Abstract
Protein nanocages are self-organized complexes of oligomers whose three-dimensional architecture can been determined in detail. These structures possess nanoscale inner cavities into which a variety of molecules, including therapeutic or diagnostic agents, can be encapsulated. These properties yield these particles suitable for a new class of drug delivery carrier, or as a bioimaging reagent that might respond to biochemical signals in many different cellular processes. We report here the design, synthesis, and biological characterization of a hepatocyte-specific nanocage carrying small heat-shock protein. These nanoscale protein cages, with a targeting peptide composed of a preS1 derivative from the hepatitis B virus on their surfaces, were prepared by genetic engineering techniques. PreS1-carrying nanocages showed lower cytotoxicity and significantly higher specificity for human hepatocyte cell lines than other cell lines in vitro. These results suggested that small heat-shock protein-based nanocages present great potential for the development of effective targeted delivery of various agents to specific cells.
Introduction
As most drugs have both beneficial and unfavorable effects pertaining to chemotherapy, which might also be tissue dependent, it is necessary to deliver therapeutic agents selectively to their target sites.Citation1–Citation4 Conventional chemotherapeutic agents diffuse non-specifically throughout the body where they affect both malignant and normal cells; unfortunately, 95% of all new potential therapeutics have poor pharmacokinetic and biopharmaceutical properties.Citation5 To overcome these problems, there is a serious need to develop effective drug delivery systems (DDS) that distribute therapeutically active drug molecules only to the desired site of action without affecting healthy organs and tissues.
Various strategies for site-specific drug delivery have led to the development of drug carriers. In particular, DDS based on liposomesCitation6,Citation7 and synthetic polymersCitation8–Citation11 have been extensively studied as novel drug-packaging strategies for cancer chemotherapy. In the last decade, nanotechnological innovations have played an important role in size control and surface modification of nanomaterials, and the resulting properties play a critical role in target specificity for tumor tissues via improved pharmacokinetics and pharmacodynamics and in allowing active intracellular delivery characteristics.Citation12 Traditional DDS materials, including liposomes and synthetic polymers, can be manufactured in bulk at low cost and with a wide diversity of backbones, surface chemistries, and molecular weights; however, their aggregate structure and compositional sequence cannot be accurately controlled and therefore such materials offer disadvantages in their poor responsiveness to cellular behavior that require sequence specificity and structural stringency to be sensitive to signals exhibited by certain cell biochemical processes or states.
On the other hand, natural or recombinant protein-based nanocapsules or nanocages are also being actively investigated as new types of nanomaterial.Citation13–Citation16 These nanocapsules are self-organized protein oligomer complexes with nanoscale inner cavities and whose three-dimensional architecture can be specified in great detail.Citation17,Citation18 Thus, they have the potential to offer special advantages in developing new functions that are responsive to biochemical signals exhibited by various cellular processes. Such specific interactions derive from the incorporated amino acid sequences that can be designed to address multiple functional requirements for biomaterial applications. Recently, we constructed stimulus-responsive nanomaterial by incorporating a genetically engineered small heat shock protein (sHSP) that can change its conformation in response to dual stimuli, a protease signal and temperature, in contrast to the wild type.Citation19
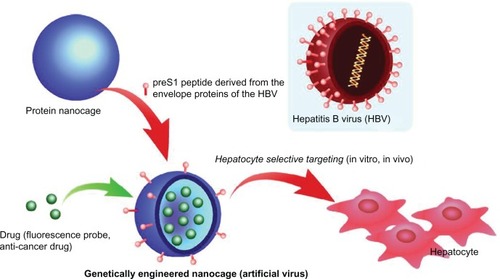
Here, a novel targeted drug delivery method is presented involving a protein-based nanocage composed of HSP16.5, a sHSP from a hyperthermophilic archaeon.Citation20–Citation22 This protein forms a 400 kDa homogeneous complex comprised of 24 subunits that self-organizes under physiological conditions to form a nanoscale, hollow, spherical capsule with small pores, contain very high thermal stability, and offer the potential for a variety of molecules, including drugs and magnetic resonance imaging contrast agents, to be encapsulated in the cavity. These unique properties yield HSP16.5 suitable for a drug carrier, but this protein exhibits no significant organ or tissue specificity in body distribution following intravenous administration in mice.Citation23 To address this problem, a recombinant fusion protein was constructed from HSP16.5 and a target-specific peptide using genetic engineering. Examinations of the crystal structure of the HSP16.5 complex have revealed that the C-terminal region is on the exterior surface of the capsule-like structure.Citation24 Thus, a recombinant HSP16.5 was designed and produced by an in-frame fusion, with the sequence for a targeting peptide added in this region. Specificity for liver cells-targeted delivery of the nanocapsule was achieved using a preS1 peptide for the targeting peptide ().Citation25–Citation27 This peptide is derived from the envelope proteins of the hepatitis B virus (HBV), with a known affinity for primary hepatocytes and hepatocyte cell lines.Citation28,Citation29 The mechanism of hepatocyte infection induced by HBV is not absolutely clear; however, specific attachment of viruses to cells is the essential first step in virus entry into cells. Several studies suggested that amino acids 21–47 of preS1 play a major role in the cell attachment.Citation30–Citation32 For example, Dash et al reported that this 27-mer preS1 peptide (21–47) binds to HepG2 cells in a dose-dependent manner with high affinity.Citation32 Furthermore, this binding is possibly tissue-specific, since only weak to insignificant levels of binding were obtained with Vero cells, HeLa cells, and human peripheral lymphocytes. This paper describes the characterization of 27-mer preS1 peptide-carrying nanocapsules and their potential for biomedical applications, including targeted drug delivery and bioimaging.
Figure 1 Schematic illustration of genetically engineered nanocages for hepatocyte selective targeting.
Materials and methods
DNA cloning and expression
Plasmid pET-HSPG41C-preS1, a bacterial expression vector containing the preS1 gene fused in frame with the HSP16.5 variant (HSPG41C), was cloned by polymerase chain reaction (PCR). The HSPG41C mutant presents unique reactive cysteine residues on the interior surface of the assembled cage for the attachment of cargo molecules. This mutant was constructed according to the guidelines noted in a previous paper,Citation19 and is used as a PCR template. The primers for HSPG41C-preS1 were 5′-taatacgactcactataggg-3′(forward primer), 5′-cgcggatccCTACGGGTTGAAGTCCCAGTC-CGGGTTGTTAGAGTTAGCACCGAAAGCCGGGTC CAGCTGGTGGTCCGGGAAGAAACCCAGCGGGCTA GCttcaatgttgattcctttcttaattgagga-3′ (reverse primer), and the latter primer contained a preS1 gene coding region (capital letters) and BamHI restriction site (underlined). The PCR-generated 0.63-kb fragment was digested with NdeI and BamHI and the resulting DNA fragment ligated into NdeI and BamHI-digested pET21a vector (Novagen; Merck Japan, Tokyo, Japan). Correct insertion of the fragment was verified by DNA sequencing and plasmids then amplified and purified by standard techniques.
The resulting vector was allowed to express HSPG41C-preS1 in Escherichia coli BL21-Gold(DE3) cells (Novagen) using a T7 expression system. Bacterial cells of BL21 gold (DE3) containing pET-HSPG41C-preS1 were grown in 2 × YT medium (Sigma-Aldrich, St Louis, MO) with 100 mg/mL ampicillin at 37°C. Recombinant protein production was initiated by addition of isopropyl thiogalacto-pyranoside (IPTG; Wako Pure Chemical Industries, Ltd, Osaka, Japan) to a final 1 mM concentration and incubation at 37°C for 4 hours.
Protein purification and characterization
Cells were pelleted by centrifugation at 4°C, the pellet resuspended in 8 mL of sample buffer (20 mM KH2PO4-KOH, pH 7.0, 2 mM DTT, and 1 mM ethylenediaminetetraacetic acid), and the resulting suspension sonicated on ice (200 watts, 45 seconds). DNase 1 and RNase A were then added to final concentrations of 5 and 1 mg/mL, respectively. The cell lysate mixture was incubated at 4°C for 30 minutes, and the insoluble material removed by centrifugation (20,000 g, 20 minutes) at 4°C. Purification of the recombinant protein was performed by ion-exchange chromatography, in which the supernatant was loaded on a HiLoad 26/10 Q Sepharose HP™ anion-exchange column (Amersham Pharmacia Biotech Inc; GE Healthcare, Little Chalfont, UK) and HSPG41C-preS1 eluted with sample buffer containing 1M NaCl. Fractions containing HSPG41C-preS1, confirmed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), were loaded onto a silica-based GPC column TSKgel G3000SW (Tosoh Corporation, Tokyo, Japan) and HSPG41C-preS1 eluted with storage buffer (25 mM NaH2PO4/Na2HPO4, pH 7.0, and 0.1 M NaCl). The resulting purified HSPG41C-preS1 fractions were analyzed by SDS-PAGE using 12% gel according to standard protocol. The recombinant HSPG41C-preS1 protein cages were characterized by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (Autoflex Speed; Bruker Daltonics Japan, Yokohama, Japan) and dynamic light scattering (Malvern Nanosizer ZS; Malvern Instruments).
Cytotoxicity assays
HepG2, Huh-7, and HeLa cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Wako Pure Chemical Industries, Ltd), supplemented with 1% (v/v) antibiotic-antimycotic mix (Gibco 15240-062; Life Technologies Japan Ltd., Tokyo, Japan) and 10% (v/v) fetal bovine serum (FBS; GE Healthcare, Little Chalfont, UK). MCF-7 cells were cultured in RPMI-1640 (Sigma-Aldrich) with 1% (v/v) antibiotics and 10% FBS and at 37°C in a humidified 5% CO2 atmosphere.
The cellular toxicity of the nanocages in vitro was assessed using the CellTiter-Glo® luminescent cell viability assay (Promega Corporation, Madison, WI) according to the manufacturer’s instructions. This assay is a homogeneous method designed to determine the viable cell numbers in culture based on quantitation of available adenosine triphosphate, signaling the presence of metabolically active cells at the assay end point. Cells (1 × 104 cells/well) were cultured in 96-well microtiter plates in complete growth medium in the presence of increasing nanocapsule concentrations. For chronic toxicity assessment, nanocapsules were applied to cells at various concentrations and incubated for 16 hours prior to assay for viable cells.
Fluorescence assay
Prior to a fluorescence assay, HSPG41C-preS1 and HSPG41C (used as a control) were labeled with fluorescent dye. Briefly, nanocapsules were incubated with a 5-fold molar excess of Alexa488-maleimide (Invitrogen; Life Technologies, Carlsbad, CA) in pH 8.0 phosphate buffer at 50°C overnight and then excess dye removed by ultrafiltration. Uptake or transfection was assessed by seeding 1 × 104 cells in 100 μL of complete growth medium per well in 96-well microtiter plates 12 hours before nanocage addition, to allow for cell attachment. After cell culture medium exchange, the cells were incubated for various times with dye-labeled nanocapsules at a final 1 μM concentration. In inhibition experiments, synthetic preS1 peptide (PLGFFPDHQLDPAFGANSNNPDWDFNP; final concentration, 25 μM) was added, as a competitive inhibitor, to the culture medium 3 hours prior to nanocage addition. After incubation, the cells were washed twice with phosphate buffered saline, the medium changed to a phenol red-free medium, and dye-labeled nanocapsule cellular uptake quantified using a multilabel counter ARVO MX (PerkinElmer Japan Co., Ltd., Yokohama, Japan) excitation and emission, 485 and 535 nm, respectively. Nanocapsule transfection efficiencies were calculated by dividing the total fluorescence intensity in each well by the total number of viable cells in the well, as determined by the cell viability assay.
For investigating nanocapsule cellular localization, HepG2, Huh-7, HeLa, and MCF-7 cells were seeded at 1 × 104 cells/well in μ-Slides (μ-Slide 6 well; ibidi GmbH, Munich, Germany), incubated with colorless DMEM containing 1% antibiotic-antimycotic mix and 10% FBS (PAA; GE Healthcare) overnight. Then, fluorescent-labeled nanocages of HSPG41C-preS1 or HSPG41C control were added to wells at a final 1 μM concentration and, after additional 6 hours of incubation, non-incorporated nanocapsules removed by washing cells twice with phosphate buffered saline and adding fresh medium. Fluorescence confocal laser scanning microscopy was performed with a Bio-Rad Radiance 2100 confocal laser scanning microscopy system (Bio-Rad Laboratories Japan, Tokyo, Japan) attached to a Nikon TE2000-U microscope (Nikon Corporation, Tokyo, Japan). All fluorescence images were acquired under the same conditions of objective lens, laser excitation intensity, photo-multiplier gain, and imaging size. Statistical significances of the differences were determined by Student’s t-test.
Results and discussion
Expression and characterization of preS1-carrying protein nanocages
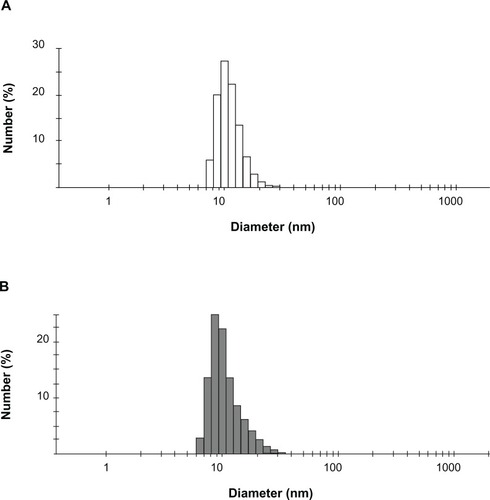
Expression vectors encoding the HSPG41C-preS1 or HSPG41C were constructed and recombinant sHSP16.5 proteins produced in E. coli BL21-Gold(DE3) and purified by sequential anion exchange chromatography followed by size exclusion chromatography under native conditions (Figure S1). Purified proteins, separated by SDS-PAGE, appeared as a single band by Coomassie blue staining. The observed molecular weight of purified HSPG41C-preS1 (m/z = 19676.2 Da), analyzed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry with a sinapic acid matrix, was largely in agreement with calculations (m/z = 19668.2 Da) (Figure S2). The protein nanocage size range and distribution were measured by means of dynamic light scattering. These results indicated that the average nanocage diameter of the HSPG41C-preS1 and HSPG41C were 14.4 and 12.7 nm, respectively, with a narrow size distribution (). These results demonstrated that HSPG41C-preS1 cages were slightly larger than HSPG41C parent cages lacking the targeting peptide.
Figure 2 Dynamic light scattering analysis of protein nanocages. (A) HSPG41C nanocage controls; (B) HSPG41C-preS1 nanocages.
In vitro cellular cytotoxicity of nanocages
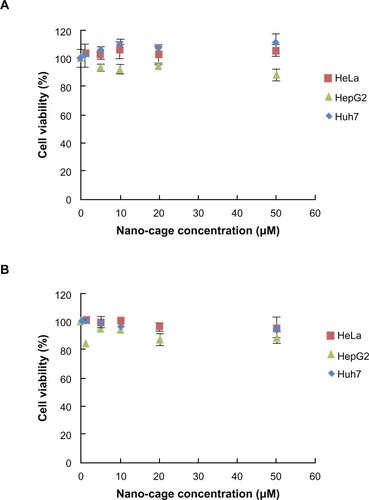
As cell cytotoxicity is an important factor in selecting materials for drug carriers, the nanocages produced here were characterized regarding their effect on cell viability under the same conditions as used for fluorescence assay of cellular uptake experiments. Neither HSPG41C nor HSPG41C-preS1 had any appreciable cytotoxic effect under these conditions (); similar results were obtained from wild type HSP16.5 protein under the same conditions (data not shown). These results showed that the single amino acid substitution mutation, contained in HSPG41C, and the cage surface modification with the preS1 peptide of wild type HSP16.5, contained on HSPG41C-preS1, did not significantly affect their cytotoxicity.
Figure 3 Cell viability measurements after treatment with protein nanocages against various cell lines. (A) HSPG41C nanocage controls; (B) HSPG41C-preS1 nanocages.
Hepatocyte specific delivery of protein nanocages in vitro
The mechanism for the entry of HBV particles into target cells, in particular into hepatocytes, is not yet understood. However, the preS1 surface antigen of HBV is known to play an important role in initial HBV attachment to hepatic cell lines, with the preS1 domain N-terminal segment believed to be essential. To demonstrate that preS1 fusion into the HSP16.5 protein nanocage produced a new nanocage binding affinity towards hepatocyte cell lines, the specific target cells of HBV preS1 molecule, transfection assays were performed.
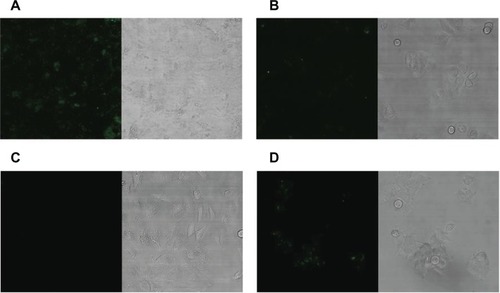
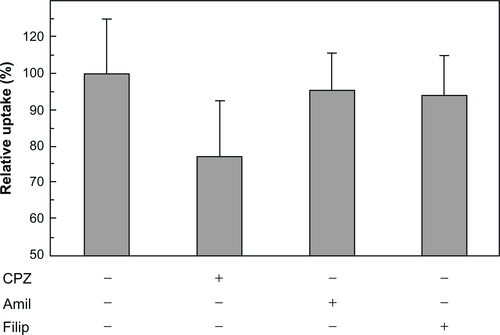
Fluorescent-labeled HSPG41C-preS1 nanocages were observed to efficiently internalize to human hepatoma cell lines HepG2 and Huh-7, which bear specific HBV receptors compared to internalization to the other cell lines (). Incubation with these nanocages resulted in specific accumulation of these proteins in hepatic cell cytoplasm but not in the nucleus. On the other hand, the control fluorescent-labeled HSPG41C nanocages were slightly taken up by Huh-7, HepG2, and MCF-7 under the same conditions (). Nanoparticles are mostly internalized into cells by endocytosis. Endocytosis can be classified into four groups: clathrin-mediated endocytosis (CME), caveolae-mediated endocytosis, macropinocytosis, and clathrin- and caveolae-independent endocyotosis.Citation33,Citation34 We investigated the endocytic pathway of HSPG41C-preS1 nanocages using three types of chemical compound that inhibited specific endocytic pathways (Figure S3). Chlorpromazine inhibits CME,Citation35 amiloride inhibits macropinocytosis,Citation36 and filipin III inhibits caveolae-mediated endocytosis.Citation37 When HepG2 cells were treated with amiloride and filipin, 5% and 6% decreases in cellular uptake of HSPG41C-preS1 nanocages were detected compared with untreated cells. Chlorpromazine treatment showed a marked decrease (23%) in the uptake of HSPG41C-preS1 nanocages. These results suggest that HSPG41C-preS1 nanocage was taken up by HepG2 cells by various types of endocytosis, but the main uptake pathway was CME.
Figure 4 Fluorescence confocal laser scanning microscopy of various cells with fluorescently-labeled HSPG41C-preS1 nanocages. Alexa488-labeled HSPG41C-preS1 nanocages added to culture media with 1 × 104 cells of Huh-7 (A), HepG2 (B), HeLa (C), and MCF7 (D), and extensively washed with PBS before CLSM analysis.
Abbreviations: PBS, phosphate buffered saline; CLSM, confocal laser scanning microscopy.

Figure 5 Fluorescence confocal laser scanning microscopy of various cells with fluorescently-labeled HSPG41C nanocages. Alexa488-labeled HSPG41C nanocages added to culture media with 1 × 104 cells of Huh-7 (A), HepG2 (B), HeLa (C), and MCF7 (D), and extensively washed with PBS before CLSM analysis.
Abbreviations: PBS, phosphate buffered saline; CLSM, confocal laser scanning microscopy.
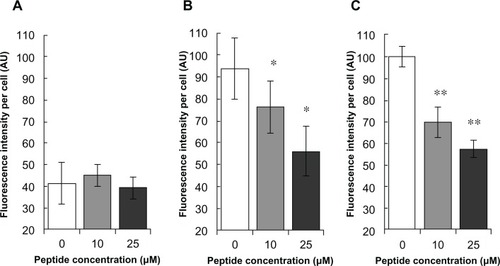
To determine the HSPG41C-preS1 nanocage internalization process into hepatoma cells, competition assays using synthetic preS1 peptide, encompassing amino acid residues 21–47 of preS1, were performed. Fluorescent-labeled HSPG41C-preS1 nanocage incorporation in the presence of various concentrations of synthetic preS1 peptides was intensity normalized to 100 for untreated Huh-7 (). In the case of human hepatoma-derived cell lines HepG2 and Huh-7, synthetic preS1 peptides inhibited incorporation of labeled HSPG41C-preS1 nanocages in a dose-dependent manner. On the other hand, the presence of synthetic preS1 peptides did not significantly influence interaction between these nanocages and HeLa cells. These results suggested that the cell binding observed for these nanocages was due specifically to the presence of the preS1 moiety on their surfaces and the internalization of the nanocages was achieved by interaction with preS1 receptors on the hepatocyte cell lines.
Figure 6 Inhibitory effects of preS1 peptide against transfection of HSPG41C-preS1 nanocages.
Notes: Cell lines HeLa, HepG2, and Huh-7 (A–C), respectively; statistical significance of differences in fluorescence intensities in absence and presence of preS1 peptide assessed by Student’s t-test; *P < 0.05; **P < 0.01.
Conclusion
In summary, genetic incorporation of hepatocyte binding preS1 peptide onto the exterior surface of nanocages conferred cell-specific targeting capabilities to this protein cage architecture. These protein nanocages were efficiently internalized into hepatocyte cell lines, mainly through clathrin-mediated endocytosis, without cytotoxicity, and were localized at cytoplasm. Furthermore, the binding of HSPG41C-preS1 nanocages to HepG2 cells was prevented by synthetic preS1-(21–47) peptide in a dose-dependent manner, suggesting that this sequence could be directly responsible for nanocage attachment to the cellular surface. In fact, it is known that the preS1 domain of HBV is modified with myristic acid at the N-terminal region and that this modification is important for efficient infectivity,Citation27,Citation28 and thus it appears that it would be necessary to conjugate the HSPG41C-preS1 nanocages with a similar lipid to enhance specificity in hepatocyte targeting in vivo. This strategy was effective for producing suitable nanocages that were directed to and taken up by specific cell types, target organs, or cancer cells, which is an important development in such applications, as ligand-mediated active binding to sites and cellular uptake are particularly valuable to therapeutic agents that are not easily taken up by cells. Furthermore, these nanocages are self-organized complexes of protein oligomers and possess nanoscale inner cavities, whose three-dimensional architecture can be specified in detail and into which a variety of molecules, including therapeutic or diagnostic agents, could be encapsulated.
Acknowledgements
This work was supported by a Health Labor Sciences Research Grant (Research on Publicly Essential Drugs and Medical Devices) from the Ministry of Health Labor and the Special Coordination Funds for Promoting Science and Technology (SCF funding program “Innovation Center for Medical Redox Navigation”), Japan.
Disclosure
The authors report no conflict of interest in this work.
Supplementary figures
Figure S1 Gel permeation chromatography purification of HSPG41C nanocages and HSPG41C-preS1 nanocages. GHSPG41C nanocages (A); HSPG41C-preS1 nanocages (B). The peaks observed at 13.31 minutes in (A) and 13.26 minutes in (B) were collected individually. The collected fractions were then analyzed by SDS-PAGE using 12% gel according to the standard protocol (C).
Note: Lane 1, molecular weight standards; lane 2, HSPG41C nanocages; lane 3, HSPG41C-preS1 nanocages.
Abbreviation: SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis.
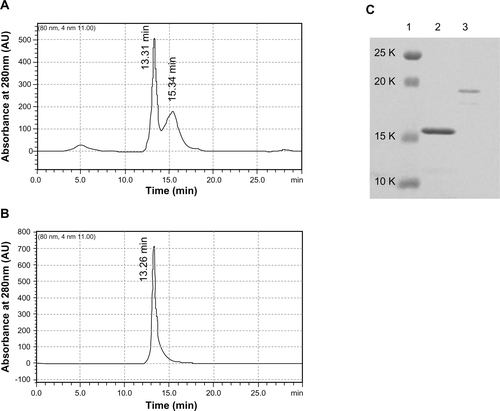
Figure S2 MALDI-TOF mass spectrum of HSPG41C nanocages and HSPG41C-preS1 nanocages. GHSPG41C nanocages (A); HSPG41C-preS1 nanocages (B).
Abbreviation: MALDI-TOF, matrix-assisted laser desorption/ionization time of flight.
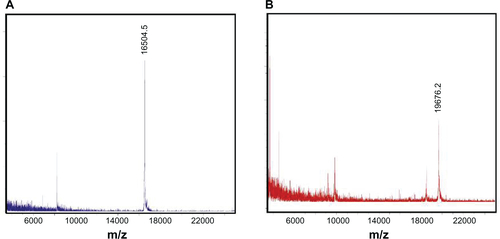
Figure S3 Inhibition of cellular uptake of HSPG41C-preS1 nanocages by endocytosis inhibitors.
Notes: HepG2 cells were harvested on poly-L-lysine-coated 48-well plates at an initial density of 50,000 cells/well and grown overnight. Cells were treated with DMEM containing chlorpromazine (10 μg/mL), amiloride (500 μM), or filipin III (10 μg/mL) for 1 hour (all from Sigma-Aldrich, St Louis, MO). Then 84 nM of fluorescent-labeled HSPG41C-preS1 nanocages solution containing the above inhibitors was added to cells. Two hours after transfection, cells were rinsed three times with PBS and then lysed in lysis buffer (pH 7.5, 20 mM Tris-HCl, 2 mM EDTA, and 0.05% Triton-X 100). Lysate solutions were replaced on black-bottomed 96-well plates and the fluorescence intensity of each sample measured using a Microplate Reader (ARVO MX 1420; Perkin Elmer Inc, Waltham, MA). Data are means ± SEM of three independent experiments.
Abbreviations: CPZ, chlorpromazine; Amil, amiloride; Filip, filipin; DMEM, Dulbecco’s Modified Eagle’s Medium; PBS, phosphate buffered saline; HCl, hydrochloride; EDTA, ethylenediaminetetraacetic acid; SEM, standard error of the mean.
References
- KangJHOishiJKimJHHepatoma-targeted gene delivery using a tumor cell-specific gene regulation system combined with a human liver cell-specific bionanocapsuleNanomedicine20106458358920138242
- PridgenEMLangerRFarokhzadOCBiodegradable, polymeric nanoparticle delivery systems for cancer therapyNanomedicine (Lond)20072566968017976029
- SawantRMHurleyJPSalmasoS“SMART” drug delivery systems: double-targeted pH-responsive pharmaceutical nanocarriersBioconjug Chem200617494394916848401
- WangMThanouMTargeting nanoparticles to cancerPharmacol Res2010622909920380880
- BraydenDJControlled release technologies for drug deliveryDrug Discov Today200382197697814643159
- AllenTMCullisPRDrug delivery systems: entering the mainstreamScience319200430356651818182215031496
- AsoSIseHTakahashiMEffective uptake of N-acetylglucosamine-conjugated liposomes by cardiomyocytes in vitroJ Control Release2007122218919817681632
- KhandareJMinkoTPolymer-drug conjugates: progress in polymeric prodrugsProg Polym Sci2006314359397
- ToitaRKangJHKimJHProtein kinase C alpha-specific peptide substrate graft-type copolymer for cancer cell-specific gene regulation systemsJ Control Release2009139213313919545594
- TsuchiyaANaritomiYKushioSImprovement in the colloidal stability of protein kinase-responsive polyplexes by PEG modificationJ Biomed Mater Res A201210051136114122337618
- YoshinoriMMasaharuMYuriHSayokoNNaoSMakotoHMolecular imaging contrast media for visualization of liver functionMagn Reson Imaging201028570871520378293
- DavisMEChenZGShinDMNanoparticle therapeutics: an emerging treatment modality for cancerNat Rev Drug Discov20087977178218758474
- FlennikenMLWillitsDAHarmsenALMelanoma and lymphocyte cell-specific targeting incorporated into a heat shock protein cage architectureChem Biol200613216117016492564
- KasuyaTKurodaSNanoparticles for human liver-specific drug and gene delivery systems: in vitro and in vivo advancesExpert Opin Drug Deliv200961395219236207
- SaoKMurataMFujisakiYA novel protease activity assay using a protease-responsive chaperone proteinBiochem Biophys Res Commun2009383329329719341711
- UchidaMKlemMTAllenMBiological containers: protein cages as multifunctional nanoplatformsAdv Mater200719810251042
- MacRaeTHStructure and function of small heat shock/alpha-crystallin proteins: established concepts and emerging ideasCell Mol Life Sci200057689991310950306
- SunYMacRaeTHSmall heat shock proteins: molecular structure and chaperone functionCell Mol Life Sci200562212460247616143830
- SaoKMurataMUmezakiKMolecular design of protein-based nanocapsules for stimulus-responsive characteristicsBioorg Med Chem2009171859319041251
- KimDRLeeIHaSCKimKKActivation mechanism of HSP16.5 from Methanococcus jannaschiiBiochem Biophys Res Commun2003307499199812878210
- KimRKimKKYokotaHKimSHSmall heat shock protein of Methanococcus jannaschii, a hyperthermophileProc Natl Acad Sci U S A19989516912991339689045
- NakamotoHVighLThe small heat shock proteins and their clientsCell Mol Life Sci200764329430617187175
- KaiserCRFlennikenMLGillitzerEBiodistribution studies of protein cage nanoparticles demonstrate broad tissue distribution and rapid clearance in vivoInt J Nanomedicine20072471573318203438
- KimKKKimRKimSHCrystal structure of a small heat-shock proteinNature199839466935955999707123
- ArgnaniRBoccafogliLMarconiPCManservigiRSpecific targeted binding of herpes simplex virus type 1 to hepatocytes via the human hepatitis B virus preS1 peptideGene Ther200411131087109815057264
- De FalcoSRuvolettoMGVerdolivaACloning and expression of a novel hepatitis B virus-binding protein from HepG2 cellsJ Biol Chem200127639366133662311389143
- ParanNCooperAShaulYInteraction of hepatitis B virus with cellsRev Med Virol200313313714312740829
- BarreraAGuerraBNotvallLLanfordREMapping of the hepatitis B virus pre-S1 domain involved in receptor recognitionJ Virol200579159786979816014940
- GlebeDUrbanSViral and cellular determinants involved in hepadnaviral entryWorld J Gastroenterol2007131223817206752
- NeurathARKentSBStrickNParkerKIdentification and chemical-synthesis of a host cell receptor binding site on hepatitis B virusCell19864634294363015414
- QiaoMMacnaughtonTBGowansEJAdsorption and penetration of hepatitis B virus in a nonpermissive cell lineVirology199420123563638184545
- DashSRaoKVPandaSKReceptor for Pre-Sl(21–47) component of hepatitis B virus on the liver cell: role in virus cell interactionJ Med Virol19923721161211629710
- SahayGAlakhovaDYKabanovAVEndocytosis of nanomedicinesJ Control Release2010145318219520226220
- ConnerSDSchmidSLRegulated portals of entry into the cellNature20034226927374412621426
- WangL-HRothbergKGAndersonRGWMis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formationJ Cell Biol19931235110711178245121
- HewlettLJPrescottARWattsCThe coated pit and macropinocytic pathways serve distinct endosome populationsJ Cell Biol199412456897038120092
- LamazeCSchmidSLThe emergence of clathrin-independent pinocytic pathwaysCurr Opin Cell Biol1995745735807495578