Abstract
The peptide vaccine clinical trials encountered limited success because of difficulties associated with stability and delivery, resulting in inefficient antigen presentation and low response rates in patients with cancer. The purpose of this study was to develop a novel delivery approach for tumor antigenic peptides in order to elicit enhanced immune responses using poly(DL-lactide-co-glycolide) nanoparticles (PLGA-NPs) encapsulating tumor antigenic peptides. PLGA-NPs were made using the double emulsion-solvent evaporation method. Artificial antigen-presenting cells were generated by human dendritic cells (DCs) loaded with PLGA-NPs encapsulating tumor antigenic peptide(s). The efficiency of the antigen presentation was measured by interferon-γ ELISpot assay (Vector Laboratories, Burlingame, CA). Antigen-specific cytotoxic T lymphocytes (CTLs) were generated and evaluated by CytoTox 96® Non-Radioactive Cytotoxicity Assay (Promega, Fitchburg, WI). The efficiency of the peptide delivery was compared between the methods of emulsification in incomplete Freund’s adjuvant and encapsulation in PLGA-NPs. Our results showed that most of the PLGA-NPs were from 150 nm to 500 nm in diameter, and were negatively charged at pH 7.4 with a mean zeta potential of −15.53 ± 0.71 mV; the PLGA-NPs could be colocalized in human DCs in 30 minutes of incubation. Human DCs loaded with PLGA-NPs encapsulating peptide induced significantly stronger CTL cytotoxicity than those pulsed with free peptide, while human DCs loaded with PLGA-NPs encapsulating a three-peptide cocktail induced a significantly greater CTL response than those encapsulating a two-peptide cocktail. Most importantly, the peptide dose encapsulated in PLGA-NPs was 63 times less than that emulsified in incomplete Freund’s adjuvant, but it induced a more powerful CTL response in vivo. These results demonstrate that the delivery of peptides encapsulated in PLGA-NPs is a promising approach to induce effective antitumor CTL responses in vivo.
Introduction
Tumor antigen-reactive T cells have been detected in the circulation of patients with cancer,Citation1 evidencing the existence of a host immune response, which unfortunately fails to prevent disease progression in most cancer patients. It has been postulated that tumor-reactive T cells are in a nonresponsive state and low in number, and need to be activated to achieve an antitumor response.Citation2 T lymphocytes recognize tumor antigens as small peptides of 8–10 amino acids in length, bound to cell surface molecules encoded by the major histocompatibility complex class I molecules. These major histocompatibility complex class I molecules are expressed in a number of cell types, including antigen-presenting cells (APCs). Dendritic cells (DCs) are widely distributed in peripheral tissues and lymphoid organs, they are believed to be the most potent APCs, and are specialized for the uptake of antigens, migration to lymph nodes, and activation of naïve T cells.Citation3
In general, immunotherapy requiring an eff icient T lymphocyte response is initiated by antigen delivery to APC.Citation4 A large number of hydrophilic molecules are poorly taken up by cells since they do not efficiently cross the lipid bilayer of the plasma membrane. The low bioavailability of peptides, proteins, and nucleic acids is due to many factors, including poor stability and susceptibility to enzymes.Citation5 This is considered to be a major limitation for their use as therapeutic agents in biomedical research and the pharmaceutical industry. Cytoplasmic delivery is particularly important for immunotherapy, because cytosolic peptides are transported across the endoplasmic reticulum membrane with the help of the ATP-dependent transporters associated with antigen processing.Citation6 Peptides complexed with major histocompatibility complex class I molecules in the endoplasmic reticulum are then transported to the cell surface for recognition by cytotoxic T lymphocytes (CTLs).Citation7 Peptide-based vaccines have been tested in clinical trials but have encountered very limited success due to difficulties associated with stability and delivery, resulting in inefficient antigen presentation. The overall objective clinical response rate of peptide vaccine trials is in the range of only 4%–8%.Citation8–Citation10 In order to improve the efficiency of peptide-based vaccines, there is a significant need to design novel peptide delivery systems capable of inducing more effective tumor antigen-specific CTLs.
Nanoparticles (NPs) have been used as adjuvantsCitation11 (added to a vaccine to augment immune responses toward antigens). Although the adjuvanticity of NPs is not completely understood, it has been suggested that NPs can enhance antigen uptake and/or stimulate APCs.Citation12 NPs have gradually garnered a global reputation as an effective delivery system for therapeutic agents since they can be designed to slip between intercellular spaces, enter cells, or transport directly through biological barriers to access targeted sites.Citation13,Citation14 NPs also encapsulate therapeutic agents offering potential protection from enzymatic degradation, metabolism, and filtration. Poly(DL-lactide-co-glycolide) (PLGA) is a copolymer that allows controlled release of drug over time or in response to a biological cue.Citation15 PLGA has many advantages over other polymers used in drug and gene delivery including biodegradability, biocompatibility, and approval for human use granted by the US Food and Drug Administration (FDA).Citation16
We have successfully delivered plasmid DNA,Citation17–Citation19 drugs,Citation13,Citation19,Citation20 and peptidesCitation21 by using PLGA-NPs in our previous work. In this report, we explore a novel strategy for effective peptide delivery that enhances the cytoplasmic delivery of peptides into DCs, eliciting a more robust tumor antigen-specific CTL response both in vitro and in vivo.
Materials and methods
Materials
Poly(D,L-lactide-co-glycolide) (PLGA, molecular weight 23,000, copolymer ratio 50:50) was purchased from Birmingham Polymers, Inc (Birmingham, AL). Polyvinyl alcohol (PVA, average molecular weight 30,000–70,000), trifluoroacetic acid, acetonitrile, lipopolysaccharide, and L-15 (Leibovitz) medium were purchased from Sigma-Aldrich (St Louis, MO). Coumarin 6 was purchased from Polyscience, Inc (Warrington, PA). β2-Microglobulin was purchased from EMD Biosciences, (San Diego, CA). Human serum albumin was purchased from Instituto Grifols (Barcelona, Spain). Granulocyte-macrophage colony-stimulating factor, interleukin (IL)-2, IL-4 and IL-7 were purchased from R and D (Minneapolis, MN). Monclonal human anti-interferon (IFN)-γ antibody was purchased from Mabtech (Cincinnati, OH). Hoechst 33342 was purchased from Invitrogen (Carlsbad, CA). CD8 MicroBeads were purchased from Miltenyi Biotec (Auburn, CA). All peptides used in this study were synthesized by GenScript Corp (Piscataway, NJ). HLA-A2 (+) blood was purchased from San Diego Blood Bank and Scripps Green Hospital (San Diego, CA). T2, EL4 and transgenic adenocarcinoma of mouse prostate (TRAMP)-C2 cell lines were purchased from ATCC (Manassas, VA). Melanoma cell lines (624 and 1351) and TIL2080 (human tumor infiltrating lymphocyte) were kindly provided by Dr John R Wunderlich (National Institutes of Health/National Cancer Institute, Bethesda, MD). ELISpot plates were purchased from Millipore (Billerica, MA). ELISpot substrate was purchased from Vector Laboratories (Burlingame, CA). Standard lactate dehydrogenase release assay was purchased from Promega (Madison, WI). All salts used in the preparation of buffers were from Fisher Scientific (Pittsburgh, PA). All aqueous solutions were prepared with distilled and deionized water (WaterPro Plus; Labconco, Kansas City, MO). C57BL/6 mice were purchased from Jackson Laboratory (Bar Harbor, ME).
Formulation and characterization of PLGA-NPs
NPs were formulated as previously describedCitation13 with minor modifications. In brief, 30 mg PLGA and 600 μg peptide (melanoma antigen recognized by T-cells [MART]-1: 27–35, AAGIGILTV;Citation22 gp100: 154–162, KTWGQYWQV;Citation23 gp100: 209–217, ITDQVPFSV;Citation24 ovalbumin (OVA): 256–264, SIINFEKL, Citation25 and mouse six-transmembrane epithelial antigen of the prostate (mSTEAP): 326–335, DVSKINRTEMCitation26) in 1 mL of chloroform was emulsified in 6 mL of 2% PVA to form an oil-in-water emulsion. The emulsification was carried out using a micro-tip probe ultrasonic sonicator set at 55 watts of energy output (XL 2015 Sonicator® ultrasonic processor; Misonix, Inc, Farmingdale, NY) for 2 minutes over an ice bath. The emulsion was stirred overnight on a magnetic stir plate to allow evaporation of chloroform and formation of PLGA-NPs. PLGA-NPs were recovered by ultracentrifugation at 30,000 rpm for 30 minutes at 4°C (Beckman Optima™ LE-80 K, Beckman Instruments, Palo Alto, CA), washed twice with sterile nano water to remove PVA and unencapsulated peptide, and then lyophilized for 48 hours (VirTis Company, Freeze Dryer, Gardiner, NY). To determine cellular uptake of PLGA-NPs, the formulation contained coumarin 6. The dye solution (50 μg of coumarin 6 in 50 μL chloroform) was added to the polymer solution prior to emulsification.
For measuring the size of the PLGA-NPs, samples were sputter coated with gold/palladium and imaged with a scanning electron microscope (Phillips XL30, FEI, OR). Size distribution was analyzed using MetaMorph (Version 7.1.0.0, Molecular Devices Inc, Sunnyvale, CA). Zeta potential of the PLGA-NPs was measured using the Zetasizer® Nano ZS90 (Malvern Instruments, Malvern, UK).
Determination of peptide loading by high-performance liquid chromatography (HPLC)
Peptides and peptide-loaded PLGA-NPs were identified using ultraviolet detection at 280 nm. Briefly, 50 μL of PLGA-NP suspension was injected into a Varian HPLC system (Varian, Walnut Creek, CA) equipped with a C18 reverse phase column (8 × 100 mm, 15 μm particle size, pore size 300 A°) and a ultraviolet detector (Waters 486) set at 210 nm and a Gilson autoinjector (Mandel Scientific, Guelph, ON, Canada). The mobile phases employed were 10% ACN in water with 0.1% trifluoroacetic acid (Solvent A) and 70% ACN in water with 0.085% trifluoroacetic acid (Solvent B). The column was equilibrated with 75% A and 25% B prior to each run. Samples were eluted on a linear gradient of 25% to 100% B over 12 minutes at a fixed flow rate of 0.8 ml/min. The amount of peptide in each sample was calculated using a standard curve generated with known concentrations of the peptide.
Generation of human DCs and intracellular uptake of PLGA-NPs
Human DCs were generated by using enriched CD14+ monocytes from HLA-A2 (+) healthy donors purchased from San Diego Blood Bank and Scripps Green Hospital with the presence of granulocyte-macrophage colony-stimulating factor (1000 U/mL) and recombinant IL-4 (400 U/mL) for 7 days to produce immature DC (imDCs). Human imDCs at 50,000/mL were incubated with PLGA-NPs in medium (100 μg/mL) in a 12-well plate for 30 minutes, respectively. Cells were then washed with cold phosphate buffered saline (PBS) to remove uninternalized PLGA-NPs. Cell nuclei were stained with trihydrochloride (Hoechst 33342; Invitrogen, Carlsbad, CA) and washed with PBS before subjected to fluorescence-activated cell sorting analysis and imaging by a confocal microscope (Leica TCS SP2; Leica Microsystems Inc, Buffalo Grove, IL).
Antigen presentation by human mature DCs (mDCs) loaded with PLGA-NPs encapsulating MART-1
Human imDCs at day 7 were incubated with control PLGA-NPs (empty-NPs), or PLGA-NPs encapsulating MART-1 peptide for 1 hour, followed by the addition of lipopolysaccharide for maturation. Mature DCs (mDCs) pulsed with MART-1 (5 μg/mL) served as the positive control. Test and control mDCs were cultured with TIL2080 cells at a ratio of 1:1 for 20 hours. The efficiency of the antigen presentation was measured by IFN-γ ELISpot assay.
ELISpot assay
To test whether human mDCs loaded with PLGA-NPs encapsulating MART-1 present antigens more efficiently to T cells than do MART-1 peptide-pulsed human mDCs, IFN-γ ELISpot assay was performed. Briefly, 96-well ELISpot plates were coated with 10 μg/mL monoclonal human anti-IFN-γ antibody (Mabtech) at 4°C overnight. The plates were then blocked with 10% human AB serum (Omega Scientific, Tarzana, CA) for 2 hours at 37°C. Human mDCs (2 × 104) loaded with PLGA-NPs encapsulating MART-1 and MART-1 peptide-pulsed human mDCs (100 ng/mL) were incubated with TIL2080 cells, which recognize MART-1 peptide, at a ratio of 1:1 in CTL medium in the ELIspot plates at 37°C for 20 hours. All cultures were carried out in triplicate. The plates were developed using 100 μL/well of substrate solution. Spots on the membrane were counted using automated image analysis system (CTL Analyzers LLC, Cleveland, OH).
Generation of tumor antigen-specific CTLs
Human mDCs loaded with PLGA-NPs encapsulating peptides were used as the APCs. In brief, NP-loaded DCs were added to enriched CD8+ cells derived from HLA-A2 (+) donors at a ratio of 1 to 10 in CTL medium (RPMI 1640 with 10% human AB serum). IL-2 (20 U/mL) and IL-7 (30 U/ mL) were added 3 days later. Restimulation was first carried out on day 7 as follows: β2-microglobulin (5 μg/mL) and corresponding peptides (5 μg/mL) were added to irradiated autologous CD8− cells and incubated for 2 hours at 37°C. Four milliliters of L-15 (Leibovitz) medium containing 1% human serum albumin, β2-microglobulin and peptide was added to the adherent cells and incubated for 1.5 hours at 25°C. Effector cells were added at 2 × 106 cells/mL in fresh CTL medium. The second restimulation was carried out on day 14. On day 21 tumor antigen-specific CTLs were tested by ELISpot and cytotoxicity assay (CytoTox 96® Non-Radioactive Cytotoxicity Assay [Promega, Fitchburg, WI]).
Cytotoxic assay
T2 cells pulsed with corresponding peptides and melanoma cells were used as target cells to test peptide-specific CTLs. EL4 pulsed with OVA, or mSTEAP peptide and TRAMP-C2 cells were used as target cells to test the immune response elicited by vaccination of PLGA-NPs encapsulating peptide and peptide emulsified in incomplete Freund’s adjuvant (IFA). For cellular cytotoxicity assays, a standard lactate dehydrogenase release assay was performed according to the manufacturer’s instructions. In brief, 2 × 104 target cells were added to the peptides-specific CTLs at the ratios of 1:12.5, 1:25, and 1:50 in a final volume of 100 μL. The plates were incubated for 3 hours and 15 minutes at 37°C after spin down. Ten microliters of 10× lysis buffer was added into the wells for maximum release, and incubation continued for 45 minutes at 37°C. Fifty microliters of supernatant was transferred to a fresh ELISA plate after spin down and 50 μL substrate was added to each well and mixed, and the plate was sealed. The mixture was incubated for 30 minutes at room temperature in the dark, and 50 μL stop solution was added to each well. The plate was read at 490 nm and the specific lysis was calculated by the following formula: % Cytotoxicity = 100 × (Experiment – Effector Spontaneous – Target Spontaneous)/ (Target Maximum – Target Spontaneous).Citation27
In vivo distribution of PLGA-NPs after intraperitoneal (IP) injection
Coumarin 6-loaded PLGA-NPs were injected into C57BL/6 mice at 500 μg in 500 μL PBS by IP injection. The mice were sacrificed 30 minutes after injection and imaged using the Olympus OV100 imaging system (Olympus Corp, Shinjuku-ku, Japan).
Comparison of peptide delivery efficiency in vivo
Peptide delivery efficiency was measured in vivo using mSTEAP (a mouse peptide with 80% homology to human STEAPCitation28) and OVA peptide as test and positive control peptides, respectively. For free peptide immunization, 100 μg of mSTEAP or OVA was emulsified in IFA and delivered via subcutaneous (SC) injection. IP injection was performed for empty-NP- and peptide- (both OVA and mSTEAP) loaded PLGA-NPs.
Thirty-five male C57BL/6 mice were randomized into the following five groups: (1) Control NP (empty-NP), (2) OVA emulsified in IFA (OVA + IFA), (3) mSTEAP emulsified in IFA (mSTEAP + IFA), (4) PLGA-NPs encapsulating OVA, and (5) PLGA-NPs encapsulating mSTEAP. All mice were immunized once on day 0 (). Ten days after immunization, three mice in each group were sacrificed followed by in vivo restimulation of splenocytes with the same peptides used for priming. Cytolytic activity of the CTLs was assayed 6 days after restimulationCitation27 using a standard cytotoxic assay with the following targets: (1) EL-4 cells; (2) EL-4 cells pulsed with OVA peptide; (3) EL-4 cells pulsed with mSTEAP peptide; and (4) TRAMP-C2 cells.
Table 1 Vaccination of C57BL/6 mice in different groups
Tumor size in immunized mice after challenge
Four C57BL/6 mice immunized in each group were challenged with 1 × 106 of TRAMP-C2 on day 10 via SC injection in the right flank, and tumor size was monitored with a digital caliper (Thermo Fisher Scientific, Pittsburgh, PA) every 3 days.
Statistical analysis
P-values were calculated to test the statistical significance between the groups of free peptides and PLGA-NPs encapsulating peptides using two-tailed unpaired Student’s t-test. The confidence interval was set at 95% and a P value of <0.05 was accepted as significant (*); P < 0.01 (**), P < 0.001 (***).
Results
Characterization of peptide-loaded PLGA-NPs
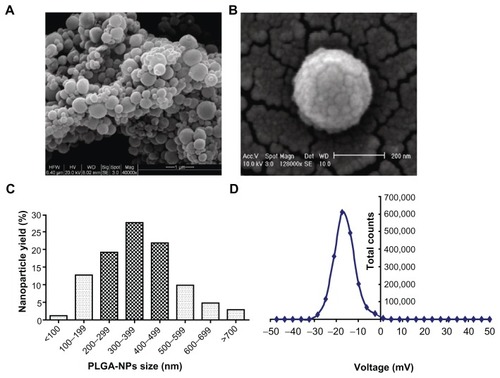
NPs are colloidal systems with a size range typically from 1 to 1000 nm in diameter. They are formulated from a polymer, in which the therapeutic agents were entrapped in the polymer matrix. The double emulsion-solvent technique is the most commonly used method to formulate polylactic acid (PLA) and PLGA NPs, with PVA to stabilize the emulsion. PLGA-NPs were sputter coated with gold/palladium and imaged using a scanning electron microscope under 20 kV () and 10 kV (), respectively. The unfractionated PLGA-NPs demonstrated a size distribution range from 70 nm to 795 nm, and 78% of these PLGA-NPs are from 150 nm to 500 nm in diameter (). The PLGA-NPs were negatively charged at pH 7.4 with a mean zeta potential of –15.53 ± 0.71 mV (, n = 3), and the polydispersity index (PDI) is 0.308 ± 0.034 (n = 3) when analyzed by Zetasizer Nano ZS90. The size and potential of the PLGA-NPs used in this study varied slightly between batches, and also varied according to the different peptide(s) encapsulated. MART-1 and gp100:154–162 peptide-loaded PLGA-NPs are the representatives, which were used in the characterization of the PLGA-NPs in .
Figure 1 Characterization of PLGA-NPs. Images were taken by a scanning electron microscope (Phillips XL 30, FEI, OR). (A) Under 20 kV (40,000×). (B) under 10 kV (128,000×). (C) Size range of unfractionated PLGA-NPs. (D) Zeta potential of PLGA-NPs.
Abbreviation: PLGA-NPs, poly(DL-lactide-co-glycolide) nanoparticles.
Peptide loading and encapsulation in PLGA-NPs
Controlling both the drug-loading efficiency and particle size of the drug-loaded NPs is very important when using NPs as drug delivery systems.Citation29 To measure the peptide loading and encapsulation efficiency, we defined peptide loading as the peptide present (μg) in 1 mg PLGA-NPs, and the encapsulation efficiency stands for the percentage of peptide entrapped in PLGA-NPs over the initial amount of loaded peptide. Based on these definitions and the HPLC-analyzed results, the PLGA-NP peptide loading is 3.176 ± 0.144 μg (n = 3), and the peptide encapsulation efficiency (%) is 82.34% ± 8.4% (n = 3).
Cellular uptake of PLGA-NPs in human DCs
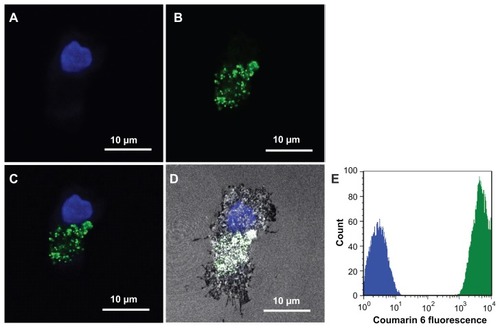
Next, we studied whether the NP-loaded DCs could present the antigenic peptides to T cells more efficiently, leading to the induction of antigen-specific CTLs for immunotherapy. Coumarin 6 loaded PLGA-NPs were incubated with human imDCs for 1 hour and cell nuclei were stained with Hoechst 33342. Images were taken under a confocal microscope, as shown in (4′,6-diamidino-2-phenylindole channel), (fluorescein isothiocyanate channel), (both 4′,6-diamidino- 2-phenylindole and fluorescein isothiocyanate channels overlaid), and (4′,6-diamidino- 2- phenylindole, fluorescein isothiocyanate, and reflection channels overlaid). Fluorescent signals were seen in the cytoplasm of human imDCs. To test uptake efficiency, human imDCs were next incubated with coumarin 6-loaded PLGA-NPs for 30 minutes, and washed twice with Hank’s buffered salt solution to remove uninternalized PLGA-NPs for fluorescence-activated cell sorting analysis. The result showed that 100% of human DCs were fluorescent positive ().
Figure 2 Colocalization of poly(DL-lactide-co-glycolide) nanoparticles in human dendritic cells. Images were taken under a confocal microscope. (A) 4′,6-Diamidino-2- phenylindole channel. (B) Fluorescein isothiocyanate channel. (C) 4′,6-Diamidino-2-phenylindole and fluorescein isothiocyanate channels overlaid. (D) 4′,6-Diamidino-2- phenylindole, fluorescein isothiocyanate, and reflection channels overlaid. (E) Fluorescence-activated cell sorting analysis result.
Antigen presentation comparison between human mDCs loaded with PLGA-NPs encapsulating peptide and those pulsed with free peptide
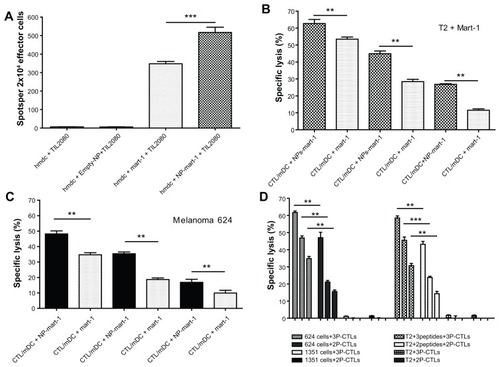
MART-1 peptide is the most well characterized and most commonly used epitope in melanoma studies, and is the target of tumor-infiltrating lymphocytes.Citation30 TIL2080 cell line was used as effector cells to recognize and kill human mDCs loaded with PLGA-NPs encapsulating MART-1, or MART-1 peptide-pulsed human mDCs while being the target cells. The results showed that human mDCs loaded with PLGA-NPs encapsulating MART-1 presented more MART-1 peptides to TIL2080 cells than those pulsed with MART-1 peptide, resulted in more release of IFNγ, as measured by ELISpot assay (, P = 0.0003).
Figure 3 Enhanced antigen presentation and cytotoxic T lymphocyte (CTL) activity comparison. (A) ELISpot assay to test interferon-γ release when human mature dendritic cells presented melanoma antigen recognized by T-cell (MART)-1 peptide to TIL2080 cells. (B) CTL activity comparison between the CTLs generated by human mature dendritic cells loaded with NPs encapsulating MART-1 peptide and free MART-1 peptide-pulsed human mature dendritic cells when the target cells were T2 cells pulsed with MART-1 peptide. (C) CTL activity comparison when the target cells were melanoma 624 cells. (D) Cytotoxic activity comparison of the CTLs generated by poly (DL-lactide-co-glycolide nanoparticles encapsulating the two-peptide (MART-1:27–35 and gp100:154–162) and three-peptide (MART-1:27–35, gp100:154–162, and gp100:209–217) cocktails.
Notes: The confidence interval was set at 95% and a P value of <0.05 was accepted as significant; **P < 0.01, ***P < 0.001.
Abbreviations: CTL, cytotoxic T lymphocyte; PLGA-NPs, poly(DL-lactide-co-glycolide) nanoparticles; hmDC, human mature dendritic cells.
Comparison of CTL cytotoxic activities
The cytototoxic activity of tumor antigen-specific CTLs generated by using both human mDCs loaded with PLGA-NPs encapsulating MART-1 and human mDCs pulsed with MART-1 peptide were tested using cytotoxic assays. All the CTLs generated using the above methods killed both T2 cells pulsed with MART-1 () and melanoma 624 cells (). The results demonstrated that the cytototoxic activity of the tumor antigen-specific CTLs generated by human mDCs loaded with PLGA-NPs encapsulating MART-1 peptide (CTL/mDC + NP-MART-1) was significantly greater than that generated by human mDCs pulsed with free peptide (CTL/mDC + MART-1) when the ratio of effector/target was both 50/1 and 25/1 (). The cytototoxic activity of CTL/mDC + NP-MART-1 was still significantly stronger than CTL/mDC-MART-1 when the ratio of effector/target was 12.5/1 while the targets were MART-1-pulsed T2 cells (), along with targets that were melanoma 624 cells (). However, these CTLs could not recognize and kill T2 cells and 1351 melanoma cells due to lack of MART-1 antigen expression on their cell surfaces (data not shown).
Most interestingly, the CTLs induced by human mDCs loaded with PLGA-NPs encapsulating a three-peptide cocktail (3P, including MART-1:27–35, gp100:154–162 and gp100: 209–217) generated significantly more robust cytotoxic activity than those encapsulating a two-peptide cocktail (2P, including MART-1:27–35 and gp100:154–162) when the targets were melanoma 624 cells and T2 cells pulsed with the peptide-cocktails at any effector/target ratio (, P < 0.01). These CTLs are not able to recognize and kill 1351 melanoma cells and T2 cells owing to lack of antigen expression on their cell surfaces ().
Distribution of the PLGA-NPs in vivo after IP injection
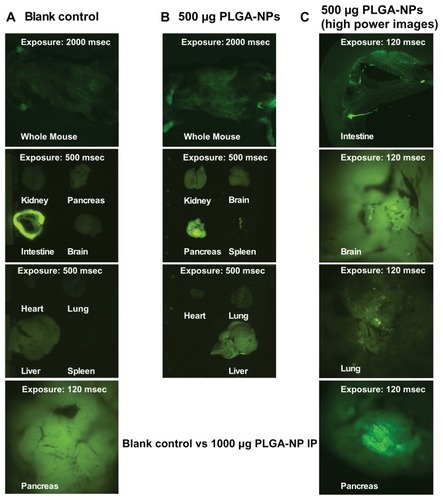
The three main types of professional APCs are macrophages, DCs, and B cells. Macrophages are predominantly located in the lungs, liver, neural tissue, bone, and spleen; imDCs are mostly located in the epithelium of the skin, the gastrointestinal tract and the respiratory tract. Most of the B cells are located in bone marrow and spleen. Thus, these organs were chosen for the biodistribution study. Fluorescent signals were compared between the blank control mouse and the mouse dosed with 500 μg coumarin 6-loaded PLGA-NPs 30 minutes after IP injection. Scattered fluorescent particles were seen under the skin all over the mouse body, and these signals were also seen in the kidney, brain, pancreas, spleen, heart, lung, and liver (), but were not seen in the blank control mouse, apart from autofluorescence in the paws and intestine (). Enlarged images clearly showed that the fluorescent PLGA-NPs are distributed to various organs, including the intestine, brain, lung, and pancreas ().
Figure 4 Distribution of poly(DL-lactide-co-glycolide) nanoparticles in vivo after intraperitoneal injection. (A) Blank control mouse. (B) Test mouse dosed with 500 μg poly(DL-lactide-co-glycolide) nanoparticles (PLGA-NPs) 30 minutes after intraperitoneal injection. (C) High-power mouse organ images. Coumarin 6-loaded poly(DL-lactideco- glycolide) nanoparticles were injected into C57BL/6 mice at 500 μg in 500 μL phosphate-buffered saline by intraperitoneal injection.
Note: The mice were sacrificed 30 minutes after injection and imaged using the Olympus OV100 imaging system (Olympus Corp, Shinjuku-ku, Japan).
Tumor suppression in mice immunized with PLGA-NPs encapsulating mSTEAP peptide
Based on observation records, tumor size measurement, and statistical data, delayed tumor development was observed in mice immunized with PLGA-NPs encapsulating mSTEAP peptide, and the tumor size was significantly smaller than in those in the control group immunized with empty-NP (P = 0.019, ). The difference in tumor size between the mice immunized with mSTEAP peptide emulsified in IFA and PBS was not significant (P = 0.361, ). Tumor size difference between the mice immunized with PLGA-NPs encapsulating mSTEAP peptide and those immunized with mSTEAP peptide emulsified in IFA did not reach significance, but there was a trend to smaller tumor size (P = 0.123) in the former.
Figure 5 Immunization with PLGA-NPs encapsulating mSTEAP peptide elicited stronger immune response. (A) Tumor suppression was observed in the mice immunized with PLGA-NPs encapsulating mouse six-transmembrane epithelial antigen of the prostate (mSTEAP) peptide (●) when compared with mSTEAP peptide emulsified in incomplete Freund’s adjuvant (○). (B) Ovalbumin (OVA)-specific CTLs derived from the mice immunized with PLGA-NPs encapsulating OVA lysed a significantly higher percentage of EL4 cells pulsed with OVA than those immunized with OVA emulsified in IFA (■ vs ○). The CTLs derived from the mice immunized with empty-NP against EL4 cells pulsed with OVA (◆). Mice immunized with PLGA-NPs encapsulating mSTEAP peptide induced a more effective immune response, not only lysed significantly higher percentages of EL4 cells pulsed with mSTEAP peptide (C), but transgenic adenocarcinoma of mouse prostate-C2 cells than those derived from the mice immunized with mSTEAP emulsified in incomplete Freund’s adjuvant (D).
Abbreviation: PLGA-NPs, poly(DL-lactide-co-glycolide) nanoparticles.
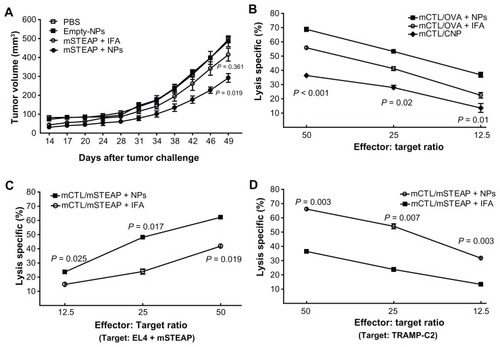
PLGA-NPs encapsulating peptides induce a significantly stronger antitumor response
We next sought to address the question of whether PLGA-NPs encapsulating peptides can be taken up by professional APCs (DCs, macrophages, and B cells) in vivo, which can then present the loaded peptides to T cells, and eventually induce an effective immune response. To answer this, 35 male C57BL/6 mice were immunized with their corresponding agents only once followed by in vitro restimulation of splenocytes with the same peptides used for priming. We evaluated the lysis patterns of each agent on target cells. The results showed that mouse CTLs derived from the mice immunized with empty-NPs, if pulsed with SIINFEKL peptide (OVA, 5 μg/mL) in vitro, were able to recognize and kill EL4 cells pulsed with OVA (◆ in ).
CTL activity in the mice immunized with PLGA-NPs encapsulating peptides was significantly higher than in those immunized with peptides emulsified in IFA. The difference in CTL activity between the groups of OVA + PLGA-NPs and OVA + IFA was 68.76% vs 55.19% (P = 0.0007), 53.25% vs 39.05% (P = 0.0185) and 34.58% vs 20.74% (P = 0.0110) when the effector/target ratio was 50/1, 25/1 and 12.5/1, respectively, while the target cells were EL4 cells pulsed with OVA peptide ().
We also compared the CTL activity between the groups of mSTEAP + PLGA-NPs and mSTEAP + IFA; the difference was 62.19% vs 41.81% (P = 0.025), 48.07% vs 23.91% (P = 0.017), and 23.6% vs 14.85% (P = 0.019) when the effector/ target ratio was 50/1, 25/1 and 12.5/1, respectively, while the target cells were EL4 cells pulsed with mSTEAP peptide (). When the target cells were TRAMP-C2 cells, the difference in CTL activity was 66.12% vs 36.41% (P = 0.003), 54.07% vs 23.79% (P = 0.007), and 31.73% vs 13.35% (P = 0.003) while the effector/target ratio was 50/1, 25/1 and 12.5/1, respectively ().
Most importantly, the peptide dose encapsulated in PLGA-NPs was 63 times less than that emulsified in IFA, but it induced a more robust CTL response. The lysis capacity of the CTLs induced by PLGA-NPs encapsulating mSTEAP peptide was 1.82–2.22-fold higher when the effector/ target ratio was 12.5/1 to 50/1 than of the CTLs induced by mSTEAP peptide emulsified in IFA while the targets were TRAMP-C2 cells.
Discussion
The successful generation of an effective immune response against tumor cells is a prerequisite for any immunotherapeutic strategy for cancer.Citation31 Boosting the immune system with tailored immunotherapies provides a promising alternative to standard chemo- and radiotherapy and will ideally yield a specific anticancer response with fewer side effects.Citation32 In the field of cancer immunotherapy, most enthusiasm has been directed to the use of cancer vaccines.Citation8 Peptide-based vaccines are more attractive than other forms including whole cell vaccines, protein vaccines, and DNA-based vaccines. However, one difficulty for peptide-based vaccines is their rather poor immunogenicity,Citation33 thus, appropriate adjuvants are greatly needed to make them more potent in eliciting effective T cell responses.
IFA serves as a conventional carrier for peptide delivery, yet the response rate of peptide emulsified in IFA trials in 246 individuals was only 3.66%.Citation8 To improve cancer prevention and treatment, specific immune-activating strategies must be investigated. Adjuvants are usually pharmacological or immunological agents which modify the effect of other agents (eg, drugs or vaccines) while having few, if any, direct effects when given alone. For standard prophylactic immunization in healthy individuals, only adjuvants that induce minimal side effects will prove to be acceptable. Adjuvants for cancer vaccines, nevertheless, need to be very potent, which may increase their toxicity and the induction of autoimmune reactions.Citation34 Particulate vaccine carriers offer a good balance between adjuvanticity and safety. In this study, we demonstrated that peptide-loaded PLGA-NPs mediated efficient peptide delivery to APC and induced tumor antigen-specific CTLs and effective prophylactic immune responses in vivo.
Particulate adjuvants have comparable dimensions to the pathogens recognized by the immune system (<5 μm), which offers a great advantage in vaccine technology.Citation35 Particles are internalized by APCs via different mechanisms, depending on their size.Citation36 Particles of 20–200 nm are usually taken up via endocytosis by DCs, while those of 0.5–5 μm are primarily taken up by macrophages via macropinocytosis or phagocytosis.Citation32 We demonstrated by fluorescence-activated cell sorting that PLGA-NPs could be taken up by 100% of human DCs within 30 minutes after incubation. Importantly, more effective tumor antigen-specific CTLs were induced by using human DCs loaded with PLGA-NPs encapsulating peptide than human DCs pulsed with free peptide.
From our study of the distribution of PLGA-NPs after IP injection, we found that fluorescent PLGA-NPs are present in various organs, including the liver, spleen, brain, lung, etc (). It was reported that PLGA NPs of 105 nm were present in the blood at higher concentrations for up to 24 hours and were able to reduce their uptake by the reticuloendothelial system, compared with that of etoposide-loaded PLGA NPs of 160 nm and pure drug.Citation37 For therapeutic targeting, PLGA-NPs should evade the reticuloendothelial system to achieve highest drug delivery efficiency. For example, PLGA-NPs of size 100 nm can be used for long-term circulation without the need for surface modification,Citation38 and the nanocarriers must be hidden from the reticuloendothelial system because it could destroy any foreign material through opsonization, followed by phagocytosis by macrophages.Citation39,Citation40 The most important finding of our study is the in vivo targeting of the APCs, which could phagocytize the antigen-loaded PLGA-NPs after IP administration and present the antigens to naïve T cells for generating an effective immune response. Interestingly, we found that PLGA-NPs cross the blood–brain barrier (BBB) in our study; another group also reported that the PLGA-NPs were concentrated mainly in the hippocampus.Citation41 The mechanism might be taking advantage of the leaky vessel walls by passive diffusion or convection, dependent on the hydrostatic and osmotic pressure differences between blood and interstitial space.
Among different polymers, PLGA and PLA have been extensively used as NP carriers in controlled-release delivery systems for many bioactive molecules due to their low toxicity, good bioavailability and biocompatibility, and FDA approval status. The bioactive molecules delivered include drugs,Citation13,Citation42–Citation44 proteins,Citation45,Citation46 DNA,Citation47–Citation49 and oligonucleotides.Citation50–Citation52 Only a few studies have shown peptide delivery by PLGA-NPs to elicit a type 1 T helper immune response.Citation53–Citation55 We used the same PLGA polymer and double emulsion-solvent evaporation method, but a different protocol and with entrapped peptides, thus producing PLGA-NPs products with different characteristics including size, potential and polydispersity, etc. In other studies, particle size ranged from 1 to 10 μm,Citation56 350 to 410 nm,Citation55 and 290 to 350 nmCitation57 in diameter. Encapsulation efficiency was 23% for OVA peptideCitation56, 5.2% ± 0.6% for TRP2,Citation55 and 67.3% ± 6.9% for 7-acyl lipid A,Citation55 and the peptide loading was 0.03 μg for OVA peptide,Citation56 0.94 ± 0.11 μg for TRP2,Citation55 and 1.79 ± 0.18 μg for 7-acyl lipid A,Citation55 respectively. Our results showed a size range for unfractionated PLGA-NPs from 70 nm to 795 nm; 78% of these PLGA-NPs sized from 150 nm to 500 nm in diameter. Based on HPLC-analyzed results, our peptide loading was 3.176 ± 0.144 μg (n = 3), and the peptide encapsulation efficiency was 82.34% ± 8.4% (n = 3). Our data are consistent with above results, but the peptide loading and encapsulation efficiency of our PLGA-NP products was much higher than that of other authors. These differences may be due to a more optimized delivery system because we found the PLGA-NPs, which were loaded with more peptide (1 mg/batch), did not function better than those loaded with less peptide (600 μg/batch, data not shown).
For tumor antigen-specific CTL induction, Hamdy et al utilized C57BL/6 mice immunized SC with 10 μg tyrosinase-related protein-2 (TRP2) encapsulated in PLGA-NPs, versus mice who received 10 mg plain PLGA-NPs (empty-NPs) SC on day 0, and boosted on day 11.Citation55 Their results showed activated TRP2-specific CD8+ T cell responses in the draining lymph nodes and spleen of mice immunized with PLGA-NPs encapsulating TRP2.Citation55
In our study, the mice were immunized only once with 500 μg PLGA-NPs encapsulating 1.588 μg mSTEAP peptide, 500 μg control PLGA-NPs by IP injection, or 100 μg mSTEAP peptide emulsified in 100 μL IFA by SC injection. Most importantly, the peptide dose encapsulated in PLGA-NPs was 63 times less than that emulsified in IFA, but it induced more robust CTLs with the lysis rate of 66.12% vs 36.41%; 55.075% vs 24.795%, and 31.73% vs 14.85% when the target cells were TRAMP-C2 cells, and the effector/target ratio was 50/1, 25/1, and 12.5/1, respectively (). We found that tumors in the mice immunized with PLGA-NPs encapsulating mSTEAP developed later and were significantly smaller than tumors in the mice immunized with mSTEAP + IFA (P = 0.019). CTL activity in the mice immunized with PLGA-NPs encapsulating peptide was much higher than that in the mice immunized with peptide emulsified in IFA.
Antigen presentation is a crucial step in the initiation of an effective immune response. DCs have a unique ability to efficiently present antigens and thus play a central role in the orchestration of the adaptive immune response. This has made these cells a major focus of interest in the conception of immunotherapeutic vaccine strategies. An immunization strategy using PLGA-NPs encapsulating antigenic peptides is an efficient approach for antigen presentation that induces more effective tumor antigen-specific immune responses than free peptide emulsified in IFA. These findings provide a good rationale for using PLGA-NPs as competent carriers for future peptide-based cancer vaccine formulations.
Conclusion
The majority of the PLGA-NPs made in this study measured from 150 nm to 500 nm in diameter, and the mean zeta potential was –15.53 mV. The PLGA-NPs were able to be colocalized in human DCs within 30 minutes of incubation. As the APCs, human DCs loaded with PLGA-NPs encapsulating peptide induced a significantly stronger CTL cytotoxicity than those pulsed with free peptide in vitro. Much higher efficiency of peptide delivery (63 times) was found in PLGA-NPs when compared with IFA. PLGA-NPs, as a peptide carrier, induce a more powerful CTL response in vivo. These findings are fundamental for the development of peptide vaccines, and the induction of an effective immune response for cancer immunotherapy.
Acknowledgments
This work was supported by grants from the Department of Defense PC041024 (B. Minev), and NCI grants U54CA132384 and U54CA132379 (Minev/Grotjahn), and RO1CA154256 (Minev/Kruse). The authors would like to thank Ronnie Fang and Liangfang Zhang at UCSD Moores Cancer Center for their excellent help on the characterization of the PLGA-NPs.
Disclosure
The authors declare that they have no competing financial interests in this work.
References
- RosenbergSAProgress in human tumour immunology and immunotherapyNature2001411683538038411357146
- SloanJMKershawMHTouloukianCEMHC class I and class II presentation of tumor antigen in retrovirally and adenovirally transduced dendritic cellsCancer Gene Ther200291194695012386833
- SteinmanRMLasker Basic Medical Research Award. Dendritic cells: versatile controllers of the immune systemNat Med200713101155115917917664
- EpaulardOToussaintBQueneeLAnti-tumor immunotherapy via antigen delivery from a live attenuated genetically engineered Pseudomonas aeruginosa type III secretion system-based vectorMol Ther200614565666117010670
- MurthyNCampbellJFaustoNHoffmanASStaytonPSBioinspired pH-responsive polymers for the intracellular delivery of biomolecular drugsBioconjug Chem200314241241912643752
- SpiesTCerundoloVColonnaMCresswellPTownsendADeMarsRPresentation of viral antigen by MHC class I molecules is dependent on a putative peptide transporter heterodimerNature199235563616446461538752
- LehnerPJCresswellPProcessing and delivery of peptides presented by MHC class I moleculesCurr Opin Immunol19968159678729447
- RosenbergSAYangJCRestifoNPCancer immunotherapy: moving beyond current vaccinesNat Med200410990991515340416
- NoguchiMMineTKomatsuNAssessment of immunological biomarkers in patients with advanced cancer treated by personalized peptide vaccinationCancer Biol Ther201110121266127920935522
- BrunsvigPFKyteJAKerstenCTelomerase peptide vaccination in NSCLC: a phase II trial in stage III patients vaccinated after chemoradiotherapy and an 8-year update on a phase I/II trialClin Cancer Res201117216847685721918169
- GreenlandJRLetvinNLChemical adjuvants for plasmid DNA vaccinesVaccine200725193731374117350735
- DobrovolskaiaMAMcNeilSEImmunological properties of engineered nanomaterialsNat Nanotechnol20072846947818654343
- SahooSKMaWLabhasetwarVEfficacy of transferrin-conjugated paclitaxel-loaded nanoparticles in a murine model of prostate cancerInt J Cancer2004112233534015352049
- YamamotoHKunoYSugimotoSTakeuchiHKawashimaYSurface- modified PLGA nanosphere with chitosan improved pulmonary delivery of calcitonin by mucoadhesion and opening of the intercellular tight junctionsJ Control Release2005102237338115653158
- ZhangNChittasuphoCDuangratCSiahaanTJBerklandCPLGA nanoparticle–peptide conjugate effectively targets intercellular cell-adhesion molecule-1Bioconjug Chem200819114515217997512
- CampolongoMJLuoDDrug delivery: old polymer learns new tractsNat Mater20098644744819458640
- PrabhaSLabhasetwarVNanoparticle-mediated wild-type p53 gene delivery results in sustained antiproliferative activity in breast cancer cellsMol Pharm20041321121915981924
- PrabhaSZhouWZPanyamJLabhasetwarVSize-dependency of nanoparticle-mediated gene transfection: studies with fractionated nanoparticlesInt J Pharm20022441–210511512204570
- PanyamJZhouWZPrabhaSSahooSKLabhasetwarVRapid endo-lysosomal escape of poly(DL-lactide-co-glycolide) nanoparticles: implications for drug and gene deliveryFASEB J200216101217122612153989
- VasirJKLabhasetwarVTargeted drug delivery in cancer therapyTechnol Cancer Res Treat20054436337416029056
- MaWSmithTBoginVEnhanced presentation of MHC class Ia, Ib and class II-restricted peptides encapsulated in biodegradable nanoparticles: a promising strategy for tumor immunotherapyJ Transl Med201193421450109
- MinevBHippJFiratHSchmidtJDLanglade-DemoyenPZanettiMCytotoxic T cell immunity against telomerase reverse transcriptase in humansProc Natl Acad Sci U S A20009794796480110759561
- de VriesIJLesterhuisWJBarentszJOMagnetic resonance tracking of dendritic cells in melanoma patients for monitoring of cellular therapyNat Biotechnol200523111407141316258544
- MailliardRBEgawaSCaiQComplementary dendritic cell-activating function of CD8+ and CD4+ T cells: helper role of CD8+ T cells in the development of T helper type 1 responsesJ Exp Med2002195447348311854360
- CraiuAAkopianTGoldbergARockKLTwo distinct proteolytic processes in the generation of a major histocompatibility complex class I-presented peptideProc Natl Acad Sci U S A1997942010850108559380723
- de Garcia-HernandezMLGrayAHubbyBKastWMIn vivo effects of vaccination with six-transmembrane epithelial antigen of the prostate: a candidate antigen for treating prostate cancerCancer Res20076731344135117283172
- MaWYuHWangQJinHSolheimJLabhasetwarVA novel approach for cancer immunotherapy: tumor cells with anchored superantigen SEA generate effective antitumor immunityJ Clin Immunol200424329430115114060
- YangDHoltGEVeldersMPKwonEDKastWMMurine six-transmembrane epithelial antigen of the prostate, prostate stem cell antigen, and prostate-specific membrane antigen: prostate-specific cell-surface antigens highly expressed in prostate cancer of transgenic adenocarcinoma mouse prostate miceCancer Res200161155857586011479226
- ItoFFujimoriHMakinoKFactors affecting the loading efficiency of water-soluble drugs in PLGA microspheresColloids Surf B Biointerfaces2008611252917719753
- RosenbergSAKawakamiYRobbinsPFWangRIdentification of the genes encoding cancer antigens: implications for cancer immunotherapyAdv Cancer Res1996701451778902056
- RewSBPeggsKSanjuanIGeneration of potent antitumor CTL from patients with multiple myeloma directed against HM1.24Clin Cancer Res20051193377338415867238
- XiangSDScalzo-InguantiKMinigoGParkAHardyCLPlebanskiMPromising particle-based vaccines in cancer therapyExpert Rev Vaccines2008771103111918767957
- MandelboimOVadaiEFridkinMRegression of established murine carcinoma metastases following vaccination with tumour-associated antigen peptidesNat Med1995111117911837584991
- MesaCFernandezLEChallenges facing adjuvants for cancer immunotherapyImmunol Cell Biol200482664465015550123
- SinghMO’HaganDAdvances in vaccine adjuvantsNat Biotechnol199917111075108110545912
- FifisTGamvrellisACrimeen-IrwinBSize-dependent immunogenicity: therapeutic and protective properties of nano-vaccines against tumorsJ Immunol200417353148315415322175
- YadavKSChuttaniKMishraAKSawantKKEffect of Size on the Biodistribution and Blood Clearance of Etoposide-Loaded PLGA NanoparticlesPDA J Pharm Sci Technol201165213113921502074
- LiSDHuangLNanoparticles evading the reticuloendothelial system: role of the supported bilayerBiochim Biophys Acta20091788102259226619595666
- AlexisFPridgenEMolnarLKFarokhzadOCFactors affecting the clearance and biodistribution of polymeric nanoparticlesMol Pharm20085450551518672949
- AcharyaSSahooSKPLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effectAdv Drug Deliv Rev201163317018320965219
- TsaiYMChienCFLinLCTsaiTHCurcumin and its nano-formulation: the kinetics of tissue distribution and blood–brain barrier penetrationInt J Pharm2011416133133821729743
- GuFZhangLTeplyBAPrecise engineering of targeted nanoparticles by using self-assembled biointegrated block copolymersProc Natl Acad Sci U S A200810572586259118272481
- DharSGuFXLangerRFarokhzadOCLippardSJTargeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticlesProc Natl Acad Sci U S A200810545173561736118978032
- BennyOMenonLGArielGLocal delivery of poly lactic-co-glycolic acid microspheres containing imatinib mesylate inhibits intracranial xenograft glioma growthClin Cancer Res20091541222123119190128
- Kaiser-SchulzGHeitAQuintanilla-MartinezLPolylactide-coglycolide microspheres co-encapsulating recombinant tandem prion protein with CpG-oligonucleotide break self-tolerance to prion protein in wild-type mice and induce CD4 and CD8 T cell responsesJ Immunol200717952797280717709493
- WangXWenkEHuXSilk coatings on PLGA and alginate microspheres for protein deliveryBiomaterials200728284161416917583788
- TseMTBlatchfordCOya AlparHEvaluation of different buffers on plasmid DNA encapsulation into PLGA microparticlesInt J Pharm20093701–2334019059325
- PeartonMAllenderCBrainKGene delivery to the epidermal cells of human skin explants using microfabricated microneedles and hydrogel formulationsPharm Res200825240741617671832
- JesselNOulad-AbdelghaniMMeyerFMultiple and time-scheduled in situ DNA delivery mediated by beta-cyclodextrin embedded in a polyelectrolyte multilayerProc Natl Acad Sci U S A2006103238618862116735471
- CholerisELittleSRMongJAPuramSVLangerRPfaffDWMicroparticle-based delivery of oxytocin receptor antisense DNA in the medial amygdala blocks social recognition in female miceProc Natl Acad Sci U S A2007104114670467517360582
- SirsiSRSchrayRCWheatleyMALutzGJFormulation of poly-lactide- co-glycolic acid nanospheres for encapsulation and sustained release of poly(ethylene imine)-poly(ethylene glycol) copolymers complexed to oligonucleotidesJ Nanobiotechnology200971119351396
- GoforthRSalemAKZhuXImmune stimulatory antigen loaded particles combined with depletion of regulatory T-cells induce potent tumor specific immunity in a mouse model of melanomaCancer Immunol Immunother200958451753018719913
- ElamanchiliPDiwanMCaoMSamuelJCharacterization of poly(D,L-lactic-co-glycolic acid) based nanoparticulate system for enhanced delivery of antigens to dendritic cellsVaccine200422192406241215193402
- LutsiakMEKwonGSSamuelJBiodegradable nanoparticle delivery of a Th2-biased peptide for induction of Th1 immune responsesJ Pharm Pharmacol200658673974716734975
- HamdySMolaviOMaZCo-delivery of cancer-associated antigen and Toll-like receptor 4 ligand in PLGA nanoparticles induces potent CD8+ T cell-mediated anti-tumor immunityVaccine200826395046505718680779
- NewmanKDSamuelJKwonGOvalbumin peptide encapsulated in poly(d,l lactic-co-glycolic acid) microspheres is capable of inducing a T helper type 1 immune responseJ Control Release199854149599741903
- DiwanMElamanchiliPLaneHGainerASamuelJBiodegradable nanoparticle mediated antigen delivery to human cord blood derived dendritic cells for induction of primary T cell responsesJ Drug Target2003118–1049550715203918