Abstract
This paper attempts to predict and emphasize molecular interactions of dopamine, levodopa, and their derivatives (Dopimid compounds) containing 2-phenyl-imidazopyridine moiety with the α-cyclodextrin dimer in order to assess and improve drug delivery to the central nervous system. The molecular docking method is used to determine the energetic profiles, hydrogen bond formation, and hydrophobic effect of 14 host–guest complexes. The results show that the “chemical branching” represented by additional ethyl-acetate residue is energetically unfavorable and promotes a conformational shift due to the high root mean square deviation levels. This phenomenon is characterized by a low number of H-bonds and a significant decrease of the host–guest hydrophobic potential surface. Finally, the overall docking procedure presents a powerful rationale for screening and analyzing various sets of promising drug-like chemical compounds in the fields of supramolecular chemistry, molecular sensing, synthetic receptors, and nanobiotechnology.
Introduction
Molecular encapsulation of chemical compounds with cyclodextrins (CDs), a form of cone-shaped oligosaccharides, has drawn much attention in recent years for its ability to improve drug pharmacokinetic and pharmacodynamic properties and to potentiate biological effects.Citation1,Citation2 Therefore, experimental and theoretical methodologies investigating the structural features of CD–drug complexes, as well as their mechanisms of interaction, are of increasing importance. Only a few studies have reported that α-and β-CDs are good binders for dopamine (3-hydroxytyramine, 3,4-dihydroxyphenethylamine [DA]), an important neurotransmitter that is widely distributed in the mammalian central nervous system (CNS).Citation3–Citation5
Newly synthesized amphiphilic α-cyclodextrin esters have the potential to serve as DA carrier moleculesCitation36 and to facilitate drug delivery to the CNS. However, many CNS-active drugs, such as levodopa (L-3,4-dihydroxyphenylalanine [L-DOPA]) and its derivatives, are characterized by low or insufficient water solubility, oxidation, and decarboxylation reactivity with intestinal and blood enzymes.Citation6,Citation7
It is already known that a lack of DA in the brain is associated with the progressive neurological ailment known as Parkinson’s disease.Citation8 In this context, the effectiveness of DA and L-DOPA derivatives to treat this disorder would be highly compromised. Some attempts have been made to bypass these problems by formulation and complexation in CD, and to promote drug absorption by extracting cholesterol and phospholipids from the membranes.Citation9,Citation36 Overall, the improvement of intestinal absorption of poorly soluble compounds by CDs is an important parameter because it allows these compounds to reach lipophilic areas in aqueous environments.Citation10 In fact, a central hydrophobic channel of CD can encompass a chemical compound as a guest molecule and can produce a host–guest complex formation, thereby enhancing the solubility rate.Citation11–Citation14 On the other hand, CDs may protect guest molecules from chemical hydrolysis by diminishing their ability to bind to plasma proteins for analyzed substances.Citation15 In this way, CDs can increase the plasma level of complexed drugs and, ultimately, increase their therapeutic effect.
The mechanisms that involve the interaction of hydrogen bonds are van der Waals’ forces and the hydrophobic effect; these mechanisms are at the core of host–guest inclusion complexation.Citation16 Considering the lipophilic interior of CDs, the binding ability of DA/L-DOPA derivatives is generally attributable to a hydrophobic interaction, and the conformational shape and size complementarities between these structures.
This study docked a new series of DA and L-DOPA derivatives with a tail-to-tail α-CD dimer to determine and characterize the supramolecular driving forces of the host–guest complexation, and to emphasize its ability to assess and improve drug delivery to the central nervous system.
Computational methods
The crystal holo-structure of a tail-to-tail α-CD dimer with a monofunctional carboxylic acid was taken from the OpenCDLig, a free web application for sharing resources about cyclodextrin complexes.Citation17 The aliphatic chain of the carboxylic acid was threaded through a α-CD dimer, resulting in a 1:2 stoichiometry.Citation32
DA and L-DOPA molecular structures were extracted from the PubChem database with reference codes CID_681 and CID_6047, respectively (). The chemical formulas of the so-called Dopimid compounds () were obtained from a research article by Denora et al.Citation27 The two-dimensional (2D) structures of these compounds were drawn using MarvinSketch software (ChemAxon, Budapest, Hungary).Citation18 After the 2D sketches were converted into three-dimensional (3D) images, explicit hydrogens were added and the structures were energetically minimized and recorded in PDB format. Log BB, human intestinal absorption (HIA), and plasma protein binding (PPB) parameters were predicted by the PreADMET workflow using the genetic functional approximation method.Citation19 This strategy is based on the calculation of molecular descriptors, which involves dividing all compounds into training, validation, and external sets.
Figure 1 Chemical structure of DA, L-DOPA, and Dopimid compounds.
Abbreviations: DA, dopamine; L-DOPA, levodopa.
The octanol–water partition coefficient (log P) as a measure of molecular hydrophobicity was determined by the robust Molinspiration method (miLogP2.2, 2005) using the group contributions. These were obtained by fitting the experimental log P values with predicted ones for a training set of more than 12,000 drug-like compounds.
The hydrophobic cavity volume was calculated using the MOLCAD module that is integrated in the Sybyl-X 1.1 software (Tripos International, St Louis, MO). The binding site () was detected in the α-CD dimer molecule using the PocketAnalyzerPCA program (Novartis Institutes for Biomedical Research, Horsham, UK).Citation20 Before starting the molecular docking, the AutoDockTools software (Molecular Graphics Laboratory Department of Molecular Biology, MB-5 The Scripps Research Institute, La Jolla, CA).Citation21 was used to optimize the guest compounds from the PDB files, add Gasteiger charges, assign polar hydrogen atoms, and set up rotatable bonds. Flexible molecular docking was applied to the center of the binding cavity with the AutoDock Vina (ADVina) docking engine (Molecular Graphics Laboratory)Citation22 using the following Cartesian coordinates: x = 8.52 Å, y = 13.94 Å, z = 8.99 Å. A docking grid with a dimension of 25 Å × 25 Å × 25 Å was used. The ADVina output results represented the docking scores as Gibbs free energy of binding (ΔG), and were further converted to the predicted equilibrium constants (Kc). The Kc values for all the docked poses were calculated from the ΔG values as follows: Kc = exp([ΔG*1000]/[R*T]), where R (gas constant) is 1.98 cal*(mol*K)−1 and T (room temperature) is 298.15 Kelvin. The molecular poses were analyzed with the PyMol AutoDock/Vina plug-in (Computational Biomolecular Dynamics Group, Max-Planck-Institute for Biophysical Chemistry, Göttingen, Germany) and the Visual Molecular Dynamics program (version 1.9; Theoretical and Computational Biophysics Group, Beckman Institute, Urbana, IL). In situ optimization and refinement of docking poses were performed with the Szybki program (OpenEye Scientific Software, Santa Fe, NM) using the Merck Molecular Force Field (MMFF94; Merck & Co, Whitehouse Station, NJ) without the solvent effect. MMFF94-optimized geometries based on the atom types, energies, and their gradients were calculated and strictly validated against Merck’s published dataset.Citation23
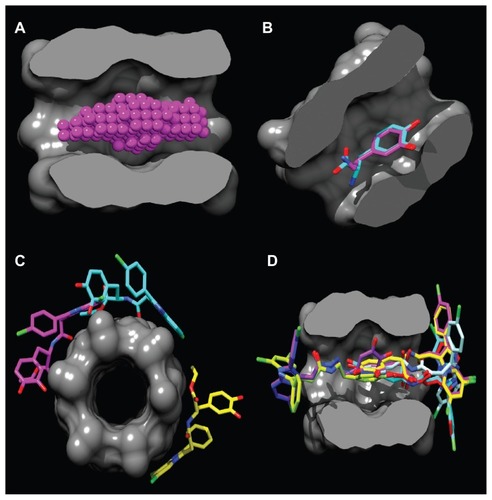
Figure 2 Binding site in the α-CD dimer cavity (A) is detected with the PocketAnalyzerPCA (Novartis Institutes for Biomedical Research, Horsham, UK). (B) Shows the docking of the DA and L-DOPA molecules and the mode of hydrophobic moiety (2,3-dihydroxyphenyl) interaction with the α-CD dimer. Out-channel docking poses are exposed outside the α-CD binding site (C), while in-channel binding modes (D) are docked inside the cavity, highlighting the phenomenon of chiral docking. The molecular surface is divided by the frontal plane to visualize a binding channel of the host. Hydrogen atoms are omitted for clarity.
Abbreviations: α-CD, α-cyclodextrin; DA, dopamine; L-DOPA, levodopa.
Molecular graphics and visualization were performed with the Chimera software (Resource for Biocomputing, Visualization, and Informatic, San Francisco, CA).Citation24 The host–guest molecular hydrophobicity potential surface (SMHP) was calculated before (MMFF94−) and after (MMFF94+) the MMFF94 optimization ( and ) and hydrogen bonds were formed. This was achieved with the PLATINUM web tool (Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, Ul., Moscow, Russia)Citation25 using the following equation: SMHP = (Sburied − SH/H)/(Stotal), where SH/H is the hydrophilic match surface, Sburied is the contact (hydrophobic) surface, and Stotal is the total ligand surface.
Table 1 Molecular properties of chemical compounds
Table 2 Predicted parameters for MMFF94− conformational set
Table 3 Predicted parameters for MMFF94+ conformational set
To assign SMHP correctly, the hydrophobic properties of a host and its guest were projected onto the molecular surface of the host. Statistical analyses were performed using a linear regression analysis followed by a 3D graphic representation using GraphPad Prism v.4. for Windows (GraphPad Software, San Diego, CS) and SigmaPlot 11.0 (Systat Software GmbH, Germany). The differences were considered statistically significant at P < 0.0001.
Results and discussion
Evaluation of physicochemical properties and the blood–brain barrier (BBB) permeability rates of guest compounds
The calculated log P and log BB values for DA, L-DOPA, and Dopimid compounds were obtained to evaluate their ability to act at the CNS level and possess optimal BBB permeation properties (). Denora et al have shown that Dopimid compounds 1–12 include log P values in the range of 3.03–5.13, which is similar to our data (1.72–5.47), which was calculated by the Molinspiration algorithm. These results indicate the lipophilic character of the examined compounds, which was considered high enough to cross the BBB with the exception of DA and L-DOPA (log P −0.05 and −2.19). However, the log P has proven insufficient for the accurate evaluation of BBB permeation, since it correlates poorly with the log BB.
According to the CNS± activity classification for different chemical datasets,Citation26 compounds with log BB > 0 can cross the BBB readily, while drugs with log BB < 0 cannot. Therefore, only Dopimid compounds 9–12 (log BB 0.01–0.4) should be considered as BBB-permeants; these results deviate from the previously shown data.Citation27 The authors provide the log BB distribution values in a range from −0.57 to −1.24 for the examined derivatives, which indicates the BBB impermeability prerequisites. The experimental log BB value for L-DOPA is approximately twofold lower than the predicted one (−0.77 versus −0.36), which is probably due to P-g efflux mechanism’sCitation28 influence on metabolic factors, or other experimental difficulties such as compound extraction and purification. The discrepancies of the log BB prediction can be explained by the fact that the authors used a simplified Clark’s model approach that relates this coefficient to the polar surface area descriptor.Citation29
PPB and human intestinal absorption rates for Dopimid compounds were more than 80% and 90%, respectively. However, the parameters for DA and L-DOPA were significantly reduced, particularly for PPB. As a control, the PPH and HIA values for L-DOPA (<10% and 40% ± 19%)Citation30,Citation31 closely matched our predicted results (2.96% and 59.66%).
Analysis of ADVina docking results and binding affinities of α-CD dimer complexation
In terms of the molecular docking studies, we set out to analyze the host–guest complexation mechanism of α-CD dimer, DA, L-DOPA molecules, and Dopimid compounds by measuring the binding affinities. Since it engulfs most of the guest molecule, the α-CD dimer can be considered more suitable for molecular docking than its monomer, which has a deep 346.604 Å3 hydrophobic cavity volume and an approximate length of 15.6 Å (174 Å3 and 7.8 Å for monomer). The dimer was kept in the form of a single conformation as provided in the crystal structure,Citation32 since it is rather rigid, and the flip-flop hydrogen bonds between the secondary hydroxyl groups restrict the flexibility of the hydrophobic core. The cavity size of the α-CD dimer must be large enough to allow the guest molecule to enter by keeping the guest molecule inside the cavity via a high stability constant, which achieves relatively low dissociation of the inclusion. Furthermore, it does not implement significant conformational alterations when complexing to different chemical compounds.
A standard molecular docking procedure yielded two main outcomes: a conformational sampling (docking pose) of guest molecules within the host binding site, and a score approximation function (ΔG) representing the strength of host–guest interaction. All docking poses were visually inspected to rank the different host conformations of the complex.
As shown in , the location of the DA and L-DOPA docking poses is roughly defined in the middle of the α-CD dimer, where the binding site is positioned. The site is stabilized by intramolecular H-bonds between the adjacent secondary hydroxyl-groups with a mean distances of 2.78 Å. The α-CD dimer encompasses the whole DA, L-DOPA molecules, and the 2,3-dihydroxyphenyl moiety of the Dopimid compounds, while the lipophilic 2-phenyl-imidazoperidyne moiety is exposed outside the α-CD binding channel. However, this hydrophobic moiety could serve as a transporter system to increase DA and L-DOPA brain concentrations by considering the high BBB permeation of the phenyl-imidazopiridyne derivatives.Citation33
Compounds 5, 6, and 7 were docked outside the α-CD dimer binding channel, representing “out-channel” noninclusion complexation with maximal ΔG values of −4.4, −4.5, and −4.3 kcal*mole−1, respectively (). The rest of the guest molecules were docked “in-channel” through the entry site at the narrow 6-rim of the dimer where primary hydroxyl groups exist (). Meanwhile, guest lipophilic phenyl groups fit better into a wider 2, 3-rim of the relatively hydrophobic cavity. Interestingly, “in-channel” docking modes were subdivided into the R-site docking for DA, L-DOPA, compounds 1–4, 8, 9, and the S-site docking for compounds 10–12, indicating the “chiral” character of this type of host–guest complexation.
Dopimid compounds 9, 10, and 11 were associated with the best binding modes, which represents the minimal ΔG because of the strong binding affinity and intermolecular force between the host and its guest.
Conformational analysis of Dopimid molecules and docking poses
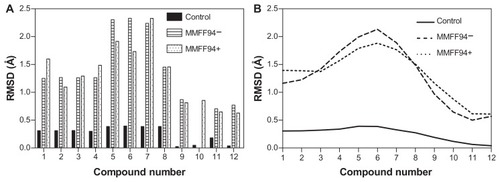
Dopimid compounds contain different numbers of chloride radical residues and functional groups (hydroxyl, carboxyl, and carbonyl groups) as a result of hydroxylation, chlorination, and other chemical modifications of the initial structure such as N-(2-phenylethyl)-2-{2-phenylimidazo[1,2-a]pyridine-3-yl}acetamide (reference molecule). To emphasize the differences between the minimized Dopimid molecules and the docking poses, we aligned these compounds with the initial structure and measured the root-mean-square deviation (RMSD) values. To achieve this task, the Dopimid modifications were preliminarily removed to reach a proper atomic pair-wise matching. Prior to the rigid alignment, Dopimid docking poses were subjected to in situ MMFF94 refinement to optimize the noncovalent host–guest interactions within the α-CD dimer active site. Although the parameters in the force field are derived initially from computational data, they perform well for a wide range of organic chemistry calculations.Citation34 MMFF94-docking poses are associated with the highest level of deviation rates. The RMSD values were even more prominent at compounds 5, 6, and 7 compared to the minimized Dopimid control substances (). These compounds have additional ethyl-acetate residue in their structure that might contribute to the conformational shift that is the result of “chemical branching.” At this point, the chemical branching is energetically unfavorable; it restricts the conformational shape of the guest molecules and prevents them from integrating into the α-CD dimer active channel. The restriction factor might be the insufficient diameter of the α-CD inner cavity (5.7 Å), which is less than the other cyclodextrins (7.8 Å for β-CD and 9.8 Å for γ-CD, respectively).
Figure 3 Reported guest RMSD values for Dopimid compound (rigid alignment was performed on the reference molecule) (A). Data smoothing was utilized to produce a fitting curve for the RMSD comparison (B).
Abbreviations: MMFF94, Merck Molecular Force Field (Merck & Co, Whitehouse Station, NJ); MMFF94−, before MMFF94 optimization; MMFF94+, after MMFF94 optimization; RMSD, root-mean-square deviation.
Estimation of hydrogen bonding and hydrophobic effect for α-CD dimer complexes
The mechanisms of noncovalent interaction in host–guest complexes cover dipolar, ionic interactions, hydrogen bonding, van der Waals interactions, and so on. Nevertheless, the driving force of inclusion complexation is hydrophobic interaction and the host–guest complementary match, which are mainly attributed to the molecular shape.Citation35
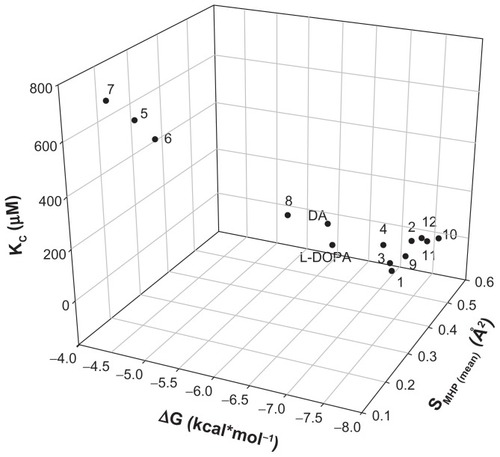
The PLATINUM methodology was applied to investigate if the hydrogen bonding and hydrophobic forces have an impact on the complexation mechanism. H-bond formations were established for all the analyzed compounds () and represented different configurations observed in the dataset including H-bonds between the host–guest functional groups (amino, hydroxyl, and carbonyl groups). demonstrates that the corresponding Dopimid compounds, such as 5 and 6, have shown the minimal number of H-bonds with the α-CD dimer after MMFF94 refinement as an outcome of the unfavorable energetic profile and the “out-channel” docking. This holds true for the same compounds as well as compound 7; the calculated SMHC (MMFF94−/MMFF94+/mean) variables would also give the minimal measurements (). Consequently, the obtained SMHC(MMFF94−), SMHC(MMFF94+), SMHC(mean) and equilibrium constants (Kc) represented a relationship of correlation by a linear regression analysis with reliable statistics (r2 = 0.73 [0.87], F = 32.56 [81.29], n = 14), and P < 0.0001. shows that of the 14 guest molecules, eight landed in one major cluster (compounds 1, 2–4, 9–12); and the rest formed two minor clusters comprising three substances each: compounds 5–7 and DA, L-DOPA, and compound 8. The clustering of the chemical compounds shows that the distribution rate occurs in an MHP-dependant manner and that high levels of ΔG and Kc correlate with low levels of the SMHP parameters.
Figure 4 H-bond formation before and after the MMFF94 optimization (A). Relationship between calculated hydrophobic descriptors (B) and Kc values (C) using the linear regression analysis to determine a clustering for examined chemical compounds.
Note: SMHP = (Sburied − SH/H)/(Stotal), where SH/H is the hydrophilic match surface, Sburied is the contact (hydrophobic) surface, and Stotal is the total ligand surface.
Abbreviations: DA, dopamine; Kc, predicted equilibrium constant; L-DOPA, levodopa; MMFF94, Merck Molecular Force Field (Merck & Co, Whitehouse Station, NJ); MMFF94−, before MMFF94 optimization; MMFF94+, after MMFF94 optimization.
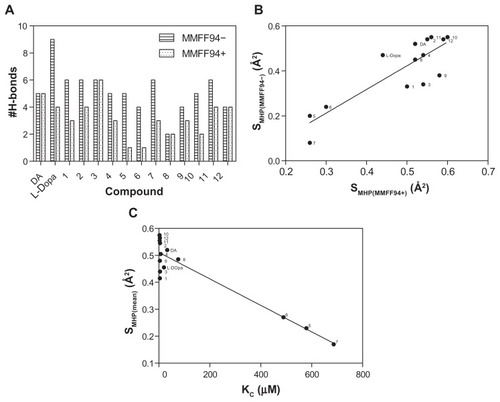
Figure 5 3D dot plot-visualization for the compounds reported in . The distribution of the cluster formation is based on the energy function, equilibrium constant, and hydrophobicity.
Notes: SMHP = (Sburied − SH/H)/(Stotal), where SH/H is the hydrophilic match surface, Sburied is the contact (hydrophobic) surface, and Stotal is the total ligand surface.
Abbreviations: ΔG, Gibbs free energy of binding; DA, dopamine; Kc, predicted equilibrium constant; L-DOPA, levodopa.
Conclusion
This study presents a molecular docking strategy to analyze the inclusion complexation of DA and L-DOPA derivatives enhanced by an appropriate 2-phenyl-imidazopyridine moiety using a α-CD dimer. This technique makes it possible to obtain conformational geometric structures of the host–guest inclusion complexes with docking energies. The guest molecules inside the immobilized α-CD dimer produced encapsulation via their size and shape complementary adaptation to a hydrophobic cavity of the dimer.
The 2-phenyl-imidazolpyridine moiety improves the lipophilicity of the Dopimid compounds and, partly, the log BB parameters of the Dopimid compounds. However, it has a negative effect on PPB and human intestinal absorption. Chemical branching is characterized by a low number of H-bond formations and a significant decrease in the hydrophobic potential surface (SMHP). From the results mentioned above, we can conclude that the ethyl-acetate residues for Dopimid compounds 5, 6, and 7 are involved in the conformational shift with very high values of ΔG and Kc. Therefore, a combination of host–guest hydrophobic interactions with energetic profiles is a novel aspect that allows more rational formulation prerequisites for applications in the supramolecular chemistry of cyclodextrins.
Acknowledgments
The authors are grateful to the IZKF (Interdisziplinäres Zentrum für Klinische Forschung der Universität Würzburg) and the BMBF (Bundesministerium für Bildung und Forschung) for their support of this work by providing grants (BMBF01, EO1004) to Carola Förster.
Disclosure
The authors report no conflicts of interest in this work.
References
- SzejtliJCyclodextrins and their Inclusion ComplexesBudapestAkadémiai Kiadó1982
- PithaJSzenteLSzejtliJMolecular encapsulation of drugs by cyclodextrins and congenersBruckSDControlled Drug Delivery1Boca Raton, FLCRC Press1983124148
- SzejtliJCyclodextrin TechnologyDordrechtKluwer Academic Press1988
- GaoZNWenXLLiHLStudy of the inclusion complexes of catecholamines with b-cyclodextrin by cyclic voltammetryPol J Chem200276710011007
- FukudaTMaedaYKitanoHStereoselective inclusion of DOPA derivatives by a self-assembled monolayer of thiolated cyclodextrin on a gold electrodeLangmuir199915518871890
- Di StefanoBMosciattiGCingolaniMDimeric L-Dopa derivatives as potential prodrugsBiorg Med Chem20011110851088
- WangHLeeJTsaiMLuHHsuWSynthesis and pharmacological activities of a novel tripeptide mimetic dopamine prodrugBiorg Med Chem1995521952198
- LiYHuangXChenYWangLLinXSimultaneous determination of dopamine and serotonin by use of covalent modification of 5-hydroxytryptophan on glassy carbon electrodeMicrochimica Acta20091641–2107112
- IrieTUekamaKPharmaceutical application of cyclodextrins: Toxycological issues and safety evaluationJ Pham Sci1997862147162
- IrieTTsunenariYUekamaKJosefPEffect of bile on the intestinal absorption of α-cyclodextrin in ratsInt J Pharm1988431–24144
- LoftsonTBrewsterMEPharmaceutical applications of cyclodextrins. 1. Drug solubilization and stabilizationJ Pharm Sci199685101710258897265
- ConnorsKAThe stability of cyclodextrin complexes in solutionChem Rev1997971325135711851454
- UekemaKHirayamaFIrieTCyclodextrin drug carrier systemsChem Rev1998982045207611848959
- StellaVJRajewskiRACyclodextrins: their future in drug formulation and deliveryPharm Res1997145565679165524
- FrijlinkHWFranssenEJEissensACOostingRLerkCFMeijerDKThe effects of cyclodextrins on the disposition of intravenously injected drugs in the ratPharm Res1991833803842052529
- SteedJWTurnerDRWallaceKCore Concepts in Supramolecular Chemistry and NanochemistryBognor RegisJohn Wiley & Sons, Ltd2007
- EspositoRErmondiGCaronGOpenCDLig: a free web application for sharing resources about cyclodextrin/ligand complexesJ Comput Aided Mol Des200923966967519533371
- PirokGMatéNVargaJMaking “real” molecules in virtual spaceJ Chem Inf Model200646256356816562984
- RogersDHopfingerAJApplication of genetic function approximation to quantitative structure-activity relationships and quantitative structure-property relationshipsJ Chem Inf Comput Sci1994344854866
- CraigIRPflegerCGohlkeHEssexJWSpiegelKPocket-space maps to identify novel binding-site conformations in proteinsJ Chem Inf Model201151102666267921910474
- MorrisGMHueyRLindstromWAutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibilityJ Comput Chem200930162785279119399780
- TrottOOlsonAJAutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreadingJ Comput Chem201031245546119499576
- WlodekSSkillmanAGNichollsALigand entropy in gas-phase, upon solvation and protein complexation. Fast estimation with Quasi-Newton HessianJ Chem Theory Comput20106721402152
- PettersenEFGoddardTDHuangCCUCSF Chimera – a visualization system for exploratory research and analysisJ Comput Chem200425131605161215264254
- PyrkovTVChugunovAOKrylovNANoldeDEEfremovRGPLATINUM: a web tool for analysis of hydrophobic/hydrophilic organization of biomolecular complexesBioinformatics20092591201120219244385
- AjayBemisGWMurckoMADesigning libraries with CNS activityJ Med Chem199942244942495110585204
- DenoraNLaquintanaVLopedotaANovel L-Dopa and dopamine prodrugs containing a 2-phenyl-imidazopyridine moietyPharm Res20072471309132417404814
- Soares-Da-SilvaPSerrãoMPOutward transfer of dopamine precursor L-3,4-dihydroxyphenylalanine (L-dopa) by native and human P-glycoprotein in LLC-PK(1) and LLC-GA5 col300 renal cellsJ Pharmacol Exp Ther2000293269770410773046
- ClarkDERapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. 2. Prediction of blood–brain barrier penetrationJ Pharm Sci199988881582110430548
- SeyffartGDrug dosage in Renal InsufficiencyBerlinSpringer-Verlag GmbH1991
- LennernäsHNilssonDAquiloniusSMAhrenstedtOKnutsonLPaalzowLKThe effect of L-leucine on the absorption of levodopa, studied by regional jejunal perfusion in manBr J Clin Pharmacol19933532432508471400
- Rodríguez-LlamazaresSYutronicNJaraPEnglertUNoyongMSimonUThe structure of the first supramolecular α-cyclodextrin complex with an aliphatic monofunctional carboxylic acidEur Journ Org Chem200742984300
- DurandAThénotJPBianchettiGMorselliPLComparative pharmacokinetic profile of two imidazopyridine drugs: zolpidem and alpidemDrug Metab Rev19922422392661576937
- HalgrenTAMerck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94J Comput Chem1996175–6490519
- LehnJMSupramolecular chemistry – scope and perspectives molecules, supermolecules, and molecular devices (Nobel Lecture)Angew Chem Int Ed Engl198827189112
- SeyediSMSadeghianHJabbariAAssadieskandarAMomeniHSynthesis of new series of α-cyclodextrin esters as dopamine carrier moleculeBioorg Med Chem201119144307431121700467