 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Background
Indirubin, isolated from the leaves of the Chinese herb Isatis tinctoria L, is a protein kinase inhibitor and promising antitumor agent. However, the poor water solubility of indirubin has limited its application. In this study, a supersaturatable self-microemulsifying drug delivery system (S-SMEDDS) was developed to improve the oral bioavailability of indirubin.
Methods
A prototype S-SMEDDS was designed using solubility studies and phase diagram construction. Precipitation inhibitors were selected from hydrophilic polymers according to their crystallization-inhibiting capacity through in vitro precipitation tests. In vitro release of indirubin from S-SMEDDS was examined to investigate its likely release behavior in vivo. The in vivo bioavailability of indirubin from S-SMEDDS and from SMEDDS was compared in rats.
Results
The prototype formulation of S-SMEDDS comprised Maisine™ 35-1:Cremophor® EL:Transcutol® P (15:40:45, w/w/w). Polyvinylpyrrolidone K17, a hydrophilic polymer, was used as a precipitation inhibitor based on its better crystallization-inhibiting capacity compared with polyethylene glycol 4000 and hydroxypropyl methylcellulose. In vitro release analysis showed more rapid drug release from S-SMEDDS than from SMEDDS. In vivo bioavailability analysis in rats indicated that improved oral absorption was achieved and that the relative bioavailability of S-SMEDDS was 129.5% compared with SMEDDS.
Conclusion
The novel S-SMEDDS developed in this study increased the dissolution rate and improved the oral bioavailability of indirubin in rats. The results suggest that S-SMEDDS is a superior means of oral delivery of indirubin.
Introduction
Indirubin () is a bisindole compound and protein kinase inhibitor, which is isolated from the leaves of the Chinese herb, Isatis tinctoria L. The drug has various pharmacological effects, including antitumor and anti-inflammatory activity.Citation1–Citation3 In the clinical setting, it has been shown to be effective in the treatment of chronic myelocytic leukemia. In addition, indirubin has been shown to have marked antiproliferative activity and to be a strong inducer of apoptosis in multiple tumor cell types, including cervical cancer, liver cancer, and lymphoma cell lines.Citation4 The reported mechanisms for the antitumor effect involve inhibition of DNA and protein synthesis and inhibition of key protein kinases.Citation5 Long-term animal studies of indirubin have detected neither bone marrow toxicity nor hematotoxicity.Citation6,Citation7 However, indirubin has poor water solubility, resulting in low oral bioavailability. Moreover, ingestion often leads to irritation in the gastrointestinal tract. Thus, although indirubin is a promising antitumor agent, its clinical use has been limited.Citation8
Figure 1 Chemical structure of indirubin.
Absorption of indirubin is reported to occur by passive diffusion. The transport of indirubin through the rat intestine membrane was not influenced by P-glycoprotein or by the multidrug resistance-associated protein.Citation9,Citation10 From the point of view of drug concentration, regardless of other issues, such as the size of the drug molecule and the absorptive surface area, the higher the drug concentration at the absorption site, the higher the fraction of absorbed drug transported by passive diffusion. Improved solubility and a higher dissolution rate for poorly water soluble drugs are key factors to enhance drug absorption.Citation11 Thus, it is considered that improvement of indirubin bioavailability may be achieved by improving its solubilization in the gastrointestinal tract and increasing its dissolution rate in aqueous solution. Recent studies have focused on the development and evaluation of a self-emulsifying drug delivery system (SEDDS) for indirubin,Citation12,Citation13 in which indirubin has a 1.57-fold improvement in bioavailability compared with commercial indirubin tablets in beagle dogs.
Over the last two decades, self-(micro)emulsifying drug delivery systems (SMEDDS) have been widely investigated as an effective means of delivering poorly water-soluble drugs via the oral route. The system is composed of oil, surfactant, and cosurfactant. An emulsion or microemulsion can be formed upon dispersion of SMEDDS in aqueous solution. However, when a drug is released from a microemulsion, precipitation often occurs due to decreased solubility, leading to decreased drug dissolution and absorption in vivo. In addition, high amounts of surfactant and cosurfactant can generate side effects, including irritation of the gastrointestinal tract, changes in intestinal membrane permeability, and toxicity.Citation14,Citation15 Nevertheless, significant amounts of surfactant and cosurfactant are required to deliver drugs in dissolved form. Thus, inhibiting drug precipitation upon mixing SMEDDS with aqueous solution is a key consideration in designing these formulations.
A supersaturation process can maintain drug solubilization above equilibrium solubility without precipitation.Citation16–Citation19 A high energy form of the drug (in comparison with crystal-line powder) in solution yielded a supersaturated state with increased chemical potential. Thus, it is a thermodynamically unstable system. When a supersaturated drug delivery system exists at the absorption site for a sufficient period of time, the higher drug concentration generated from the supersaturated state may enhance drug absorption.Citation20 Hydrophilic polymers such as hydroxypropyl methylcellulose (HPMC) and polyvinylpyrrolidone (PVP) can be used in SMEDDS formulations as precipitation inhibitors to form supersaturatable self-(micro)emulsifying drug delivery systems (S-SMEDDS). When S-SMEDDS come into contact with the aqueous environment of the gastrointestinal tract, the preparations are first emulsified, and an emulsion or microemulsion is formed immediately. The drug may be dissolved in free form or incorporated in emulsion or microemulsion droplets. Precipitation inhibitors may increase the solubility of the free drug or the drug in microemulsion and further increase the concentration gradient of the drug across the intestinal membrane, which may significantly improve the water solubility of the drug and enhance oral absorption. Hence, more recently, S-SMEDDS have been developed to improve oral absorption of poorly water-soluble drugs.Citation21–Citation26 Generation of drug supersaturation in the gastrointestinal tract seems a promising way to improve the oral bioavailability of poorly water-soluble drugs.
The aim of this study was to develop an S-SMEDDS to improve the oral bioavailability of indirubin. First, prototype S-SMEDDS formulations were prepared. Hydrophilic polymers PVP, HPMC, and polyethylene glycol (PEG) 4000 were used as precipitation inhibitors, and their crystallization-inhibiting capacities were evaluated. The mechanism of inhibition of crystallization by the precipitation inhibitors was explored. Preparations were then assessed based on drug release and bioavailability.
Materials and methods
Materials
Indirubin was obtained from Nanjing Zelang Medical Technology Co Ltd, Nanjing, China. Glyceryl monolinoleate (Maisine™ 35-1), diethylene glycol monoethyl ether (Transcutol ® P), linoleoyl polyoxyl-6 glycerides (Labrafil ® M2125CS), medium-chain triglycerides (Labrafac™ Lipophile WL1349) and caprylocaproyl macrogol- 8 glycerides (Labrasol ®) were donated by Gattefossè, Gennevilliers, France. PEG 400, PEG 200, PEG 4000, and isopropyl myristate were purchased from Shanghai Chemical Reagent Corporation, Shanghai, China. Olive oil was sourced from Sictia, Marseille, France. Caprylic/capric triglyceride (GTCC) was obtained from Guangdong Indwell Industrial and Trading Co, Ltd, Guangdong, China. Polyoxyl 40 hydrogenated castor oil (Cremophor® RH 40), polyoxyl castor oil (Cremophor EL), and povidone K17 (PVP K17) were from BASF, Ludwigshafen, Germany. Methocel E6 premium HPMC was purchased from Colorcon, Shanghai, China. The other chemicals were of analytical grade.
Solubility studies
The solubility of indirubin in various oils, surfactants, and cosurfactants was determined as follows. Vehicle (1 g) was added to capped tubes containing an excess of indirubin. After the test tubes were sealed, the mixtures in the test tubes were shaken for 72 hours at 100 strokes per minutes in a water bath maintained at 25°C, and the tubes were then centrifuged at 6700 g for 5 minutes. The supernatant was dissolved in ethyl acetate in a volumetric flask, and methanol was added to make up the volume. After filtration through a 0.45 μm membrane filter, the concentration of indirubin was determined by HPLC. HPLC analysis was carried out using a LC-2010A HT liquid chromatography system (Shimadzu Corporation, Kyoto, Japan) as follows: Kromasil®-5C18 column (250 mm × 4.6 mm, 5 μm); mobile phase of methanol:water:phosphoric acid (90:10:0.1, v/v/v); column temperature of 40°C; detection wavelength 225 nm; flow rate of 1 mL/minute.
Phase diagram construction
The ternary phase diagrams of mixtures of oil, surfactant, and cosurfactant at certain ratios were constructed as previously described.Citation27 After dilution and gentle agitation of the mixtures in simulated gastric fluid, the emulsification behavior was observed. The boundaries of the self-microemulsification regions in the phase diagrams were determined by connecting the points representing formation of the microemulsion.
Preparation of SMEDDS
The compounds used in the indirubin SMEDDS formulations are shown in . Indirubin were prepared by dissolving the drug in the mixture of Transcutol P, Cremophor EL, and Maisine 35-1 with continuous stirring. SMEDDS was formed when the dispersion became transparent. PVP K17 was then introduced into the SMEDDS preparation and dispersed by vortexing.
Table 1 Solubility of indirubin in various vehicles at 25°C
Determination of droplet size and zeta potential
Formulations were diluted in distilled water before analysis. Droplet size and distribution was measured by photocorrelation spectroscopy using a Nicomp 380/ZLS system (PSS, Port Richey, FL). Each sample was analyzed in triplicate. Zeta potential was determined in the same manner.
In vitro evaluation of precipitation
To investigate the precipitation-inhibiting capacity of hydrophilic polymers, in vitro precipitation assays were conducted using a RC 806 dissolution tester (Tianjin Tianda Tianfa Technology Co, Ltd, Tianjin, China) with 250 mL round vessels at 37°C ± 0.5°C and a stirring speed of 100 rpm. The formulations including SMEDDS and SMEDDS containing various hydrophilic polymers were mixed with simulated gastric fluid to a total volume of 100 mL. At predetermined times, 0.5 mL of sample was withdrawn and centrifuged at 13,000 rpm for 3 minutes. Supernatant (0.2 mL) was mixed with 0.4 mL of methanol, and the concentration of indirubin was analyzed by HPLC.
In vitro release
In vitro release assays were carried out according to the release test method described in the Chinese Pharmacopeia (2010 edition, small cup method).Citation28 Indirubin S-SMEDDS (0.5 g) was added to a size zero capsule. The capsule was then put into the basket in 100 mL of simulated gastric fluid. Samples (0.5 mL) were removed at 5, 10, 20, 30, 40, 60, 120, 180, 240, and 420 minutes after addition of drug, and the volume was replaced by adding 0.5 mL of fresh medium. The indirubin concentration was measured as described above. The cumulative released percentage was calculated as shown in EquationEquation 1(1) :
in which Q is the cumulative percentage released; Ct is the indirubin concentration at time t; V represents the volume of release medium (V = 100 mL); Va is the volume of sampling aliquot (Va = 0.5 mL); and W is the initial amount of indirubin in the capsule.
Bioavailability study
The animal experiments were approved by the institutional animal ethics committee of Shanghai University of Traditional Chinese Medicine. Male Sprague-Dawley rats (weight 200 ± 20 g) were divided into three groups of six animals. Prior to drug administration, the animals were fasted for 12 hours with free access to water. The tested preparations were SMEDDS, S-SMEDDS, and the suspensions. SMEDDS was prepared using the same method as described for preparation of S-SMEDDS but with PVP K17 omitted. Suspensions were prepared by dispersing indirubin in sodium carboxymethyl cellulose solution with glycerin as a wetting agent. Rats were administered a indirubin dose of 2.58 mg/kg for each preparation by gavage, followed by 1 mL of physiological saline. Blood samples (0.5 mL) were collected into heparinized tubes via retro-orbital puncture at hours 0.5, 1, 2, 3, 4, 5, 6, 8, 10, 12, and 24 post dose. Blood samples were centrifuged at 2000 g for 5 minutes. Plasma was kept at −20°C until analysis, as described below.
A 0.2 mL sample of plasma was mixed with an equal volume of saturated ammonium sulfate solution and vortexed for 10 seconds. Ethyl acetate (0.6 mL) was added, and the mixture was vortexed for 3 minutes. After centrifugation at 10,000 rpm for 3 minutes, the upper layer was removed and dried under gentle flowing nitrogen. Residuals were reconstituted in 100 μL of methanol. The mixture was vortexed for 20 seconds and then centrifuged at 13,000 rpm for 3 minutes. The supernatant was used for content analysis.
Indirubin concentrations in rat plasma were determined using a Waters Acquity UPLC-Micromass ZQ 2000 system (Waters Corporation, Milford, MA). An Acquity UPLC R BEH C18 column (1.7 μm, 2.1 × 50 mm) was used at 30°C. The mobile phase A was 0.1% formic acid in water. The mobile phase B was methanol. The gradient program was set as follows: 0–6 minutes: 40% A, 60% B; 6–7 minutes: 10% A, 90% B; 7–9 minutes: 40% A, 60% B. The flow rate of the mobile phase was kept at 0.3 mL/minute. The injection volume was 10 μL. Triple-quadruple tandem mass spectrometric detection was operated in positive ionization mode with m/z of 262.3, a source temperature of 120°C, capillary voltage of 3000 V, desolvation flow rate of 500 L/hour, and desolvation temperature of 250°C.
Data analysis
Pharmacokinetic parameters were obtained using DAS 2.0 software. Statistical analysis for the pharmacokinetic parameters was carried out using one-way analysis of variance. A P value of <0.05 was considered to be statistically significant.
Results and discussion
SMEDDS formulation
Initially, SMEDDS were developed as prototypes of S-SMEDDS. To obtain a SMEDDS formulation, we determined the solubility of indirubin in various oils, surfactants, and cosurfactants. The results are presented in . The solubility of indirubin in the tested oils, in decreasing order, was as follows: Maisine 35-1 > Labrafil M2125CS > Labrafac Lipophile WL1349 > GTCC > olive oil > isopropyl myristate. Solubility of indirubin in Cremophor EL (surfactant) and Transcutol P (cosurfactant) was markedly higher than in other vehicles, indicating superior dissolving capability for indirubin. Based on these results, we selected Maisine 35-1 for the oil phase, Cremophor EL as the surfactant, and Transcutol P as the cosurfactant for further investigation.
A phase diagram was constructed to identify the self-microemulsification regions. A series of formulations containing Maisine 35-1, Cremophor EL, and Transcutol P were prepared and diluted 200-fold in simulated gastric fluid at 37°C. A dispersion with a transparent appearance was regarded as a microemulsion. The phase diagram is shown in .
Figure 2 Ternary system containing Maisine 35-1/Cremophor EL/Transcutol P. Region of efficient self-microemulsification is bound by the solid line, and the filled circles represent compositions that were evaluated.
Based on analysis of self-microemulsifying regions, three formulations were selected for further assessment of droplet size, polydispersibility, and equilibrium solubility. As shown in , the droplet size of formulation C was clearly smaller than that of the other formulations. In addition, formulation C had better solubilizing capacity than the other formulations. In a previous study, we found that both the oil percentage and surfactant to cosurfactant ratio influence equilibrium solubility.Citation29 As shown in , the Transcutol P cosurfactant had better dissolving capability for indirubin than Cremophor EL. This may explain the fact that when the oil percentage was the same, formulation C (with higher Transcutol P content) exhibited better drug solubility than the other two formulations. Based on these findings, formulation C was selected as the final prototype S-SMEDDS.
Table 2 Droplet size, polydispersibility, and solubility for the three tested SMEDDS formulations
Screening for a precipitation inhibitor
A hydrophilic polymer is an essential excipient in a S-SMEDDS formulation. Hydrophilic polymers, such as PVP, HPMC, and PEG, have been found to be useful as precipitation inhibitors.Citation18,Citation30–Citation34 In this study, HPMC, PEG 4000, and PVP K17 were assayed for inhibition of crystallization in vitro.
As reported previously, evaluation of the true free drug concentration in preparations dispersing in aqueous systems is difficult, mainly due to the complex drug state and dynamic processes during dispersion.Citation21–Citation23 When SMEDDS is dispersed into aqueous solution, the drug may exist in three main states, namely, as a free molecule in solution, solubilized in microemulsion or similar vehicles, and/or precipitated as solid particles. To evaluate drug precipitation behavior accurately, it is important to separate precipitated drug from the dispersion system upon mixing SMEDDS with an aqueous solution. Syringe filters method and centrifugation method are generally used methods.Citation24,Citation25,Citation35 The syringe filter method may separate a solution with precipitated drugs by filtration, and the centrifugal method achieves separation by centrifugation. In addition, Gao et al reported an improved approach using focused beam reflectance measurement technology.Citation23 Their method is a fast and convenient in situ measurement technology that may detect particles within the size range of 1–1000 μm.Citation23 However, it was noted that focused beam reflectance measurement cannot detect particles less than 1 μm in size. In this study, we compared membrane filtration and centrifugation by evaluating the recovery and repeatability of each method. We found that the centrifugation method yielded higher recovery and repeatability than filtration, perhaps by eliminating loss by adsorption to the filter (data not shown). It should also be noted that in addition to free molecules in solution and solubilized drug in microemulsion or other vehicles, nanosized solid drug particles may also be present in supernatants after centrifugation.
The indirubin concentrations in simulated gastric fluid after dilution of SMEDDS (without hydrophilic polymers) and S-SMEDDS formulations are shown in . shows the indirubin concentration-time profile using varying amounts of PEG 4000 as a precipitating inhibitor. The results indicate that the formulation with 2% PEG 4000 achieved the highest drug concentration compared with the other formulations. PEG 4000 was finely dissolved in SMEDDS, but as the amount of PEG 4000 increased, the dispersions became increasingly viscous. At 5% PEG 4000, the dispersions became gel-like due to high viscosity, which may hinder self-microemulsification.
Figure 3 In vitro indirubin concentration-time profiles from the SMEDDS formulations and S-SMEDDS with hydrophilic polymers of (A) PEG 4000, (B) HPMC, and (C) PVP K17 (n = 5).
Abbreviations: SMEDDS, self-microemulsifying drug delivery system; S-SMEDDS, supersaturatable self-microemulsifying drug delivery system; PEG, polyethylene glycol; PVP, polyvinylpyrrolidone; HPMC, hydroxypropyl methylcellulose.
In the case of HPMC, compared with HPMC-free SMEDDS, there was no marked enhancement of drug release in the presence of 0.5%, 2%, and 5% HPMC (). This result indicated that HPMC appeared less effective as a precipitation inhibitor. As shown in , the highest release was achieved when 0.5% PVP K17 was used. In addition, PVP K17 was well dispersed in SMEDDS, and the addition of PVP K17 to SMEDDS had no significant influence on the viscosity of the dispersions. Based on these results, we concluded that the precipitation-inhibiting capacity of the three hydrophilic polymers was in the order PVP K17 > PEG 4000 > HPMC, and that 0.5% PVP K17 can efficiently retard precipitation and maintain a higher indirubin concentration for approximately 2 hours or longer. Therefore, 0.5% of PVP K17 was selected to add to SMEDDS. The final S-SMEDDS formulation composition was Maisine 35-1:Cremophor EL:Transcutol P 15%:40%:45% (w/w/w) with 0.5% PVP K17.
The indirubin molecule is formed using a π-conjugated system. There are strong hydrogen bonds between an oxygen atom and adjacent hydrogen atom. Hydrogen bonds are responsible for the stability of the molecule. One hexatomic ring can be formed by intramolecular hydrogen bonds, and therefore lower the ring strain and the molecular total system energy.Citation36 Therefore, the indirubin molecule is stable and has low water solubility. We found that both PEG 4000 and indirubin were well dissolved in SMEDDS. We propose that when each PEG 4000 molecule () opened the two parallel helical bonds, the indirubin molecules were incorporated into the coiling chain to cause molecular dispersion. Meanwhile, the intramolecular hydrogen bond was activated upon dissolving of indirubin in SMEDDS, and exchanging of a hydrogen bond with PEG 4000 may occur (). In a previous report, it was proposed that the adsorption of the polymer onto the crystal surface may play a role in inhibition of crystallization, caused by a hydrogen bond between the drug and the hydrophilic polymer.Citation37–Citation39 Therefore, from this perspective, the hydrogen bonds between indirubin and PEG 4000 may contribute to the retarded precipitation of indirubin after dilution of SMEDDS.
Figure 4 Molecular structures of (A) PEG 4000, (B) PVP, (C) HPMC and schematic diagram of hydrogen bonds between indirubin, (D) PEG 4000, and (E) PVP.
Abbreviations: PEG, polyethylene glycol; PVP, polyvinylpyrrolidone; HPMC, hydroxypropyl methylcellulose.
Another factor responsible for the role of the precipitation inhibitor is thought to be the incorporation of drug molecules into polymer aggregates.Citation40 In the case of PVP K17 (), after PVP K17 and indirubin were dissolved in SMEDDS, PVP K17 showed reticulate structures, and indirubin molecules may be incorporated into the reticulate structures resulting in molecular dispersion. Indirubin-PVP K17 hydrogen bonds were formed between the carbonyl group of the pyrrolidone ring of PVP K17 and the amino group in the pyrrole ring as well as the amido hydrogen of indirubin (). Hydrogen bonding and incorporation of indirubin into reticulate structures, as well as high viscosity, may efficiently inhibit the aggregation and crystallization of indirubin. Further study of the indirubin-PVP K17 interaction using infrared spectroscopy or other analytic strategies may clarify these matters.
HPMC (), a cellulosic polymer, has been reported to be an efficient crystal growth inhibitor for many drugs in aqueous dispersions.Citation18,Citation33,Citation39 Approximately 60% of the hydroxyl groups in HPMC are not substituted. HPMC can thus form both intramolecular and intermolecular hydrogen bonds. Thus, HPMC is more hydrophobic than PVP K17 and PEG 4000. Once the hydrogen bonds are disrupted in the dissolved state, HPMC may interact with a drug and form hydrogen bonds, which is likely to retard drug precipitation. Furthermore, based on the molecular structure, HPMC may provide more hydrogen bonding groups per monomer unit compared with PVP K17. (Interaction of indirubin and HPMC is not described herein due to the complex substituent group leading to uncertain hydrogen bond formation). However, in this study, we found that HPMC cannot be dissolved in indirubin SMEDDS. Thus, it appears that HPMC exerted no precipitation-inhibiting action on indirubin SMEDDS.
In vitro release study
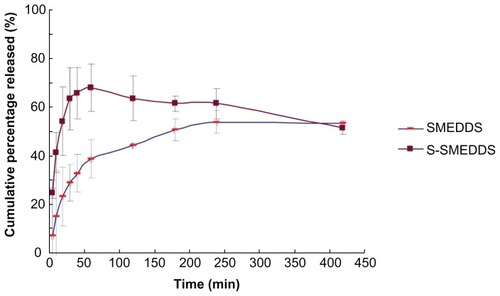
The in vitro release assay () showed that 70% of the drugs was released from S-SMEDDS in 60 minutes, while approximately 50% of the drugs was released from SMEDDS in 120 minutes. S-SMEDDS exhibited faster release behavior than SMEDDS. However, the percentage released in the case of S-SMEDDS decreased slightly after 60 minutes, and decreased slowly to approximately 60% at 240 minutes. The released drug was detected in the dissolved form either in solution or in the microemulsion-like vehicles. Thus, the decrease in the amount of dissolved drug indicated that precipitation had occurred. A supersaturatable system is thermodynamically unstable, and a supersaturatable drug solution has a tendency to return to its equilibrium state via drug precipitation.Citation20 Thus, the supersaturation process may only keep the drug dissolved for a limited period of time. With increasing time, drug precipitation may occur, which results in a decreased amount of dissolved drug. Because there is no supersaturation process involved in SMEDDS, the percentage of drug released increased steadily with the passage of time. Almost the same percentage of drug release was achieved for the two preparations at 420 minutes. Based on the results of our in vitro release study, we surmise that S-SMEDDS would have higher bioavailability in vivo than SMEDDS.
Figure 5 Cumulative release profile of indirubin SMEDDS and S-SMEDDS.
Abbreviations: SMEDDS, self-microemulsifying drug delivery system; S-SMEDDS, supersaturatable self-microemulsifying drug delivery system.
Bioavailability study
Ultra performance liquid chromatography/mass spectrometry chromatograms are shown in , which indicate that endogenous substances in plasma did not affect detection of indirubin. shows the mean plasma concentration profile of indirubin from the suspension, SMEDDS, and S-SMEDDS after oral administration to rats. The plasma concentration of indirubin after dosing of the indirubin suspension could not be detected due to poor oral absorption in vivo. S-SMEDDS containing indirubin provided higher plasma drug concentrations than SMEDDS containing indirubin.
Figure 6 Ultra performance liquid chromatography/mass spectrometry chromatograms of (A) blank plasma, (B) blank plasma spiked with indirubin, and (C) plasma sample after oral administration of indirubin to a rat.
Figure 7 Plasma concentration profile of indirubin after oral administration of SMEDDS and S-SMEDDS in rats (n = 5).
Abbreviations: SMEDDS, self-microemulsifying drug delivery system; S-SMEDDS, supersaturatable self-microemulsifying drug delivery system.
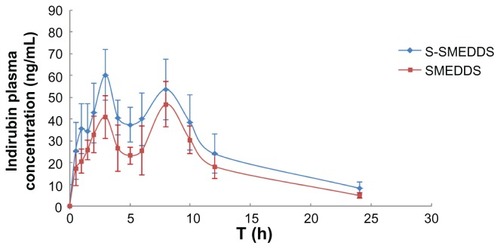
Mean area under the curve (AUC), peak plasma concentration, and relative bioavailability are listed in . Parameters for the suspensions were not analyzed because the plasma concentration could not be detected. The relative bioavailability was calculated by dividing AUC0–24 h (S-SMEDDS) by AUC0–24 h (SMEDDS). The peak plasma concentration and mean residence time values for indirubin from S-SMEDDS were not significantly different than those from SMEDDS. However, the AUC for S-SMEDDS was significant larger than that of SMEDDS (P < 0.05).
Table 3 Pharmacokinetic parameters for S-SMEDDS and SMEDDS (n = 5)
These results suggest that S-SMEDDS might be useful for enhancing the oral absorption of indirubin. Furthermore, the results of the in vivo bioavailability study appear to correlate with those of the in vitro release study, which indicates a faster release rate from S-SMEDDS than from SMEDDS. For poorly water soluble drugs, dissolution is generally the rate-limiting step to drug absorption. A higher release rate might play an important role in enhancing bioavailability. In addition, S-SMEDDS provides indirubin in supersaturated conditions upon mixing with aqueous dispersions, allowing indirubin to dissolve rather than precipitate, and further enhancing bioavailability.
Conclusion
The novel S-SMEDDS developed in this study increased the dissolution rate and improved the oral absorption of indirubin. The S-SMEDDS is a promising way to deliver indirubin by the oral route.
Acknowledgments
This work was financially supported by a grant (J50302) from the Shanghai Education Committee, a grant (10XD14303900) from the Science and Technology Commission of Shanghai Municipality, and by grants (NCET08-0898 and IRT1071) from the State Education Ministry, People’s Republic of China.
Disclosure
The authors report no conflicts of interest in this work.
References
- BradburyJFrom Chinese medicine to anticancer drugsDrug Discov Today200510171131113216182200
- WuQWGeZLGaoYZhangLInhibitory effect of indirubin on growth of some cancer cells and its mechanismTianjin Journal of Traditional Chinese Medicine20082515558 Chinese
- RuiHRecent progress of traditional Chinese medicine and herbal medicine for the treatment and prevention of cancerGan To Kagaku Ryoho20022916775 Japanese11890117
- EisenbrandGHippeFJakobsSMuehlbeyerSMolecular mechanisms of indirubin and its derivatives: novel anticancer molecules with their origin in traditional Chinese phytomedicineJ Cancer Res Clin Oncol20041301162763515340840
- ZhangAQuYZhangBThe different effects of indirubin on effector and CD4+CD25+ regulatory T cells in mice: potential implication for the treatment of autoimmune diseasesJ Mol Med (Berl)200785111263127017639287
- JiXJZhangFRLeiJLXuYTStudies on the antineoplastic effect and toxicity of synthetic indirubinYao Xue Xue Bao1981162146148 Chinese7304182
- WanJHYouYCMiJXYingHGEffect of indirubin on hematopoietic cell productionActa Pharmacol Sin198124241244
- JiXJZhangFRStudies on antineoplastic action of indirubin derivatives and analogues and their structure-activity relationshipsYao Xue Xue Bao1985202137139 Chinese4036618
- WuZHZhouJPingQNZhangYHWangZLZhouHGut transport mechanism of indirubinJournal of China Pharmaceutical University2006376494498 Chinese
- LiJKJiangXHAbsorption mechanism of indirubin in rat in situChung Kuo Yao Hsueh Tsa Chih2007421713061311 Chinese
- SinghAWorkuZAVan den MooterGOral formulation strategies to improve solubility of poorly water-soluble drugs. ChineseExpert Opin Drug Deliv20118101361137821810062
- WuZHZhouJPingQNZhangYHWangZLZhouHTransport of indirubin SEDDS across the intestinal mucosa of rat and the mono-layer of Caco-2 cell and its pharmacokinetics in Beagle dogsChinese Journal of New Drugs2007161713681372 Chinese
- LeiJWuZHPingQNZhouJIn vitro evaluation of indirubin self-emulsifying drug delivery systemChinese Traditional Patent Medicine2008307968972 Chinese
- PoelmaFGBreäsRTukkerJJCrommelinDJIntestinal absorption of drugs. The influence of mixed micelles on the disappearance kinetics of drugs from the small intestine of the ratJ Pharm Pharmacol19914353173241680173
- ChiuYYHigakiKNeudeckBLBarnettJLWelageLSAmidonGLHuman jejunal permeability of cyclosporin A: influence of surfactants on P-glycoprotein efflux in Caco-2 cellsPharm Res200320574975612751630
- PellettaMACastellanobSHadgraftaJDavisAFThe penetration of supersaturated solutions of piroxicam across silicone membranes and human skin in vitroJ Control Release1997463205214
- IervolinoMRaghavanSLHadgraftJMembrane penetration enhancement of ibuprofen using supersaturationInt J Pharm2000198222923810767571
- FusaoUKazukoMAkiraKKenjiNKeijiYInhibitory effects of water-soluble polymers on precipitation of RS-8359Int J Pharm199715415966
- AugustijnsPBrewsterMESupersaturating drug delivery systems: fast is not necessarily good enoughJ Pharm Sci201210117921953470
- BrouwersJBrewsterMEAugustijnsPSupersaturating drug delivery systems: the answer to solubility-limited oral bioavailability?J Pharm Sci20099882549257219373886
- GaoPMorozowichWDevelopment of supersaturatable self-emulsifying drug delivery system formulations for improving the oral absorption of poorly soluble drugsExpert Opin Drug Deliv2006319711016370943
- GaoPRushBDPfundWPDevelopment of a supersaturatable SEDDS (S-SEDDS) formulation of paclitaxel with improved oral bioavailabilityJ Pharm Sci200392122386239814603484
- GaoPAkramiAAlvarezFCharacterization and optimization of AMG 517 supersaturatable self-emulsifying drug delivery system (S-SEDDS) for improved oral absorptionJ Pharm Sci200998251652818543293
- ZhangNZhangWDJinYHQuanDQStudies on preparation of carbamazepine (CBZ) supersaturatable self-microemulsifying (S-SMEDDS) formulation and relative bioavailability in beagle dogsPharm Dev Technol201116441542120433250
- GaoPGuytonMEHuangTBauerJMStefanskiKJLuQEnhanced oral bioavailability of a poorly water soluble drug PNU- 91325 by supersaturatable formulationsDrug Dev Ind Pharm200430222122915089057
- ChenYChenCZhengJChenZShiQLiuHDevelopment of a solid supersaturatable self-emulsifying drug delivery system of docetaxel with improved dissolution and bioavailabilityBiol Pharm Bull201134227828621415541
- WeiLLSunPNNieSFPanWPreparation and evaluation of SEDDS and SMEDDS containing carvedilolDrug Dev Ind Pharm200531878579416221613
- YangFBiGWLongXYYuZMYangWSFengLPThe preparation and the in vitro release of OANO-1 microspheresZhongguo Zhong Yao Za Zhi20053013992994 Chinese16161426
- LiuYZhangPFengNPZhangXWuSZhaoJHOptimization and in situ intestinal absorption of self-microemulsifying drug delivery system of oridoninInt J Pharm20093651–213614218782611
- O’DriscollKMCorriganOIChlorothiazide-polyvinylpyrrolidone (PVP) interactions: influence on membrane permeation (everted rat intestine) and dissolutionDrug Dev Ind Pharm198284547564
- MaXHTawJChiangCMControl of drug crystallization in transdermal matrix systemsInt J Pharm19961421115119
- SchwarbFPImanidisGSmithEWHaighJMSurberCEffect of concentration and degree of saturation of topical fluocinonide formulations on in vitro membrane transport and in vivo availability on human skinPharm Res199916690991510397613
- RaqhavanSLTrividicADavisAFHadgraftJEffect of cellulose polymers on supersaturation and in vitro membrane transport of hydrocortisone acetateInt J Pharm2000193223123710606787
- SuzukiHSunadaHComparison of nicotinamide, ethylurea and polyethylene glycol as carriers for nifedipine solid dispersion systemsChem Pharm Bull19974510168816939353898
- CuratoloWNightingaleJAHerbigSMUtility of hydroxypropy-lmethylcellulose acetate succinate (HPMCAS) for initiation and maintenance of drug supersaturation in the GI milieuPharm Res20092661419143119277850
- WangYLiuYMLiangXQYangMTheoretical study of indigo and its isomerJournal of Sichuan University (Natural Science Edition)2004411143147 Chinese
- RaghavanSLTrividicADavisAFCrystallization of hydrocortisone acetate: influence of polymersInt J Pharm2001212221322111165079
- RaghavanSLKiepferBDavisAFKazarianSGHadgraftJMembrane transport of hydrocortisone acetate from supersaturated solutions: the role of polymersInt J Pharm20012211–29510511397571
- ZillerKHRupprechtHControl of crystal growth in drug suspensionsDrug Dev Ind Pharm1988145–1723412370
- KimHJLeeSHLimEAKimJSFormulation optimization of solid dispersion of mosapride hydrochlorideArch Pharm Res20113491467147521975808