 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
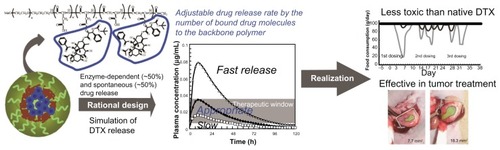
Drug release rate is an important factor in determining efficacy and toxicity of nanoscale drug delivery systems. However, optimization of the release rate in polymeric micellar nanoscale drug delivery systems has not been fully investigated. In this study NC-6301, a poly(ethylene glycol)-poly(aspartate) block copolymer with docetaxel (DTX) covalently bound via ester link, was synthesized with various numbers of DTX molecules bound to the polymer backbone. The number of DTX molecules was determined up to 14 to achieve an optimal release rate, based upon the authors’ own pharmacokinetic model using known patient data. Efficacy and toxicity of the formulation was then tested in animals. When administered three times at 4-day intervals, the maximum tolerated doses of NC-6301 and native DTX were 50 and 10 mg/kg, respectively, in nude mice. Tissue distribution studies of NC-6301 in mice at 50 mg/kg revealed prolonged release of free DTX in the tumor for at least 120 hours, thus supporting its effectiveness. Furthermore, in cynomolgus monkeys, NC-6301 at 6 mg/kg three times at 2-week intervals showed marginal toxicity, whereas native DTX, at 3 mg/kg with the same schedule, induced significant decrease of food consumption and neutrophil count. NC-6301 at 50 mg/kg in mice also regressed a xenografted tumor of MDA-MB-231 human breast cancer. Native DTX, on the other hand, produced only transient and slight regression of the same tumor xenograft. NC-6301 also significantly inhibited growth of OCUM-2MLN human scirrhous gastric carcinoma in an orthotopic mouse model. Total weight of metastatic lymph nodes was also reduced. In conclusion, NC-6301 with an optimized release rate improved the potency of DTX while reducing its toxicity.
Keywords:
Introduction
Various formations of a nanoscale drug delivery system (nanoDDS), of around 100 nm in diameter, have been developed and have been brought into clinical use. Of these, polymeric, micelle-based, anticancer drugs were originally developed by Kataoka et al.Citation1–Citation3 The assumed bases of higher effectiveness and lower toxicity of anticancer nanoDDSs are the “enhanced permeability and retention (EPR) effect”.Citation4
In addition to the EPR effect, the drug release rate from macromolecular DDSs is also critical to efficacy,Citation5,Citation6 for the following two reasons: (1) time course of plasma concentration of the naked drug is important, since it is the loaded drug that has toxic effects for both normal and tumor cells; (2) not all nanoDDSs reach their target tissue, releasing the loaded drugs in plasma. The release rate of loaded drugs with polymeric micelles can be optimized by various methods such as redesigning the chemical bond between polymer and drug; however, with liposomes, it is not easy to control release rate, because they have a stable lamella of lipid bilayer on the surface to contain drugs.
Docetaxel (DTX) is an antineoplastic agent used for a number of cancers.Citation7–Citation10 Although both DTX and paclitaxel (PTX) are taxanes with antimicrotubular properties, they have different toxicity profiles. The authors chose DTX because of its lower neurotoxicity than and its superiority to PTX.Citation11 It has also been shown to be superior to PTX in a number of preclinical models; this may be because of improved cellular uptake and increased potency of microtubule stabilization. In addition DTX has a significantly lower clinical incidence of neurotoxicity than PTX.Citation12 PTX produces serious gastrointestinal side effects,Citation12,Citation13 while anorexia and fatigue are the most common nonhematologic effects of DTX. Even so, the effects of DTX are less serious than those produced by PTX.Citation12,Citation13
The authors designed NC-6301 as a polymer micelle-based nanoDDS, consisting of poly(ethylene glycol) (PEG)-poly(aspartate) (pAsp) block copolymerCitation14 and ester-linked DTX.Citation15 The authors hypothesized that the linkage of DTX to polymer should increase in vivo half-life in comparison with a formulation of DTX made using hydrophobic interactions, and the increased half-life should lead to a better therapeutic effect because tumors are exposed to the drug for longer. In fact, longer exposure of tumor cells to DTX in vitro did produce more potent inhibition.Citation11
NC-6301 has a unique drug release profile compared with other copolymer formulations of PTX instead of DTX. These have already been characterized and include copolymers of D, L-lactide and PEG,Citation16 and poly(L-glutamic acid)-PTX.Citation17 Furthermore, NC-6301 was designed to release DTX for longer using a known effective plasma concentration in patients.Citation18 This concentration is based on the authors’ pharmacokinetic model simulation that uses published clinical data on DTX.Citation18,Citation19
The present study was designed to determine the number of DTX molecules required to achieve the optimal release rate and to investigate the in vivo efficacy of the formulation as compared with native DTX. The authors first confirmed release of DTX in human serum in vitro. Antitumor activity and tissue distribution of NC-6301, in nude mice xenografts of MDA-MB-231 human triple-negative breast tumor, were examined. Efficacy and tissue concentrations of NC-6301 were compared with those of native DTX. The toxicity profile of NC-6301 was evaluated in cynomolgus monkeys. Furthermore, the therapeutic effect of NC-6301 was assessed in an animal model of scirrhous gastric cancer with lymph node metastasis. The results supported the therapeutic advantage of using NC-6301 rather than native DTX.
Material and methods
Synthesis of NC-6301
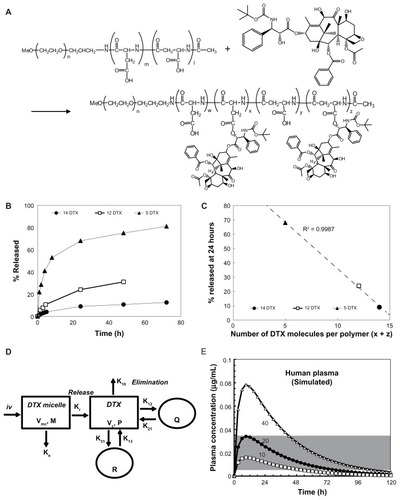
shows the chemical structure of NC-6301. The conjugate with 14 DTX molecules per polymer was synthesized as follows:Citation15 protective benzyl groups of the β-benzyl L-aspartate units were removed by alkaline hydrolysis to obtain PEG-pAsp-Ac (acetyl). The block copolymer (500 mg or 0.035 mmol) was dissolved in 10 mL of dehydrated N,N-dimethylformamide. Then DTX (1.12 g or 1.4 mmol), N,N′-diisopropylcarbodiimide (220 μL or 1.43 mmol), and N,N-dimethyl-4-aminopyridine (170 mg or 1.4 mmol) were added under argon. After stirring overnight at room temperature, the reaction mixture was poured into 300 mL of mixed hexane and ethyl acetate (1:1) to obtain the precipitate. This was dissolved in 100 mL of purified water to spontaneously form polymeric micelles. The micelles were purified by ultrafiltration using a membrane with a molecular weight limit of 100 kDa (Millipore Corporation, Billerica, MA) followed by lyophilization. The lyophilizate was dissolved in 10 mL of dehydrated N,N-dimethylformamide and poured into 300 mL of hexane and ethyl acetate mixture (1:1) to precipitate NC-6301. To purify NC-6301, the precipitate was treated a second time. Finally NC-6301 (520 mg) was obtained after drying under vacuum. The conjugate with twelve DTX molecules per polymer was obtained using PEG-pAsp-Ac containing 100 mol% of the α-amide form of aspartic acid residues, whereas the conjugate with five DTX was obtained using DTX trihydrate instead of DTX.Citation15 The final number of DTX molecules per polymer in a conjugate was determined by absorbance at 233 nm in 50% ethanol solution. All of the chemicals were reagent-grade products obtained commercially.
Figure 1 Determination of the optimum number of docetaxel (DTX) molecules per polymer. (A) Synthesis and chemical structure of NC-6301. Weight-average molecular weight of poly(ethylene glycol) is 10 kDa (n = 227). The number of aspartate residues (ie, w + x + y + z) was 40. The figure is a reproduction from the authors’ patent application with additional detailed information for the compound used in this study. (B) Time-dependent release of DTX to total DTX conjugated (%), with formulations of 14 (●), 12 (□), and 5 (▴) DTX molecules per polymer. (C) Plot of the values of released DTX (%) at 24 hours shown in (B) as a function of the number of DTX molecules per polymer; correlation efficient (R2) was 0.9987. (D) Scheme of the proposed release model of DTX from NC-6301 in vivo. The NC-6301 dose enters the central compartment (Vmc) via intravenous administration. NC-6301 in the central compartment (M) can be eliminated via linear (Ke) and release (Kr) pathways. The released DTX (P) in the central compartment (Vc) can distribute to the peripheral shallow (Q) and deep (R) compartments according to a three-compartment model. (E) The simulated plasma concentrations of released DTX after intravenous administration of NC-6301 based on the pharmacokinetic model, using parameters in humans. Open squares (□), closed circles (●), and open triangles (Δ) represent the time-course of released DTX when the first-order release constant of DTX from NC-6301 corresponds to 10%, 20% and 40% release in 24 hours, respectively. The dose of NC-6301 was set to be 100 mg/m2. The shaded area represents the range of concentrations required to inhibit cell growth by 50% for several cell lines.Citation18,Citation23,Citation24
Micelle preparation
The NC-6301 was first dissolved in purified water to allow spontaneous formation of micelles (5 mg of DTX/mL determined by absorbance at 233 nm). The micelles were passed through a 0.22 μm filter (Millex®-GP PES Express; Millipore Corporation) and frozen at −80°C after addition of 10% (w/v) sucrose. After dilution with water (×10), particle size was measured using the cumulative method with a Malvern Zetasizer 3000HSA (Malvern Instruments, Malvern, Worcestershire, UK) at 25°C.
Pharmacokinetic model of released DTX after administration of NC-6301
The plasma concentrations of released DTX after intravenous administration of NC-6301 in humans were simulated with a compartment model, using curve fitting software (SCIENTIST 3.0; Micromath, Saint Louis, MO). In terms of released DTX, this model has first-order input and output, corresponding to release of DTX from NC-6301 in the central compartment, and elimination of the resultant DTX. A three-compartment model was used to simulate elimination of DTX.Citation18 The differential equations describing this compartment model are (Equation1(1) ) and (Equation2
(2) ) for the central compartment and (Equation3
(3) ) and (Equation4
(4) ) for the peripheral compartment ():
M and P are the concentrations of NC-6301 and released DTX in the central compartment. Vmc and Vc are the distribution volumes of the central compartment for NC-6301 and released DTX. Ke and K10 are the first-order elimination constants of respective NC-6301 and released DTX from the central compartment. Kr is the first-order release constant of DTX from NC-6301 in the central compartment. K12 and K21 are the first-order transfer constants of released DTX between the central and peripheral shallow compartments. K13 and K31 are the first-order transfer constants of released DTX between the central and peripheral deep compartments. Q and R are the amounts of DTX in the peripheral shallow and deep compartments. All DTX parameter values are as reported elsewhere.Citation18,Citation19
Plasma concentrations of released DTX in humans were simulated using different values of Kr corresponding to 10%, 20%, and 40% release over 24 hours (0.00439, 0.00929, and 0.0213 h−1, respectively) under the following conditions: (a) half-life of NC-6301 is 24 hours and corresponds to the sum of Kr and Ke, is constant and equal to 0.0289 h−1; (b) the distribution volume of NC-6301 is limited and close to plasma volume: Vmc is 1.6 L/m2.Citation20
Dose translation between species
Dosage of DTX in humans using body surface area (mg/m2) was converted to weight (mg/kg) in mice and monkeys using US Food and Drug Administration guidelines.Citation21 The authors assumed 175 cm in height and 75 kg in weight for humans, and 0.02 and 3.0 kg in body weight in mice and monkeys, respectively. For example, 35 mg/m2 in humans, a weekly dose currently used in clinical setting, is equivalent to approximately 10 mg/kg in mice, and 3 mg/kg in monkeys (). The pharmacokinetics of small molecule compounds and those of nanoparticles are different.
Table 1 Dose conversion between species and units: mg/kg and mg/m2
Cells and animals
An MDA-MB-231 human triple-negative breast tumor was purchased from the European Collection of Cell Cultures through DS Pharma Biomedical (Osaka, Japan). An OCUM-2MLN cell line was established from human scirrhous gastric carcinoma, as previously described.Citation22 A PC-3 human prostate tumor was obtained from American Type Culture Collection through Summit Pharmaceuticals International (Tokyo, Japan). Nude mice (BALB/c nu/nu) and Crlj:CD1 (ICR) mice were purchased from Charles River, Japan (Yokohama, Japan). Cynomolgus monkeys, 3–4 years old, were obtained from Ina Research, the Philippines (Binan, Laguna, Philippines). All of the experiments involving animals were performed following national ethical conduct guidelines for use of animals in research.
Release of DTX from NC-6301 in fresh human serum or phosphate buffer
NC-6301 in saline (200 μL) was added to 0.1 M sodium phosphate buffer (pH 7.4) or fresh human serum (800 μL) prewarmed at 37°C with a final DTX concentration of 25 μg/mL. The fresh human serum used in this experiment was harvested, with informed consent, from the blood of healthy volunteers by Ina Research Inc (Ina, Japan). The mixtures were kept at 37°C. At given time intervals, aliquots of the samples (50 μL) were taken. The released DTX was measured by high-performance liquid chromatography (HPLC) after extraction with ethyl acetate. The collection of blood from donors was performed in compliance with national guidelines and was approved by the ethics committee of Ina Research Inc.
Effect of carboxy esterase inhibitor and protease inhibitor cocktail
Bis (p-nitrophenyl) phosphate sodium salt (BNPP), an inhibitor of carboxyl esterase, was obtained from Sigma-Aldrich (Tokyo, Japan) and stocked in dimethyl sulfoxide. Protease inhibitor cocktail (PIC) in dimethyl sulfoxide was obtained from Thermo Scientif ic (Rockford, IL). NC-6301 in saline was added, at a final DTX concentration of 25 μg/mL, into bovine serum containing BNPP or PIC at various concentrations. The mixture was kept at 37°C with a final bovine serum concentration of 80% (v/v). Six hours after start of incubation, an aliquot of the samples (50 μL) was taken and the released DTX concentration determined. DTX release inhibition ratio was calculated as follows: release inhibition ratio% = (1 − DTX released in the presence of inhibitor/DTX released in the absence of inhibitor) × 100.
In vitro cytotoxicity
The PC-3 and MDA-MB-231 cells were maintained in RPMI1640 media supplemented with 10% heat-inactivated fetal bovine serum at 37°C under an atmosphere of 5% carbon dioxide. Exponentially growing cells were typically seeded in 96-well plates (5000 cells/well) and cultured in media overnight. Cells were then treated with either NC-6301 or native DTX for 72 hours. A WST-8 reagent (Dojindo Laboratories, Kumamoto, Japan) was added to each well and cell viability was calculated according to the manufacturer’s instructions. The drug concentration required to inhibit cell growth by 50% (GI50) was determined by dose-response curves.
Toxicity dose-finding studies
Male nude mice were intravenously administered either NC-6301 or native DTX three times at 4-day intervals. Doses were 30–75 mg/kg for NC-6301, and 5–15 mg/kg for native DTX (n = 2). Hereafter, doses of NC-6301 are expressed as DTX equivalents, mg/kg of body weight, per injection. Mice were observed for 32 days after first administration. The body weight was monitored two or three times a week. The maximum tolerated dose (MTD) was defined as that which was not lethal, and caused body weight loss of <20% of original weight.
Tissue distribution of NC-6301 in tumor-bearing mice
The MDA-MB-231 cells were suspended in a 50% Matrigel® (Becton, Dickinson and Company, Franklin Lakes, NJ) and inoculated subcutaneously into the backs of female 5-week-old nude mice at 3 × 106 cells/100 μL/animal. When tumor volumes reached approximately 400 mm3, the mice were treated intravenously with either native DTX (10 mg/kg) or NC-6301 (50 mg/kg). For each drug, 15 mice were divided into five groups (n = 3), corresponding to 1, 24, 48, 72 and 120 hours for NC-6301, and 0.083, 1, 6, 24, and 48 hours for native DTX. Immediately before sampling, the mice were anesthetized with ether and blood was collected from the heart using a heparinized syringe. Plasma was harvested by centrifugation at 4°C. Tumor, liver, spleen, kidney, heart, and lung were removed, rinsed with saline, and stored at −30°C until analysis.
Determination of drug concentration in plasma and tissue
Tissue samples were suspended in phosphate buffered saline (pH 7.4) at a concentration of 25% w/w and homogenized on ice with a Polytron PT3100 mixer (Kinematica, Lucerne, Switzerland). Using aliquots of the homogenates and plasma (50 μL), both released DTX (that released in vivo from NC-6301) and total DTX (that released from NC-6301 plus remaining NC-6301) concentrations were determined by HPLC. Reversed-phase HPLC was performed at 40°C on a Tosoh TSK-gel ODS-80TM (4.6ϕ × 150 mm) with a Tosoh ODS-80TM guard cartridge (Tokyo, Japan). DTX was eluted with 50% acetonitrile using a Waters Alliance System at a flow rate of 1.0 mL/minute. Detection was carried out using a Waters 2487 absorbance detector at 233 nm.
To determine released DTX concentration, PTX (20 μg/ mL)/50% acetonitrile (20 μL) was added to the homogenates and plasma samples (50 μL) as an internal standard. The sample was then treated with ethyl acetate (300 μL) to extract the drug. After vortexing for 10 seconds, the sample was centrifuged for 1 minute at 5000 × g at 4°C and the clear supernatant obtained. The residue was treated with ethyl acetate once again. The recovered supernatant was combined with the previous one and dried under vacuum using an IWAKI VEC-310 vacuum evaporator centrifuge (AGC Techno Glass, Funabashi, Japan) for 30 minutes at 40°C. The dried residue was dissolved with 50% acetonitrile (100 μL) and analyzed using HPLC.
To determine total DTX concentration, the samples (50 μL) were treated with acetonitrile (150 μL) to precipitate proteins. After centrifugation for 1 minute at 5000 g at 4°C, the recovered supernatant (100 μL) was alkalif ied with 50 mg/mL sodium bicarbonate (200 μL). Then 30% hydrogen peroxide (400 μL) and dichloromethane (300 μL) was added and mixed for 3 hours at room temperature to facilitate release of DTX from NC-6301 and to extract the drug. The organic phase was harvested by centrifugation for 1 minute at 5000 g at 4°C. The resultant mixture was extracted with dichloromethane (300 μL) once again. The recovered organic phase was combined with the previous one and dried. PTX (20 μg/mL)/50% acetonitrile (100 μL) was added to the dry residue. The prepared mixture was analyzed using HPLC.
Evaluation of antitumor activity in animal models
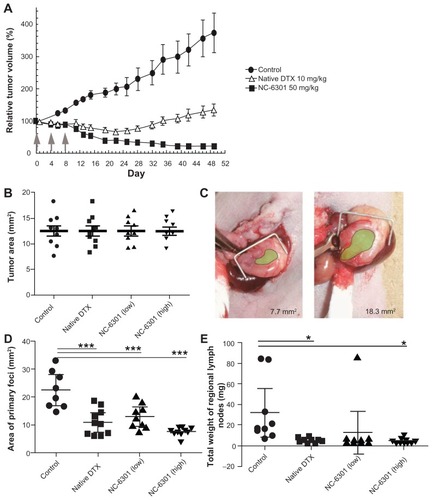
MDA-MB-231 human breast tumor model The MDA-MB-231 cells were inoculated subcutaneously into female nude mice, as described earlier. Tumor volume was calculated using the following formula: tumor volume = L × W2/2, where L and W are the major and minor dimensions, respectively. Drug treatment with seven mice per group started on day 27 after inoculation, when tumor volume reached 218 ± 20 mm3 (average plus or minus standard error). DTX at 10 mg/kg and NC-6301 at 50 mg/kg were administered by tail vein injection three times at 4-day intervals. The control group was untreated.
OCUM-2MLN human scirrhous gastric carcinoma model
A total of 5 × 106 OCUM-2MLN cells in 50 μL of phosphate buffered saline were inoculated into the subserosa of the gastric walls of male nude mice while under ether anesthesia. Two weeks after inoculation, the mice were examined in vivo for primary foci by opening the abdomen under deep anesthesia. They were then assigned to the following treatment groups (n = 10) for intravenous administration of (1) normal saline as the vehicle control, (2) 10 mg/kg of DTX, (3) 50 mg/kg of NC-6301 as the lower dose, and (4) 65 mg/kg of NC-6301 as the higher dose, on days 0, 4, and 8 after the first administration. The mice were sacrificed 4 weeks after inoculation and were examined for primary foci size, while all metastatic lymph nodes were collected and measured for total weight.
Toxicology studies in mice
Myelotoxicity and hepatotoxicity were assessed with male mice (Crlj:CD1 (ICR), n = 10) intravenously administered (three times at 4-day intervals) with (1) NC-6301 at 50 mg/kg or (2) DTX at 10 mg/kg. General condition and body weight of the animals were observed continuously for 49 days after first administration. Animals from each group were sacrificed one by one for blood collection and autopsy (macroscopic observation of liver and bone marrow). Hematology and blood biochemistry tests were carried out using an ADVIA 120 automated hematology analyzer (Siemens Healthcare Diagnostics, Tokyo, Japan) and a 7180 clinical analyzer (Hitachi High-Technologies, Tokyo, Japan), respectively.
Toxicology study in cynomolgus monkeys
Myelotoxicity was assessed, with male cynomolgus monkeys aged 3–4 years (n = 3; 2.5–3.7 kg body weight) intravenously administered, three times at 2-week intervals, with NC-6301 at 3 or 6 mg/kg. Native DTX was administered as a comparison at 3 mg/kg. Food consumption, general conditions, and body weight of animals were all observed continuously for 42 days after first administration. Blood (1 mL per time point) was collected weekly. Hematological testing was carried out at the same time.
Statistical analysis
Statistical analysis, where applicable, was performed using Excel software (Microsoft, Redmond, WA). Results were compared by Student’s t-test and were expressed as mean values with standard error. Differences were considered statistically significant at P < 0.05. All statistical tests were two-sided. Effects of NC-6301 on the growth of OCUM-2MLN were evaluated by multivariate analysis of variance testing using JMP software (SAS Institute, Cary, NC).
Results
Determination of release rate
The authors first analyzed relations between release rates of DTX and the number of DTX molecules (x + z in ) per polymer, 14, 12, and 5 (). The analysis revealed that the number of molecules conjugated and the release rate after 24 hours were inversely correlated (R2 = 0.9987, ). Next, time-dependent plasma concentration of DTX in humans was simulated. Calculations were performed using the pharmacokinetic model shown in , by assuming a dose of NC-6301 at 100 mg/m2, the MTD at a once-every-3-week schedule for native DTX in humans,Citation18 and release rates at 24 hours of 10%, 20%, or 40% (). Other variables are described in the Material and Methods section. According to the reported appropriate range of plasma DTX concentration in humansCitation18,Citation23,Citation24 (, gray area), the authors selected a formulation with 14 DTX molecules per polymer molecule.
Properties of the formulation with 14 DTX molecules per polymer, NC-6301
The release of DTX in NC-6301 with 14 DTX per polymer, of which the average diameter was 120–130 nm (), from the PEG-pAsp copolymer backbone was compared by incubating NC-6301 at a concentration of 25 μg/mL in fresh human serum and sodium phosphate buffer (pH 7.4) at 37°C. Slow release was confirmed in both media (). Approximately 20% and 10% of the released DTX were observed at 24 hours in human serum and phosphate buffer, respectively (). This supported the selection of this conjugate, and also allowed the authors to estimate that spontaneous release contributed approximately a half to total drug release in human serum.
Figure 2 Properties of NC-6301 with 14 docetaxel (DTX) molecules per polymer: (A) size distribution – the average diameter was 120–130 nm, with minor disparities; (B) release of DTX as a function of time during incubation in fresh human serum (●) or sodium phosphate buffer (pH 7.4) (○) at 37°C*; (C) concentration dependency of carboxy esterase inhibitor (bis (p-nitrophenyl) phosphate sodium salt [BNPP], ▪) and protease inhibitor cocktail (PIC, ●) on the release of DTX. NC-6301 was incubated in bovine serum in the presence of each inhibitor for 6 hours at 37°C. An aliquot of the samples was taken and the released DTX concentration was determined by high-performance liquid chromatography.** (D) Actual plasma DTX level in tumor-bearing mice compared with the simulated values (dashed line for total DTX and solid line for released DTX) based on the model shown in . For actual data, plasma concentration-time profiles of total (●) and released DTX (□) after intravenous administration of NC-6301 to nude mice bearing MDA-MB-231 breast tumor at 50 mg of DTX/kg. Profiles of DTX after DTX injection at 10 mg/kg (Δ) are also shown for comparison. (E–G) Body weight changes: healthy nude mice received either DTX (E) or NC-6301 (F) intravenously three times with a 4-day interval (indicated by arrows) for determining the maximum tolerated dose.*** Body weight changes in nude mice bearing MDA-MB-231 tumor xenograft (G). The animals were also tested with regimens indicated at a 4-day interval, with seven mice per condition.****
Notes: *Each point represents the mean for three samples, and bars indicate standard deviation; **the concentration units of BNPP and PIC are mM and % (v/v), respectively, and each point represents the mean for two or three separate experiments; ***each point represents the mean for two mice; ****each point represents the mean for seven mice; bars, standard errors.
![Figure 2 Properties of NC-6301 with 14 docetaxel (DTX) molecules per polymer: (A) size distribution – the average diameter was 120–130 nm, with minor disparities; (B) release of DTX as a function of time during incubation in fresh human serum (●) or sodium phosphate buffer (pH 7.4) (○) at 37°C*; (C) concentration dependency of carboxy esterase inhibitor (bis (p-nitrophenyl) phosphate sodium salt [BNPP], ▪) and protease inhibitor cocktail (PIC, ●) on the release of DTX. NC-6301 was incubated in bovine serum in the presence of each inhibitor for 6 hours at 37°C. An aliquot of the samples was taken and the released DTX concentration was determined by high-performance liquid chromatography.** (D) Actual plasma DTX level in tumor-bearing mice compared with the simulated values (dashed line for total DTX and solid line for released DTX) based on the model shown in Figure 1D. For actual data, plasma concentration-time profiles of total (●) and released DTX (□) after intravenous administration of NC-6301 to nude mice bearing MDA-MB-231 breast tumor at 50 mg of DTX/kg. Profiles of DTX after DTX injection at 10 mg/kg (Δ) are also shown for comparison. (E–G) Body weight changes: healthy nude mice received either DTX (E) or NC-6301 (F) intravenously three times with a 4-day interval (indicated by arrows) for determining the maximum tolerated dose.*** Body weight changes in nude mice bearing MDA-MB-231 tumor xenograft (G). The animals were also tested with regimens indicated at a 4-day interval, with seven mice per condition.****Notes: *Each point represents the mean for three samples, and bars indicate standard deviation; **the concentration units of BNPP and PIC are mM and % (v/v), respectively, and each point represents the mean for two or three separate experiments; ***each point represents the mean for two mice; ****each point represents the mean for seven mice; bars, standard errors.](/cms/asset/d12230e6-b195-4e69-9f76-0948c3eae702/dijn_a_31247_f0002_c.jpg)
To elucidate key factors of DTX release from NC-6301, the release rate was examined in bovine serum in the presence of enzyme inhibitors. shows the concentration dependency of inhibitors on DTX release from NC-6301. The PIC exhibited dose-dependent inhibition, while BNPP did not. This suggests that DTX release from NC-6301 in serum is predominantly dependent on metabolism of NC-6301 via proteases.
The authors next tested actual plasma concentrations in vivo of total and released DTX in tumor-bearing mice administered NC-6301 at 50 mg/kg (), with native DTX. The NC-6301 data points closely followed the simulated concentration, based on the pharmacokinetic model (). This supports the simulation model, where calculation for was done with murine data for parameters, and assuming a dose of 50 mg/kg. A release rate of 60% for NC-6301 was also assumed, which was data measured in murine serum for the formulation with 14 DTX molecules per polymer after 24 hours (data not shown).
With regard to total DTX and native DTX (), total DTX in plasma remained over 0.1 μg/mL for 72 hours after administration with NC-6301. With native DTX, on the other hand, it remained less than 5 hours. This illustrates in a clear way the longer circulation of NC-6301 as compared with native DTX. In fact, NC-6301 increased area under the concentration-time curve (AUC) of total DTX plasma approximately 156-fold more than native DTX at equivalent doses ().
Table 2 Area under the concentration-time curve (AUC) values in tissues and plasma after administration of either NC-6301 at 50 mg/kg or docetaxel (DTX) at 10 mg/kg in nude mice xenografted with MDA-MB-231 human breast tumor
The authors then analyzed toxicity of NC-6301 with regard to body weight in healthy nude mice and determined the MTD of NC-6301 and native DTX. To do this, the authors intravenously administered native DTX at a dose of 5, 7, 10, 12, or 15 mg/kg or NC-6301 at a dose of 30, 40, 50, 60, or 75 mg/kg three times every 4 days () and observed body weight for 32 days. Mice administered native DTX of 15 mg/kg (equivalent to 45.2 mg/m2 or 1.13 mg/kg in humans) had a significant decrease in body weight compared with those administered less than 15 mg/kg. With NC-6301, mice administered 50 mg/kg or more had significant body weight loss compared with those administered less than 50 mg/kg. Following these results, the authors considered the MTDs of native DTX and NC-6301 to be 10 mg/kg (equivalent to 30.3 mg/m2 or 0.76 mg/kg in humans) and 50 mg/kg, respectively, when administered three times at 4-day intervals. This MTD value in the study with mice for native DTX is approximately one-third of the reported MTD at a once-every-3-week schedule in humans,Citation18 although the MTD varies according to the schedule of drug administration.Citation25,Citation26 Therefore NC-6301 at 50 mg/kg and DTX at 10 mg/kg were used in the following studies. Maximum body weight loss of treated animals was slight for NC-6301 even in mice bearing MDA-MB-231 tumor xenograft, but approximately 10% with native DTX ().
Tissue distribution and toxicity of NC-6301 in mice
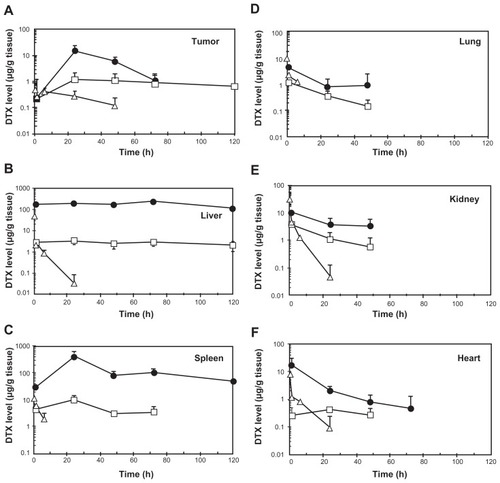
To check whether toxicity and efficacy for NC-6301 and native DTX correlated with tissue drug concentrations, in addition to those in plasma (), the authors investigated the kinetics of NC-6301 and native DTX in mice bearing MDA-MB-231 tumor xenografts. The time courses and AUC values in several tissues are summarized in and , respectively. In tumor tissue () total DTX peaked at 24 hours and declined slowly thereafter. DTX concentrations in the tumor were about 1 μg/g for at least 120 hours. This data supports the superior efficacy of NC-6301 in vivo. Large AUC values of DTX were observed in the liver and the spleen (). However, the release ratios were very restricted. In contrast, relatively high release ratios were observed in the lung and the kidney (), although AUC values of released DTX were comparable with those of native DTX. In the heart, AUC value of released DTX was relatively limited ().
Figure 3 Tissue concentration-time profiles of total (●) and released docetaxel (DTX) (□) after intravenous administration of NC-6301 to nude mice bearing MDA-MB-231 breast tumor at 50 mg of DTX/kg. Profiles of DTX after DTX injection at 10 mg/kg (Δ) are also shown for comparison.
Notes: Data points indicate mean measurement for three mice; bars (standard deviation), if not shown, are within range of the symbol plots.
We also checked in mice whether liver- or kidney-related blood test markers were affected by three injections of NC-6301 at 50 mg/kg or native DTX at 10 mg/kg for 49 days after initial administration ( and ). No significant change in the blood test markers was observed with either drug, aside from transient increases in aspartate transaminase and alanine transaminase with NC-6301. Hepatotoxicity was reversible and marginal, although certain levels of released drug were observed in spleen and liver after a single administration of NC-6301 (). Furthermore, myelosuppression was mild and transient for both drugs (data not shown).
Table 3 Changes in activity of aspartate transaminase (AST) and alanine transaminase (ALT) in mice receiving intravenous NC-6301 at 50 mg/kg or intravenous docetaxel (DTX) at 10 mg/kg three times at 4-day intervals
Table 4 Changes in concentrations of creatinine (CRE) and urea nitrogen (UN) in mice receiving intravenous NC-6301 at 50 mg/kg, or intravenous docetaxel (DTX) at 10 mg/kg three times at 4-day intervals
Figure 4 Antitumor effect of NC-6301 in murine models of tumors: (A) efficacy of NC-6301 and docetaxel (DTX) against MDA-MB-231 human breast tumor. MDA-MB-231 tumor cells (3 × 106 cells/animal) were inoculated subcutaneously into the back (day −27), and intravenous administration three times at 4-day intervals (indicated by arrows) started on day 0 when tumor volumes were 218 ± 20 mm3 (average plus or minus standard error). The dose of NC-6301 is expressed as equivalent to DTX.# (B–E) Efficacy of NC-6301 and DTX against metastatic scirrhous gastric carcinoma: (B) Tumor area at the start of drug administration (day 0) – OCUM-2MLN tumor cells (5 × 106 cells/ animal) were inoculated into the subserosa of gastric walls of the mice (day −14), and intravenous administration three times at a 4-day interval started on day 0, when tumor areas were 13 ± 3 mm2 in all conditions; (C) photos of the smallest and the largest tumors on day 0 (green shading represents the tumor area); (D) tumor area of primary foci on day 14 – the doses of NC-6301 low and high represent 50 and 65 mg/kg/injection, respectively; (E) total weight of regional lymph nodes on day 14.##
Notes: #Each point represents the mean measurement for seven mice, and bars indicate the standard error; ##each point represents the individual value, each bold line represents the mean measurement for ten mice, and bars indicate the standard error; control and treated groups were compared statistically. *P < 0.05; ***P < 0.001 (Student’s t-test).
Toxicology study in cynomolgus monkeys
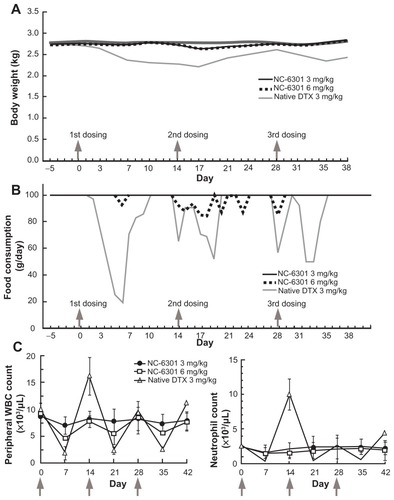
To evaluate toxicity of NC-6301 in a species close to humans, the authors used cynomolgus monkeys. Although the monkey experiments in this work were limited in scope, especially in terms of dosing, the data have been added for reference purposes. Based on the preliminary results determining the MTD in this animal (data not shown), the authors determined drug dose in cynomolgus monkeys as follows: 3 mg/kg three times at 2-week intervals for native DTX and NC-6301, and a higher dose of 6 mg/kg only for NC-6301 with the same schedule (3 mg/kg in cynomolgus monkeys is equivalent to 36.6 mg/m2, or to 12.2 mg/kg in mice and 0.92 mg/kg in humans). The higher dose of 6 mg/kg for NC-6301 was determined on the basis of the human MTD of a formulation encapsulating PTX,Citation27 where the MTD was approximately the same as native PTX but not as high as in mice. The cynomolgus monkeys exhibited adverse effects including gastrointestinal symptoms, especially in those administered native DTX at 3 mg/kg. The animals treated with native DTX lost up to 20% more weight than those administered NC-6301 (). A critical decrease in food consumption was observed in animals treated with native DTX, whereas the decrease was marginal in those treated with NC-6301 (). Bone marrow suppression was milder with NC-6301 compared with native DTX (). Both white blood cell count and neutrophil count were significantly decreased only by administration of native DTX.
Figure 5 Toxicological tests in cynomolgus monkeys treated with intravenously administered NC-6301. Male cynomolgus monkeys received NC-6301 intravenously three times with a 2-week interval (indicated by arrows) at either 3 or 6 mg/kg. Native docetaxel (DTX) was administered similarly as a control at 3 mg/kg. (A) Average body weight change; (B) average change in food consumption; (C) changes in number of white blood cells (WBCs) and number of neutrophils.
Notes: Data points indicate mean measurements for three monkeys; bars indicate standard deviation.
Antitumor effect of NC-6301
In vitro cytotoxicity was first examined using MDA-MB-231 and PC-3 tumor cells. The authors found that the GI50 (equivalent to DTX) of NC-6301 versus MDA-MB-231 was 0.01 μg/mL, while that of DTX was 0.004 μg/mL. Similarly, for PC-3 the GI50 measurements for NC-6301 and DTX were 0.01 and 0.004 μg/mL, respectively. This is because DTX release from NC-6301 is limited, although still pharmacologically active.
Next, the authors tested whether NC-6301 exhibited adequate antitumor effects compared with native DTX in a xenograft model using MDA-MB-231 triple-negative human breast cancer cells. As shown in , three injections of NC-6301 at 50 mg/kg effectively suppressed tumor growth. In contrast, native DTX at 10 mg/kg with the same schedule resulted only in transient and slight regression of the tumor. These results suggest the superior efficacy of NC-6301 with fewer side effects ().
We also confirmed NC-6301 efficacy with an orthotopic mouse model of scirrhous gastric cancer, one of the most devastating cancers accompanied by lymphatic metastasis.Citation28,Citation29 We used OCUM-2MLN cells derived from patients with scirrhous gastric cancer (). It would be therapeutically advantageous if we could increase the anticancer efficacy of DTX and decrease its side effects by using NC-6301 in this carcinoma, since monotherapy with native DTX produces response rates of only 20% in patients with advanced gastric cancer.Citation30 show the tumor area of primary foci and the total weight of regional lymph nodes, on day 14 after first drug treatment. Native DTX at 10 mg/kg and NC-6301 at the lower dose (50 mg/kg) showed similar effects with primary foci, while NC-6301 at a higher dose (65 mg/kg) effectively reduced the tumor volume. Both native DTX and NC-6301, at the higher dose, significantly suppressed lymphatic metastases, compared with the no-drug control. It is noteworthy that body weight loss in animals treated with DTX was worse than in those treated with NC-6301 at the higher dose (data not shown). These results support the potential advantage of NC-6301 over native DTX, even for devastating cancers such as metastatic scirrhous gastric carcinoma.
Discussion
NC-6301 was designed to release DTX from the polymer backbone at an appropriate rate, thereby obtaining better efficacy and tolerance than native DTX. The best DTX release rate was obtained with the formulation of 14 DTX molecules per polymer, predicted on the basis of the authors’ own pharmacokinetic model () and effective plasma concentrations published elsewhere.Citation18 The usability of this pharmacokinetic model, as a guide to human use, was confirmed by simulating plasma concentrations of DTX released in mice administered NC-6301 ().
An effective concentration of DTX in humans is suggested by GI50 values obtained against the cell lines shown in . With regard to toxic plasma concentration of DTX in patients, neutropenia and increased fluid retention correlate with extended concentrations >0.2 μM (approximately 0.16 μg/mL),Citation31 whereas neutropenia with PTX, another taxane, correlates significantly with duration of PTX plasma concentration of ≥0.05 μM (approximately 0.04 μg/mL).Citation32 When considering longer plasma exposure of DTX at an effective concentration, the simulation results () suggest a conjugate of 14 DTX molecules per polymer, which had 20% release over 24 hours in human serum, as the best candidate. In the present study, the authors have shown the potential therapeutic advantages of this formulation of NC-6301 over native DTX. Its antitumor activity, tissue distribution, and toxicology in various models was investigated. The results promise increased efficacy of NC-6301 with fewer side effects, in comparison with native DTX, as preclinical proof.
NC-6301 has a unique drug release profile compared with PTX-polymer conjugates using ester linkage. The ester between DTX and the pAsp backbone is thought to undergo half spontaneous release. The other half of the ester may undergo enzyme-dependent release (). Although not extensively studied, enzyme-mediated drug release is dependent on proteases (). Here, it should be noted that enzyme-independent drug release may reduce individual differences in release of DTX from NC-6301. On the contrary, PTX poliglumex, a clinically evaluated conjugate of PTX and poly(L-glutamic acid) via an ester linkage, is thought to release conjugated PTX through the action of lysosomal protease cathepsin B.Citation17 This suggests that the polymer backbone impacts the release mode of the conjugated drug, and thus supports the authors’ preliminary observation that a similar DTX conjugate, with a backbone of poly(L-glutamic acid), showed marginal DTX release in serum (data not shown).
The effectiveness of NC-6301 in tumors probably depends on a number of factors. Based on the pharmacokinetic data of this study, and other information already available, the following can be suggested. Both tumor targeting of NC-6301, attributable to the EPR effect, and prolonged intratumoral release of DTX at high concentration demarcate drug efficacy. In the MDA-MB-231 tumor model, total DTX AUC was eight times greater than native DTX at equivalent doses (). At equitoxic doses, released DTX AUC in tumor was approximately eight times greater than that of native DTX, supporting the superior efficacy of NC-6301.
The potent anticancer effect of NC-6301 may also be due to the sustained exposure of tumor tissue to released DTX. The intratumoral concentration of DTX at 120 hours after administration of NC-6301 was approximately 1 μg/g (). According to the authors’ in vitro experiments this concentration is greater than the GI50 value of DTX against MDA-MB-231 tumor cells with a 72-hour exposure time (4 ng/mL). The increased efficacy of NC-6301 may also be explained by the sustained DTX level in blood () as with the orthotopic model of scirrhous gastric cancer. Aside from the EPR effect, drug release may be another determinant of nanoDDS efficacy. The authors have previously shown that an even smaller polymer micelle of 65 nm in diameter did not independently inhibit tumor growth in the same model,Citation33 whereas NC-6301 of 120–130 nm did, as shown in this work. As well as sizes, another difference relates to drug release: the 65 nm micelle does not release drug into the blood stream, whereas NC-6301 does. Therefore, nanoDDS size may not be the only determinant of efficacy: drug release into the blood stream may be another factor. To support this, the required dose in the MDA-MB-231 tumor model was 50 mg/kg, less than the 65 mg/kg needed for metastatic lesions in the OCUM-2MLN model. It may thus be a combination of drug release and the EPR effect that is important. The MDA-MB-231 model exhibits sufficient EPR effect because of abundant leaky tumor vasculature.Citation34,Citation35 However, further study is needed to clarify the mechanisms behind these observations.
Although NC-6301 has a tendency to accumulate in the liver (), its side effects are less disruptive than native DTX. Lower doses of native DTX resulted in more body weight loss than higher doses of NC-6301 ( and ). This may be because use of nanoDDSs avoids a concentration “surge” of DTX in blood ( and ) and/or in other tissues () immediately after administration. Furthermore, aspartate transaminase and alanine transaminase values for healthy mice receiving NC-6301 at 50 mg/kg three times at 4-day intervals, increased transiently. However, the change was marginal and reversible (), even though a fivefold larger dose of NC-6301 was used when compared with native DTX.
Comparison with the few reports of nanoDDSs incorporating DTXCitation36–Citation38 will help understand the features of NC-6301. A PEG-DTX conjugate was prepared using PEG with a molecular weight of 2000.Citation36 One molecule of DTX binds to PEG via ester linkage. The main purpose of this conjugate is to avoid use of the excipient polysorbate 80 as a solubilizer for Taxotere® (Sanofi-Aventis US LLC, Bridgewater, NJ), the commercially available formulation of DTX. The excipient present in the formulation causes hypersensitivity reactions and peripheral neurotoxicity, the main side effect of Taxotere. This PEG-DTX formation, physically entrapping free DTX, increased total DTX equivalent AUC in plasma 1.8-fold that of native DTX in mice. NC-6301, on the other hand, increased total AUC 156-fold that of native DTX (). Thus NC-6301 is likely to stay in blood longer and at a higher concentration. There is another PEGylated DTX, NKTR-105, produced by Nektar Therapeutics (San Francisco, CA).Citation39 Although in phase I, its chemical structure has not been reported, but based on the data available, we believe NC-6301 has a number of advantages over NKTR-105. While body weight loss with NKTR-105 was approximately 19% at a dose effective with a tumor model, body weight loss with NC-6301 was approximately 9% at a comparable dose in an orthotopic gastric cancer model. AUC of free DTX in tumor tissue at the MTD was twofold more than free DTX solution with NKTR-105, whereas that with NC-6301 was eightfold.
Conclusion
In conclusion, NC-6301 is therapeutically superior to native DTX, with low toxicity and other advantages, and the authors believe it has promise as an antitumor agent. The pharmacokinetic model used may also provide a useful tool for the design of a polymeric micelle with conjugated drugs of established clinical efficacy ().
Figure 6 Graphical summary: polymeric micelle incorporating docetaxel (DTX), NC-6301, was rationally designed based on a pharmacokinetic model and proved in animal models to be effective and less toxic than native DTX.
Acknowledgments
The authors thank Prof Michael W Miller (Miller Takemoto and Partners) for help with the manuscript. The authors also thank Tsubasa Kondo, Tomomi Chijiiwa, and Masami Tsuchiya for their technical assistance.
Disclosure
M Harada, H Saito, K Ishii, T Hayashi, and Y Kato are employees of NanoCarrier Co, Ltd, Japan, and MR Kano is supported by a research grant from NanoCarrier Co, Ltd. The listed authors in the current work from NanoCarrier Co, Ltd, have been involved in designing the study; in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the paper for publication.
References
- KataokaKKwonGSYokoyamaMOkanoTSakuraiYBlock copolymer micelles as vehicles for drug deliveryJ Control Release1993241–3119132
- KwonGSKataokaKBlock copolymer micelles as long-circulating drug vehiclesAdv Drug Deliv Rev1995162–3295309
- KataokaKHaradaANagasakiYBlock copolymer micelles for drug delivery: design, characterization and biological significanceAdv Drug Deliv Rev200147111313111251249
- MatsumuraYMaedaHA new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancsCancer Res19864612 Pt 1638763922946403
- VeroneseFMSchiavonOPasutGPEG-doxorubicin conjugates: influence of polymer structure on drug release, in vitro cytotoxicity, biodistribution, and antitumor activityBioconjug Chem200516477578416029018
- HaradaMBobeISaitoHImproved anti-tumor activity of stabilized anthracycline polymeric micelle formulation, NC-6300Cancer Sci2011102119219921040218
- SwainSMJeongJHGeyerCEJrLonger therapy, iatrogenic amenorrhea, and survival in early breast cancerN Engl J Med2010362222053206520519679
- NaitoSTsukamotoTKogaHDocetaxel plus prednisolone for the treatment of metastatic hormone-refractory prostate cancer: a multicenter phase II trial in JapanJpn J Clin Oncol200838536537218417502
- PosnerMRHershockDMBlajmanCRCisplatin and fluorouracil alone or with docetaxel in head and neck cancerN Engl J Med2007357171705171517960013
- OzdemirNYAbaliHOksüzoğuBThe efficacy and safety of reduced-dose docetaxel, cisplatin, and 5-fluorouracil in the first-line treatment of advanced stage gastric adenocarcinomaMed Oncol201027368068419633962
- DorrRTPharmacology of the taxanesPharmacotherapy1997175 Pt 296S104S9322876
- KatsumataNNodaKNozawaSPhase II trial of docetaxel in advanced or metastatic endometrial cancer: a Japanese cooperative studyBr J Cancer2005939999100416234823
- OkadaSSakataYMatsunoSPhase II study of docetaxel in patients with metastatic pancreatic cancer: a Japanese cooperative studyBr J Cancer1999803–443844310408850
- YokoyamaMMiyauchiMYamadaNPolymer micelles as novel drug carrier: Adriamycin-conjugated poly(ethylene glycol)-poly(aspartic acid) block copolymerJ Control Release1990111–3269278
- HaradaMSaitoHKatoYNanoCarrier Co, LtdDocetaxel polymer derivative, method for producing same and use of same PCT patent application WO 2009/142326 A12009
- JacksonJKGleaveMEYagoVBeraldiEHunterWLBurtHMThe suppression of human prostate tumor growth in mice by the intratumoral injection of a slow-release polymeric paste formulation of paclitaxelCancer Res200060154146415110945622
- ChipmanSDOldhamFBPezzoniGSingerJWBiological and clinical characterization of paclitaxel poliglumex (PPX, CT-2103), a macromolecular polymer-drug conjugateInt J Nanomedicine20061437538317722272
- ClarkeSJRivoryLPClinical pharmacokinetics of docetaxelClin Pharmacokinet19993629911410092957
- Sanofi-Aventis [homepage on the Internet] Available from: http://di.sanofi-aventis.co.jp/interview/taxotere.pdf
- PlummerRWilsonRHCalvertHA phase I clinical study of cisplatin-incorporated polymeric micelles (NC-6004) in patients with solid tumoursBr J Cancer2011104459359821285987
- US Food and Drug Administration (FDA)Oncology tools: dose calculator [online tool]Silver Spring, MDFDA Available from: http://www.accessdata.fda.gov/scripts/cder/onctools/animalquery.cfm
- FujiharaTSawadaTHirakawaKEstablishment of lymph node metastatic model for human gastric cancer in nude mice and analysis of factors associated with metastasisClin Exp Metastasis19981643893989626818
- HillBTWhelanRDShellardSAMcCleanSHoskingLKDifferential cytotoxic effects of docetaxel in a range of mammalian tumor cell lines and certain drug resistant sublines in vitroInvest New Drugs19941231691827896535
- LavelleFBisseryMCCombeauCRiouJFVrignaudPAndréSPreclinical evaluation of docetaxel (Taxotere)Semin Oncol1995222 Suppl 4316
- LokichJAndersonNDose intensity for bolus versus infusion chemotherapy administration: review of the literature for 27 anti-neoplastic agentsAnn Oncol19978115259093703
- Bradshaw-PierceELEckhardtSGGustafsonDLA physiologically based pharmacokinetic model of docetaxel disposition: from mouse to manClin Cancer Res20071392768277617473210
- HamaguchiTKatoKYasuiHA phase I and pharmacokinetic study of NK105, a paclitaxel-incorporating micellar nanoparticle formulationBr J Cancer200797217017617595665
- HippoYYashiroMIshiiMDifferential gene expression profiles of scirrhous gastric cancer cells with high metastatic potential to peritoneum or lymph nodesCancer Res200161388989511221876
- YashiroMHirakawaKCancer-stromal interactions in scirrhous gastric carcinomaCancer Microenviron20103112713521209779
- NishiyamaMWadaSDocetaxel: its role in current and future treatments for advanced gastric cancerGastric Cancer200912313214119890692
- BrunoRHilleDRivaAPopulation pharmacokinetics/pharmacodynamics of docetaxel in phase II studies in patients with cancerJ Clin Oncol19981611871969440742
- GianniLKearnsCMGianiANonlinear pharmacokinetics and metabolism of paclitaxel and its pharmacokinetic/pharmacodynamic relationships in humansJ Clin Oncol19951311801907799018
- KanoMRBaeYIwataCImprovement of cancer-targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF-beta signalingProc Natl Acad Sci U S A200710493460346517307870
- ChabottauxVSounniNEPenningtonCJMembrane-type 4 matrix metalloproteinase promotes breast cancer growth and metastasesCancer Res200666105165517216707440
- HervéM-AButeau-LozanoHVassyROverexpression of vascular endothelial growth factor 189 in breast cancer cells leads to delayed tumor uptake with dilated intratumoral vesselsAm J Pathol2008172116717818079435
- LiuJZahediPZengFAllenCNano-sized assemblies of a PEG-docetaxel conjugate as a formulation strategy for docetaxelJ Pharm Sci20089783274329018064681
- EsmaeiliFDinarvandRGhahremaniMHDocetaxel-albumin conjugates: preparation, in vitro evaluation and biodistribution studiesJ Pharm Sci20099882718273018972321
- MikhailASAllenCPoly(ethylene glycol)-b-poly(epsilon-caprolactone) micelles containing chemically conjugated and physically entrapped docetaxel: synthesis, characterization, and the influence of the drug on micelle morphologyBiomacromolecules20101151273128020369884
- Nektar Therapeutics [homepage on the Internet] Available from: http://www.nektar.com/product_pipeline/oncology_nktr-105.htmlaccessed July 21, 2010