taking the supernatant and designating it according to hydrodynamic sizes of 327 nm (without fractionation, TiO2P25–300), 133 nm (fractionated at 4000 rpm, TiO2P25–130), and 74 nm (fractionated at 6000 rpm, TiO2P25–70). The TiO2P25–300 fraction was mainly composed of heavily agglomerated/aggregated particles, while TiO2P25–130 and TiO2P25–70 were composed of much smaller agglomerates/aggregates (). The sub-100 nm fraction (ie, TiO2P25–70) was found to be stable without agglomeration and sedimentation for more than a week, whereas the TiO2P25–300 and TiO2P25–130 fractions were easily aggregated and sedimented in slight acidic conditions (pH 4–5), However, exposure of TiO2P25–70 to typical in vitro cell culture medium (eg, RPMI 1640 or Dulbecco’s Modified Eagle’s Medium) instantly caused strong agglomeration and sedimentation, which has been previously reported to cause problems in performing accurate and reproducible toxicity assessments of nanoparticles.5,23–27 To overcome this problem of colloidal instability, nanoparticles are often stabilized with various surface-modifying ligands.Citation19 In this study, rather than adding additional surface-modifying compounds, fetal bovine serum (a supplement used for in vitro cell culture) was used as an efficient stabilizing agent.Citation17 By monitoring the hydrodynamic size distributions of all three fractions of TiO2 in RPMI media supplemented with fetal bovine serum for 48 hours, we observed that the unfractionated TiO2P25–300 increased in hydrodynamic size with aggregation over time and formed micron-sized particles with heavy sedimentation (), whereas the hydrodynamic size of TiO2P25–130 and TiO2P25–70 were not changed with time, indicating no aggregation of TiO2 (). Zeta potential measurements were also performed for all three types of TiO2 in RPMI media with fetal bovine serum, yielding values in the range of −9.42 to −10.4 mV. In addition, the impact of exposure to ultraviolet A irradiation on the physicochemical properties of TiO2 was also investigated. However, no significant changes in hydrodynamic size, zeta potential, or colloidal stability were observed after exposure to 0.75 J/hour ultraviolet A irradiation for 48 hours.
Figure 1 Representative scanning electron microscopic images of fractionated TiO2 in distilled water. TiO2 suspension in deionized water was fractionated to three different hydrodynamic sizes by differential speed of centrifugation. The supernatant was carefully taken and designated as TiO2P25–300 (A), TiO2P25–130 (B), or TiO2P25–70 (C) after centrifugation for 20 minutes at 0 rpm, 4000 rpm (6570 g), or 8000 rpm (9870 g), respectively.
Note: Scale bars indicate 500 nm (upper) and 100 nm (lower).
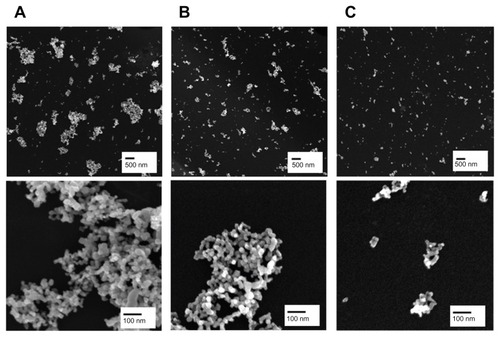
Figure 2 Temporal variations of hydrodynamic size distributions of TiO2 in media supplemented with fetal bovine serum. In the cell culture media, ie, RPMI with 10% fetal bovine serum, hydrodynamic sizes of TiO2 nanoparticles were monitored for 48 hours. The hydrodynamic size was increased in the unfractionated TiO2P25–300 (A) with time and caused micron-sized particles with heavy sedimentation, whereas the hydrodynamic size of TiO2P25–130 (B) and TiO2P25–70 (C) were not changed with time, indicating no aggregation.
TiO2 induces hydrodynamic size-dependent apoptosis in normal cells
To investigate whether TiO2 influences cell death in normal cells, we treated a Chang (normal liver) cell line with TiO2 nanoparticles of different hydrodynamic size distributions (TiO2P25–70, TiO2P25–130, and TiO2P25–300) at various concentrations from 0 to 400 ppm for 24 hours and measured cell death by flow cytometry with propidium iodide staining. As shown in Figure S1, treatment of Chang cells with TiO2 nanoparticles at a concentration lower than 200 ppm did not cause significant cell death, while treatment with a higher concentration (200 and 400 ppm) caused dramatic cell death at any TiO2 nanoparticle size (Figure S1).
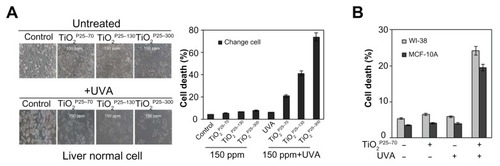
In previous studies, TiO2 has been excited by ultraviolet irradiation and showed cellular toxicity.Citation18 Because treatment with TiO2 nanoparticles at a concentration of 150 ppm did not induce cell death, we went on to investigate the synergistic effect of TiO2 nanoparticles and ultraviolet irradiation on cell death in a Chang line. To examine this possibility, we treated the Chang cells with TiO2 nanoparticles (TiO2P25–70, TiO2P25–130, and TiO2P25–300) in combination with ultraviolet A irradiation (0.75 J/hour), cultured the cells for 24 hours, and then measured cell death by flow cytometry with propidium iodide staining. As shown in , treatment with TiO2 at a concentration of 150 ppm, at which there was no significant cell death (Figure S1), in combination with ultraviolet A, induced cell death in Chang cells in a size-dependent manner; almost 20% of Chang cells underwent cell death within 24 hours after combined treatment with TiO2P25–70 and ultraviolet A irradiation; 50% and 80% underwent cell death after treatment with TiO2P25–130 and TiO2P25–300 in combination with ultraviolet A irradiation, respectively (). However, treatment with TiO2 at 150 ppm or ultraviolet A irradiation alone did not cause significant cell death. In addition, combined treatment with TiO2 and ultraviolet A irradiation induced cell death in normal breast epithelial cells (MCF-10A) and lung fibroblasts (WI38, ). Thereafter, we used TiO2 nanoparticles at a concentration of 150 ppm in all experiments. Also, although all three fractions of TiO2 showed cellular toxicity, we undertook further investigations of the mechanism of nanotoxicity only with the smallest sized nanoparticles, ie, the TiO2P25–70 fraction, because this fraction may minimize the influence of increased cellular exposure via gravitational settling of the nanoparticles as well as the toxicity mechanisms caused by the heavily agglomerated micron-sized particles.
Figure 3 TiO2 sensitizes normal cells to ultraviolet A irradiation leading to apoptotic cell death (A and B) Measurement of cell death by FACS analysis after propidium iodide staining. (A) Treatment of Chang cells with TiO2 in combination with UVA irradiation caused cell death in proportion to the size of TiO2 (TiO2P25–70, TiO2P25–130, TiO2P25–300), while treatment with TiO2 alone did not induce cell death. (B) Treatment of WI-38 and MCF10A cells with TiO2P25–70 in combination with ultraviolet A irradiation caused cell death, while treatment with either TiO2 or ultraviolet A alone did not induce cell death.
Abbreviations: UVA, ultraviolet A; WI-38, normal lung fibroblast cell line; MCF10A, normal breast epithelial cell line.
TiO2P25–70 induces apoptosis via upregulation of Fas and activation of caspases
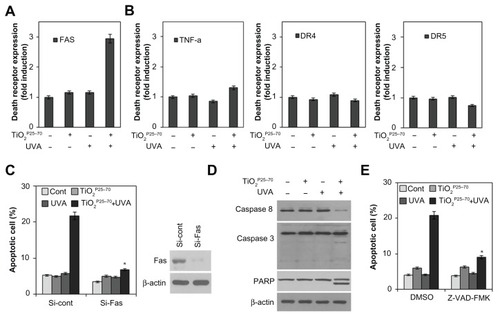
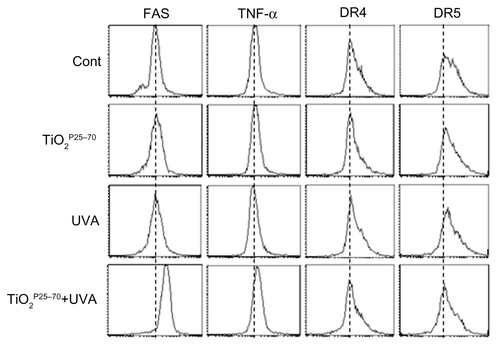
To determine whether death receptors are involved in the cell death induced by TiO2P25–70, we examined changes in death receptor expression levels, including DR4, DR5, Fas, and tumor necrosis factor receptors, in response to TiO2P25–70 treatment. As shown in and Figure S2, flow cytometric analysis clearly revealed that the protein levels of Fas were selectively increased by TiO2P25–70, but we failed to detect any changes in the expression of tumor necrosis factor receptor or the death receptor ( and Figure S2). Moreover, siRNA targeting of Fas clearly attenuated the apoptotic cell death induced by TiO2P25–70 (). In addition, cyclohexamide, a protein synthesis inhibitor, completely inhibited the Fas expression induced by TiO2P25–70 (data not shown), indicating that the Fas protein level was increased via de novo synthesis after treatment with TiO2P25–70. Because caspase-8 is a key player in Fas-mediated apoptotic cell death, we next investigated whether treatment with TiO2P25–70 activates caspase-8. As shown in , TiO2P25–70 treatment together with ultraviolet A irradiation significantly induced activation of caspase-8 and subsequently induced activation of caspase-3 and cleavage of PARP-1. In addition, a broad-spectrum caspase inhibitor, z-VAD-fmk, completely attenuated the apoptotic cell death induced by TiO2P25–70 (). Taken together, these results suggest that TiO2P25–70 induces apoptotic cell death in normal cells via Fas upregulation and subsequent caspase activation.
Figure 4 TiO2P25–70 induces apoptotic cell death through upregulation of Fas and subsequent activation of caspases. (A) Flow cytometry analysis reveals upregulation of Fas expression in Chang cells treated with TiO2P25–70 in combination with ultraviolet A irradiation, while treatment with either TiO2P25–70 or ultraviolet A alone did not upregulate the level of Fas expression. (B) Expression levels of other death receptors, f, TNFR, DR4, and DR5, were not altered in Chang cells by combined treatment with TiO2P25–70 and ultraviolet A. (C) Measurement of cell death by FACS analysis after double staining with propidium iodide and Annexin V. Pretreatment with siRNA targeted to Fas suppressed TiO2P25–70-induced cell death. (D) Treatment of Chang cells with TiO2P25–70 in combination with ultraviolet A decreased the proform of caspase-8 and increased the cleaved form of caspase-3 and PARP. (E) Measurement of cell death by FACS analysis after double staining with propidium iodide and Annexin V. Pretreatment with a broad-spectrum caspase inhibitor, z-VAD-fmk, attenuated cell death induced by combined treatment with TiO2P25–70 and ultraviolet A.
Notes: *P < 0.005. Error bars represent the mean ± standard deviation of triplicate samples.
Abbreviations: DMSO, dimethylsulfoxide; TNF, tumor necrosis factor; DR, death receptor; UVA, ultraviolet; PARP, poly (ADP-ribose) polymerase.
Conformational activation of Bax is required for mitochondrial dysfunction and apoptotic cell death induced by TiO2P25–70
To determine whether the mitochondrial pathway is involved in the cell death caused by TiO2P25–70, we examined changes in mitochondrial membrane potential and release of proapoptotic molecules from the mitochondria upon treatment with TiO2P25–70. As shown in , treatment with TiO2P25–70 under ultraviolet A irradiation induced signif icant loss of mitochondrial membrane potential. Cytosolic cytochrome c and apoptosis-inducing factor were markedly increased in response to treatment with TiO2P25–70 (), coinciding with changes in the mitochondrial membrane potential. Moreover, caspase-9 was activated by treatment with TiO2P25–70 (). These results indicate that TiO2P25–70 induces dissipation of the mitochondrial membrane potential and release of proapoptotic molecules.
Figure 5 Conformational activation of Bax is required for TiO2P25–70-induced mitochondrial dysfunction and apoptotic cell death. Combined treatment with TiO2P25–70 and ultraviolet A induced mitochondrial membrane potential loss (A) and release of cytochrome c and apoptosis-inducing factor to cytosol (B) and the decrease of proform caspase-9 (C) in Chang cells. Mitochondrial membrane potential of cells was determined by retention of DiOC6(3). The amount of retained DiOC6(3) was measured by flow cytometry. (D) Measurement of Bax and Bak expression levels by FACS and western blot analysis. Bax expression was induced in mitochondria-enriched membrane fractions of Chang cells when treated with combination of TiO2P25–70 and ultraviolet A irradiation, whereas Bak was not induced. (E) Pretreatment with siRNA targeting of Bax attenuated mitochondrial membrane potential loss (upper) and cell death (lower) induced by combination treatment of TiO2P25–70 and ultraviolet A irradiation. Cell death was measured by FACS analysis after double staining with propidium iodide and Annexin V.
Notes: *P < 0.005. Error bars represent the mean ± standard deviation of triplicate samples.
Abbreviations: UVA, ultraviolet A; AIF, apoptosis-inducing factor; MMP, matrix metalloproteinase.
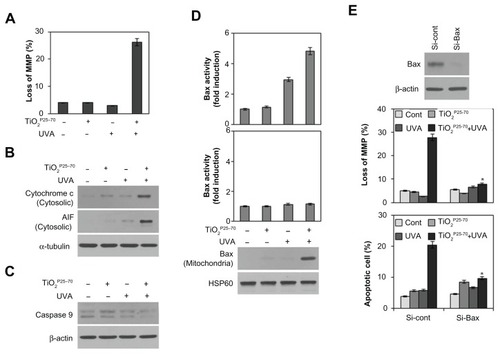
Because it has been demonstrated that proapoptotic Bcl-2 family members, especially Bax and Bak, are crucial to mitochondrial cell death pathways,Citation28,Citation29 we next analyzed activation of Bax and/or Bak after treatment with TiO2P25–70. By flow cytometry analysis with antibodies recognizing N-terminal epitopes of Bax or Bak, we found that treatment comprising TiO2P25–70 and ultraviolet A irradiation induces conformational change and mitochondrial relocalization of Bax from the cytosol (), but not Bak (). Moreover, knockdown of Bax with specific siRNA attenuated the apoptotic cell death induced by TiO2P25–70 (). These results indicate that conformational activation of Bax plays a crucial role in mitochondrial potential loss and subsequent cell death induced by TiO2P25–70.
Reactive oxygen species are involved in Fas upregulation, Bax activation, and apoptosis-induced by TiO2P25–70
Oxidative damage is often implicated in apoptotic cell death.Citation30,Citation31 We therefore subsequently examined changes in intracellular reactive oxygen species levels in Chang cells treated with TiO2P25–70. As shown in , treatment with TiO2P25–70 under ultraviolet A irradiation led to an approximately three-fold increase in mean 2′,7′-dichlorofluorescein fluorescence, indicating an increase of intracellular reactive oxygen species. To confirm further a link between elevation of intracellular reactive oxygen species and mitochondrial cell death, the cells were preincubated with N-acetyl-L-cysteine, an antioxidant, prior to TiO2P25–70 treatment. Treatment of TiO2P25–70 did not induce cell death in the presence of N-acetyl-L-cysteine (). Pretreatment of N-acetyl-L-cysteine also inhibited TiO2P25–70-induced Fas upregulation and caspase-8 activation (). Moreover, N-acetyl-L-cysteine clearly inhibited conformational change and mitochondrial redistribution of Bax induced by TiO2P25–70 (), mitochondrial membrane potential loss (), and apoptosis-inducing factor and cytochrome c release (). These observations suggest that an increase in intracellular reactive oxygen species is critically required for TiO2P25–70-induced Fas upregulation-mediated and Bax activation-mediated apoptotic cell death.
Figure 6 Reactive oxygen species are critically involved in the TiO2P25–70-induced Fas upregulation, Bax activation, and apoptotic cell death. (A) Measurement of intracellular reactive oxygen species levels by FACS analysis. Combined treatment of Chang cells with TiO2P25–70 and ultraviolet A irradiation increased intracellular reactive oxygen species levels, whereas single treatment with either TiO2P25–70 or ultraviolet A had no effect on intracellular reactive oxygen species levels. (B) Pretreatment with the antioxidant, N-acetyl-L-cysteine, attenuated cell death induced by combined treatment with TiO2P25–70 and ultraviolet A irradiation. Cell death was measured by FACS analysis after double staining with propidium iodide and Annexin V. (C) Pretreatment with antioxidant, N-acetyl-L-cysteine attenuated FAS expression (upper) and suppressed the increase of proform, caspase-8 (lower). (D) Pretreatment with N-acetyl-L-cysteine attenuated oligomerization of Bax in mitochondria-enriched membrane fractions of Chang cells treated with combination of TiO2P25–70 and ultraviolet A. (E) Pretreatment with N-acetyl-L-cysteine suppressed mitochondrial membrane potential loss (upper), the release of cytochrome c and apoptosis-inducing factor to cytosol (lower). (F) Visualization of intracellular reactive oxygen species levels in fluorescence microscopy by staining with DCFA and mitochondria specific MitoSox Red. Chang cells treated with combination of TiO2P25–70 and ultraviolet A irradiation show increased intracellular reactive oxygen species levels on both staining with DCFA and MitoSox Red.
Notes: *P < 0.005. Error bars represent the mean ± standard deviation of triplicate samples.
Abbreviations: AIF, UVA, ultraviolet A; DMSO, dimethylsulfoxide; AIF, apoptosis-inducing factor; HSP, heat shock protein; NAC, N-acetyl-L-cysteine.
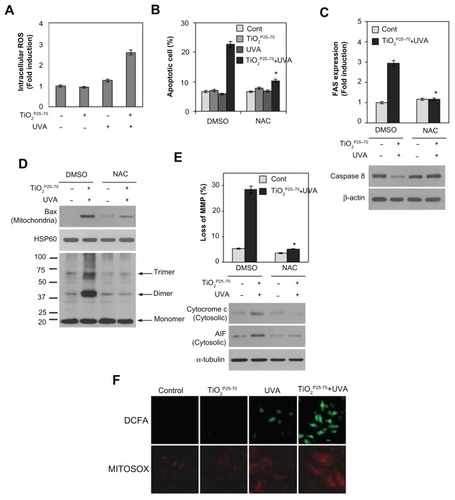
To determine further whether the mitochondria contribute to increased intracellular reactive oxygen species generation induced by TiO2P25–70, we utilized MitoSox Red, a mitochondrial reactive oxygen species detection reagent, in conjunction with fluorescence microscopy. As shown in , TiO2P25–70 together with ultraviolet A irradiation induced an increase in mitochondrial fluorescence, which is very similar to the observed increase in CM-H2DCFDA fluorescence. The stippled linear pattern of fluorescence in Chang cells loaded with MitoSox Red is consistent with mitochondrial localization of reactive oxygen species. Taken together, these results suggest that the mitochondria may contribute to the overall elevation in reactive oxygen species production in Chang cells treated with TiO2P25–70.
Recently, some nanosized particles have been shown to have considerable biological activity and a cytotoxic effect in normal cells. However, the molecular basis for the cytotoxicity of these nanoparticles has yet to be defined. In this study, we investigated the molecular basis for nanosized TiO2-induced cell death in normal human cells.
Interestingly, as shown in , treatment of TiO2 nanoparticles with ultraviolet A irradiation induced death of Chang cells in a hydrodynamic size-dependent manner. This observation provides us with some information on factors affecting the cytotoxicity of nanoparticles. According to Teeguarden et al, agglomeration and sedimentation of nanoparticles in cell culture media can cause a significant decrease in the nanoparticle dose delivered.Citation14 Therefore, even when the same nanoparticle dose is administered, the dose of nanoparticles delivered (ie, the effective cellular dose delivered to adherent cells on the bottom) can vary significantly. Highly agglomerated and sedimented nanoparticles (eg, TiO2P25–300) will result in a higher dose delivered to adherent cells via gravitational settling and diffusion processes, while well dispersed nanoparticles (eg, TiO2P25–70) may result in a smaller dose being delivered to adherent cells because they are transported to adherent cells mostly via diffusion. Further, due to significant differences in their hydrodynamic size, the highly agglomerated nano-particles sedimented on top of adherent cells may have cellular uptake and toxicity mechanisms which are completely different to those of well suspended nanoparticles. Citation16,Citation17 Therefore, the 20% cell death observed for TiO2P25–70 can be attributed solely to the cytotoxicity of TiO2 caused by diffusion-controlled delivery of sub-100 nm sized nanoparticles, while the other two cases (ie, TiO2P25–130 and TiO2P25–300 with cell death rates of 50% and 80%, respectively) may have more complex delivery pathways as well as physicochemical processes, which may involve multiple cytotoxicity mechanisms.
We have shown that treatment with TiO2P25–70 under ultraviolet A irradiation induces apoptotic cell death through reactive oxygen species-mediated upregulation of the death receptor, Fas, and activation of the preapoptotic protein, Bax. Fas is a death receptor on the cell surface of a wide variety of cell types, and mediates rapid apoptosis. Although Fas is constitutively expressed in a variety of cell types, the role of Fas has also been evoked recently in apoptosis of various cell types in response to certain stimuli;Citation32–Citation34 ultraviolet irradiation, viral infection, and chemotherapeutic agents effectively increase Fas transcription and, in turn, upregulation of Fas is involved in apoptotic cell death.Citation35 We also provide further evidence that nanosized TiO2P25–70 together with ultraviolet A irradiation induces upregulation of Fas and subsequent caspase-8 activation. Moreover, siRNA targeting of Fas attenuated TiO2P25–70-induced apoptotic cell death, suggesting that upregulation of Fas is involved in apoptotic cell death after treatment with TiO2P25–70. In response to stimuli such as etoposide, staurosporine, transforming growth factor-β, and ultraviolet irradiation, which require a mitochondria-dependent pathway for apoptosis, Bax is activated, translocated to the outer membrane of mitochondria, oligomerized therein,Citation36,Citation37 and, mitochondrial membranes are permeabilized, thereby releasing mitochondrial apoptogenic molecules into the cytosol. Similarly, we also found that TiO2P25–70 treatment under ultraviolet A irradiation redistributed Bax from the cytosol to the mitochondria. Moreover, siRNA targeting of Bax effectively attenuated mitochondrial membrane potential loss and apoptotic cell death caused by combined treatment with TiO2P25–70 and ultraviolet A irradiation. These results suggest that mitochondrial redistribution of Bax may trigger mitochondrial membrane potential loss and cause subsequent apoptotic cell death following treatment with TiO2P25–70. Accumulation of intracellular reactive oxygen species in response to diverse stimuli has been shown to lead the mitochondrial membrane permeability transition and subsequent activation of cell death machinery.Citation9 Moreover, recent studies also provide evidence for the role of reactive oxygen species as potential inducers of mitochondrial dysfunction during apoptotic cell death in response to a variety of stimuli.Citation38–Citation40 Here, we provide further evidence that reactive oxygen species are essential for the mitochondrial membrane potential loss and subsequent cell death by TiO2P25–70 treatment. We show that an antioxidant, N-acetyl-L-cysteine, completely attenuates the Bax relocalization induced by TiO2P25–70 and mitochondrial cell death. Recently, several mechanisms have been proposed for reactive oxygen species-associated Bax activation. In nonstressed cells, apoptosis signal-regulating kinase 1 (ASK1) is known to be associated with reactive oxygen species-mediated apoptotic cell death.Citation41,Citation42 However, in our study, we failed to observe the involvement of ASK-1 in response to TiO2P25–70. Moreover, siRNA targeting of ASK-1 did not affect Bax activation and cell death (data not shown), indicating that reactive oxygen species-dependent Bax activation by TiO2P25–70 is independent of ASK-1 signaling. Therefore, the more precise mechanisms by which reactive oxygen species-dependent activation of Bax occurs in response to TiO2P25–70 remain to be elucidated. In summary, we have demonstrated in this study that TiO2P25–70 induces apoptotic cell death through reactive oxygen species-dependent Fas upregulation and Bax activation in normal human liver cells. Elucidating the molecular mechanisms by which nanosized particles induce activation of cell death signaling pathways will be critical for the development of prevention strategies to minimize the cytotoxicity of nanomaterials.
Acknowledgment
This work was supported by the Korea Ministry of Environment and The Eco-Technopia 21 Project (091-091-081).
Disclosure
The authors report no conflicts of interest in this work.
Supplementary figures
Figure S1 Measurement of cell death by FACS analysis after propidium iodide staining. Chang cells were treated with TiO2 nanoparticles of different hydrodynamic size distributions (TiO2P25–70, TiO2P25–130 and TiO2P25–300) at each concentration (0, 50, 100, 150 200, 400 ppm) for 24 hours, and cell death was measured by flow cytometry with propidium iodide staining. Treatment with a lower concentration of TiO2 nanoparticles than 200 ppm did not cause significant cell death, while treatment with a higher concentration (200 and 400 ppm) resulted in dramatic cell death at any TiO2 nanoparticle size.
Note: Error bars represent the mean ± standard deviation of triplicate samples.
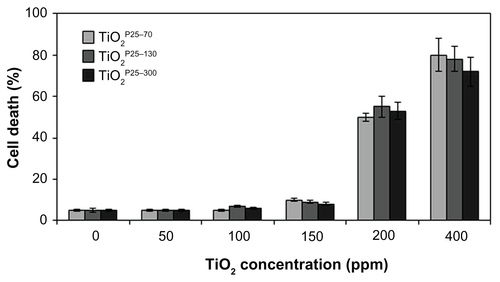
Figure S2 Representative FACS plots for measurement of cell death receptor expression levels. FACS analysis reveals upregulation of Fas expression in Chang cells treated with TiO2P25–70 in combination with ultraviolet A irradiation, while single treatment with either TiO2P25–70 or ultraviolet A did not upregulate the level of Fas expression. Expression levels of other death receptors, ie, TNF, DR4, and DR5, were not altered in Chang cells by combined treatment with TiO2P25–70 and ultraviolet A irradiation.
Abbreviations: TNF, tumor necrosis factor; DR, death receptor; UVA, ultraviolet A.
References
- ShanYMaSNieLSize-dependent endocytosis of single gold nanoparticlesChem Commun (Camb)201147288091809321687845
- WatariFTakashiNYokoyamaAMaterial nanosizing effect on living organisms: non-specific, biointeractive, physical size effectsJ R Soc Interface20096Suppl 3S37138819364724
- LiYSunLJinMSize-dependent cytotoxicity of amorphous silica nanoparticles in human hepatoma HepG2 cellsToxicol In Vitro20112571343135221575712
- FrohlichEKueznikTSambergerCRobleggEWrightonCPieberTRSize-dependent effects of nanoparticles on the activity of cytochrome P450 isoenzymesToxicol Appl Pharmacol2010242332633219909766
- NabeshiHYoshikawaTMatsuyamaKSize-dependent cytotoxic effects of amorphous silica nanoparticles on Langerhans cellsPharmazie201065319920120383940
- ItohTTerazawaRKojimaKCisplatin induces production of reactive oxygen species via NADPH oxidase activation in human prostate cancer cellsFree Radic Res20114591033103921682664
- MizutaniHMechanism of DNA damage and apoptosis induced by anticancer drugs through generation of reactive oxygen speciesYakugaku Zasshi20071271118371842 Japan17978559
- FryeRERossignolDAMitochondrial dysfunction can connect the diverse medical symptoms associated with autism spectrum disordersPediatr Res2011695 Pt 241R47R
- FleuryCMignotteBVayssiereJLMitochondrial reactive oxygen species in cell death signalingBiochimie2002842–3131141 French12022944
- ChanWHWuCCYuJSCurcumin inhibits UV irradiation-induced oxidative stress and apoptotic biochemical changes in human epidermoid carcinoma A431 cellsJ Cell Biochem200390232733814505349
- OberdorsterGMaynardADonaldsonKPrinciples for characterizing the potential human health effects from exposure to nanomaterials: elements of a screening strategyPart Fibre Toxicol20052816209704
- LimbachLKLiYGrassRNOxide nanoparticle uptake in human lung fibroblasts: effects of particle size, agglomeration, and diffusion at low concentrationsEnviron Sci Technol200539239370937616382966
- OberdorsterGOberdorsterEOberdorsterJConcepts of nanoparticle dose metric and response metricEnviron Health Perspect20071156A29017589571
- TeeguardenJGHinderliterPMOrrGThrallBDPoundsJGParticokinetics in vitro: dosimetry considerations for in vitro nanoparticle toxicity assessmentsToxicol Sci200795230031217098817
- SandhuKKMcIntoshCMSimardJMSmithSWRotelloVMGold nanoparticle-mediated transfection of mammalian cellsBioconjug Chem20021313611792172
- ChithraniBDGhazaniAAChanWCDetermining the size and shape dependence of gold nanoparticle uptake into mammalian cellsNano Lett20066466266816608261
- ZhaoFZhaoYLiuYChangXChenCCellular uptake, intracellular trafficking, and cytotoxicity of nanomaterialsSmall20117101322133721520409
- CaiRKubotaYShuinTSakaiHHashimotoKFujishimaAInduction of cytotoxicity by photoexcited TiO2 particlesCancer Res1992528234623481559237
- KwonDLeeSHKimJYoonTHDispersion, fractionation and characterization of sub-100 nm P25 TiO2 nanoparticles in aqueous mediaToxicol Environ Health Sci2010217885
- LongTCSalehNTiltonRDLowryGVVeronesiBTitanium dioxide (P25) produces reactive oxygen species in immortalized brain microglia (BV2): implications for nanoparticle neurotoxicityEnviron Sci Technol200640144346435216903269
- KangSJKimBMLeeYJChungHWTitanium dioxide nanoparticles trigger p53-mediated damage response in peripheral blood lymphocytesEnviron Mol Mutagen200849539940518418868
- DharSMurawalaPShirasAPokharkarVPrasadBLGellan gum capped silver nanoparticle dispersions and hydrogels: cytotoxicity and in vitro diffusion studiesNanoscale20124256356722134682
- FrenchRAJacobsonARKimBIsleySLPennRLBaveyePCInfluence of ionic strength, pH, and cation valence on aggregation kinetics of titanium dioxide nanoparticlesEnviron Sci Technol20094351354135919350903
- MurdockRCBraydich-StolleLSchrandAMSchlagerJJHussainSMCharacterization of nanomaterial dispersion in solution prior to in vitro exposure using dynamic light scattering techniqueToxicol Sci2008101223925317872897
- WohllebenWSchulzeCKrollANot ready to use – overcoming pitfalls when dispersing nanoparticles in physiological mediaNanotoxicology2008225161
- ZhangXYinLTangMPuYOptimized method for preparation of TiO2 nanoparticles dispersion for biological studyJ Nanosci Nanotechnol20101085213521921125873
- JiZJinXGeorgeSDispersion and stability optimization of TiO2 nanoparticles in cell culture mediaEnviron Sci Technol201044197309731420536146
- GrossAMcDonnellJMKorsmeyerSJBCL-2 family members and the mitochondria in apoptosisGenes Dev199913151899191110444588
- GustafssonABGottliebRABcl-2 family members and apoptosis, taken to heartAm J Physiol Cell Physiol20072921C455116943242
- AhamedMAkhtarMJSiddiquiMAOxidative stress mediated apoptosis induced by nickel ferrite nanoparticles in cultured A549 cellsToxicology20112832–310110821382431
- LiuSXuLZhangTRenGYangZOxidative stress and apoptosis induced by nanosized titanium dioxide in PC12 cellsToxicology20102671–317217719922763
- BogatkevichGSGustiloEOatesJCDistinct PKC isoforms mediate cell survival and DNA synthesis in thrombin-induced myofibroblastsAm J Physiol Lung Cell Mol Physiol20052881L19020115447940
- GengLZhuBDaiBHA let-7/Fas double-negative feedback loop regulates human colon carcinoma cells sensitivity to Fas-related apoptosisBiochem Biophys Res Commun2011408349449921530489
- JelaskaAKornJHRole of apoptosis and transforming growth factor beta1 in fibroblast selection and activation in systemic sclerosisArthritis Rheum200043102230223911037882
- MatsudaNYamamotoSTakanoKSilencing of fas-associated death domain protects mice from septic lung inflammation and apoptosisAm J Respir Crit Care Med2009179980681519201926
- DesagherSOsen-SandANicholsABid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosisJ Cell Biol1999144589190110085289
- EskesRDesagherSAntonssonBMartinouJCBid induces the oligomerization and insertion of Bax into the outer mitochondrial membraneMol Cell Biol200020392993510629050
- ChoiEMLeeYSMitochondrial defects and cytotoxicity by antimycin A on cultured osteoblastic MC3T3-E1 cellsFood Chem Toxicol20114992459246321601611
- PiaoMJKangKALeeIKSilver nanoparticles induce oxidative cell damage in human liver cells through inhibition of reduced glutathione and induction of mitochondria-involved apoptosisToxicol Lett201120119210021182908
- WuPPLiuKCHuangWWTriptolide induces apoptosis in human adrenal cancer NCI-H295 cells through a mitochondrial-dependent pathwayOncol Rep201125255155721152873
- PanJChangQWangXReactive oxygen species-activated Akt/ASK1/p38 signaling pathway in nickel compound-induced apoptosis in BEAS 2B cellsChem Res Toxicol201023356857720112989
- YangWHFongYCLeeCYEpigallocatechin-3-gallate induces cell apoptosis of human chondrosarcoma cells through apoptosis signal-regulating kinase 1 pathwayJ Cell Biochem201111261601161121328612