 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Background
Lipid-based liquid crystalline nanoparticles (LCNPs) have attracted growing interest as novel drug-delivery systems for improving the bioavailability of both hydrophilic and hydrophobic drugs. However, their cellular interaction and in vivo behavior have not been fully developed and characterized.
Methods
In this study, self-assembled LCNPs prepared from soy phosphatidylcholine and glycerol dioleate were developed as a platform for oral delivery of paclitaxel. The particle size of empty LCNPs and paclitaxel-loaded LCNPs was around 80 nm. The phase behavior of the liquid crystalline matrix was characterized using crossed polarized light microscopy and small-angle X-ray scattering, and showed both reversed cubic and hexagonal phase in the liquid crystalline matrix. Transmission electron microscopy and cryofield emission scanning electron microscopy analysis revealed an inner winding water channel in LCNPs and a “ ball-like”/“hexagonal” morphology.
Results
Cellular uptake of LCNPs in Caco-2 cells was found to be concentration-dependent and time-dependent, with involvement of both clathrin and caveolae/lipid raft-mediated endocytosis. Under confocal laser scanning microscopy, soy phosphatidylcholine was observed to segregate from the internalized LCNPs and to fuse with the cell membrane. An in vivo pharmacokinetic study showed that the oral bioavailability of paclitaxel-loaded LCNPs (13.16%) was 2.1 times that of Taxol® (the commercial formulation of paclitaxel, 6.39%).
Conclusion
The findings of this study suggest that this LCNP delivery system may be a promising candidate for improving the oral bioavailability of poorly water-soluble agents.
Background
Delivery of poorly water-soluble drugs with clinically acceptable bioavailability is still one of the major challenges in pharmaceutical science. Nearly 40% of new drug candidates have low water solubility, leading to decreased absorption in the gastrointestinal lumen, resulting in poor oral bioavailability.Citation1 Paclitaxel is an example of this problem. As a class IV agent in the biopharmaceutics classification system,Citation2 the clinical application of paclitaxel is greatly hindered by poor water solubility (<1 mg/L) and low permeability across the intestinal barrier.Citation3 Paclitaxel is usually administered intravenously as a concentrated solution under the trade name Taxol®, which contains paclitaxel 6 mg/mL in a mixed solvent composed of Cremophor® EL and ethanol (1:1, v/v). However, Cremophor EL is not generally well tolerated and frequently causes side effects, including hypersensitivity and neurotoxicity.Citation4 Because cancer patients require long-term treatment, development of oral formulations for paclitaxel is urgently needed to improve patient compliance and preserve quality of life.
Presently, a great deal of research effort has been devoted to increasing the water solubility of paclitaxel and improving its oral bioavailability. Modification of the chemical structure and development of new drug-delivery systems, such as self-emulsifying formulations, liposomes, nanoparticles, and polymeric micelles, has shown promising potential to replace the Cremophor EL-based vehicle for delivery of paclitaxel.Citation2,Citation5–Citation9
Lipid-based liquid crystalline nanoparticles (LCNPs) are attracting growing interest as nanocarrier systems due to their potential to improve the bioavailability of both hydrophilic and hydrophobic drugs.Citation10,Citation11 LCNPs, which are self-assembled from polar amphiphilic lipids in the presence of excess water, are generally prepared by dispersing the nonlamellar liquid crystalline matrix formed into water phase using high-energy fragmentation, such as ultrasonication, microfluidization, or homogenization.Citation12 Depending on the nature of the different lipids and the various additives and solution conditions, the structures formed may include lamellar (Lα), bicontinuous cubic (Q2), and reversed hexagonal (H2) phases, and in some particular cases, a discrete micellar cubic phase (I2).Citation13 The discrete/bicontinuous aqueous channel and lipid domains inside LCNPs make it possible to encapsulate drugs with different polarity.Citation14 In addition, despite their huge surface area, LCNPs have unique two-dimensional and three-dimensional thermodynamically stable properties.Citation15 Furthermore, lipid-based LCNPs are biocompatible, digestible, and bioadhesive. Therefore, it is believed that LCNPs might serve as novel drug-delivery systems combining the advantages of liposomes and nanoparticles by providing protection and enabling sustained release of agents solubilized in LCNPs.
Recently, a new lipid liquid crystalline matrix prepared from soy phosphatidylcholine and glycerol dioleate was reported,Citation16 in which self-assembled reversed micellar cubic (I2) and hexagonal (H2) phases were observed when soy phosphatidylcholine and glycerol dioleate were mixed in equal proportions, with addition of 10%–30% water. In previous studies, soy phosphatidylcholine/glycerol dioleate/polysorbate 80-based LCNPs showed an extended terminal half-life for intravenous delivery of both hydrophobic and hydrophilic drugs.Citation17,Citation18 Nevertheless, application of this LCNP system for oral drug administration has barely been explored. In addition, the cellular interaction between LCNPs and enterocytes remains to be defined.
In this research, a soy phosphatidylcholine/glycerol dioleate-based LCNP system was developed as an oral delivery platform for poorly water-soluble drugs. Paclitaxel, a class IV drug in the biopharmaceutics classification system, was chosen as the model agent. The phase behavior of bulk liquid crystalline matrices was characterized via crossed polarized light microscopy and small-angle X-ray scattering. The morphology of the nanoparticles was examined under a transmission electron microscope and a cryofield field emission scanning electron microscope (cryo-FESEM). Cytotoxicity, membrane fluidity, and cellular uptake of LCNPs were carried out using Caco-2 cells as the endothelial cell model. Nile red and N-(7-nitrobenz-2-oxa-1, 3-diazol-4-yl)-1, 2-dihexadecanoyl-sn-glycero-3- phosphoethanolamine (NBD-PE) were used as fluorescent probes to show the intracellular behavior of LCNPs under confocal laser scanning microscopy. An in vivo pharmacokinetic study was conducted to evaluate the ability of this LCNP system to deliver hydrophobic agents orally.
Materials and methods
Materials and animals
Soy phosphatidylcholine, Lipoid S100 (containing phosphatidylcholine 95.2%) was purchased from Toshisun (Lipoid, Ludwigshafen, Germany). Glycerol dioleate was obtained from J and K (Beijing, China). Paclitaxel was purchased from Xi’an Sanjiang Biological Engineering Co, Ltd (Xi’an, China) and Taxol from Bristol-Myers Squibb (Princeton, NJ). Nile red and 1, 6-diphenyl-1,3,5-hexatriene (DPH) were obtained from Sigma-Aldrich (Shanghai, China). NBD-PE was obtained from Invitrogen (Burlington, ON), and polysorbate-80 from Dazhong Co, Ltd (Shanghai, China). All other reagents were of analytical or chromatographic pure grade and purchased from Sinopharm Chemical Reagent Co, Ltd (Shanghai, China).
Cell counting kit-8 (CCK-8) solution was provided by Dojindo (Kumamoto, Japan). Dulbecco’s modified Eagle’s medium (DMEM) was obtained from Hyclone (Logan, US). Fetal bovine serum, 0.25% trypsin-ethylenediamine tetraacetic acid, penicillin-streptomycin, and nonessential amino acids were purchased from Gibco (Beijing, China).
Male Sprague Dawley rats (weighing 200 ± 20 g) were obtained from the Experimental Animal Center of Fudan University and housed at 22°C ± 2°C with access to food and water ad libitum. The protocol for animal experiments was approved by the Animal Experimentation Ethics Committee of Fudan University.
Preparation of LCNPs
LCNPs were prepared using a modified solvent precursor method.Citation19 Soy phosphatidylcholine, glycerol dioleate, and polysorbate 80, with or without paclitaxel, were mixed with the hydrotrope (10% ethanol to total additives, w/w) and stirred for 3 hours to form a uniform and clear oil phase. HEPES (N′-a-hydroxythylpiperazine-N′-ethanesulfanic acid) solution (containing 20 mM HEPES, pH 7.4, 20% to lipid, w/w) was gently added to the oil phase. The mixture was then incubated under stirring at room temperature for at least 24 hours (entrapment of air bubbles was carefully prevented by adjusting the stirring speed) to form a bulk liquid crystalline matrix. This liquid crystalline precursor was then injected into HEPES solution under magnetic stirring at 60°C to form a coarse dispersion, and subsequently homogenized with a Microfluidizer (Nano DeBEE, South Easton, MA) at a pressure of 10,000 psi for three cycles and 30,000 psi for two cycles. Nile red and/or NBD-PE were incorporated as fluorescent probes, and prepared using the same procedures as those used to prepare the paclitaxel-loaded LCNPs.
Phase behavior of bulk liquid crystalline matrix
The phase behavior of the bulk liquid crystalline matrix was characterized using crossed polarized light microscopy with a Zeiss Axiovert 40 MAT microscope (Carl Zeiss, Oberkochen, Germany) fitted with an AxioCam camera. Small-angle X-ray scattering (SAXSess mc2, Anton-Paar, Austria) was utilized to confirm further the internal structure of the liquid crystalline phase.
Characterization of LCNPs
The mean diameters of the nanoparticles were determined by dynamic light scattering using a Zeta Potential/Particle Sizer 380 ZLS (PSS Nicomp, Santa Barbara, CA). Morphological examination of LCNPs was carried out using a transmission electron microscope (H-600, Hitachi, Japan) following negative staining with sodium phosphotungstate solution. To study further the surface morphology of the LCNPs, the samples were examined under cryo-FESEM (Hitachi, S-4800, Japan) with liquid nitrogen freezing and gold sputtering (−100°C).
Encapsulating efficiency, loading capacity, and in vitro drug release
The encapsulating efficiency (EE%) and loading capacity (LC%) of paclitaxel in LCNPs were determined by gel filtration chromatography, as described elsewhere.Citation20 The eluate containing LCNPs was dissolved in methanol to release paclitaxel. The content of paclitaxel was analyzed using a high-performance liquid chromatography system (Shimadzu, Tokyo, Japan). Methanol/water (75/25, v/v) was used as the mobile phase at a flow rate of 1.2 mL per minute. Paclitaxel was separated by a C18 column (Diamonsil, 5 μm, 4.6 × 200 mm; Dikma, Beijing, China) and detected at 227 nm. The EE% and LC% were calculated as indicated below (n = 3),
In vitro drug release from the LCNPs was performed using an equilibrium dialysis method. Paclitaxel-loaded LCNPs and Taxol (containing 0.03 mg paclitaxel) were added into a sealed dialysis bag (molecular weight cutoff 9 kDa) with a plastic cork, immersed in 30 mL of release medium (HEPES solution with 0.1% v/v polysorbate 80 for providing sink conditions throughout the release test, pH 6.8) and incubated at 37°C with a shaking speed of 100 rpm. A 0.2 mL volume of release medium was sampled at 0, 0.5, 1, 2, 3, 4, 6, 8, 16, 24, 48, 72, and 96 hours, followed by immediate addition of an equal volume of fresh release medium. Samples were analyzed using the high-performance liquid chromatography method mentioned above.
Caco-2 cell culture
A human colon adenocarcinoma (Caco-2) cell line was obtained from the American Type Culture Collection (Rockville, MD) and grown in a 5% CO2 humidified atmosphere at 37°C in DMEM medium supplemented with 10% fetal bovine serum, 1% nonessential amino acids, penicillin 100 U/mL, and streptomycin 100 mg/L.
CCK-8 cell viability analysis
In vitro proliferation of the Caco-2 cells was accessed using a CCK-8 assay to evaluate the cytotoxicity of LCNPs. Caco-2 cells were seeded into a 96-well plate at a density of 5000 cells/well and cultured for 24 hours. Following that, the cells were incubated with LCNPs for 24 hours at concentrations of 100 mg/L, 200 mg/L, 500 mg/L, 1 mg/mL, 2 mg/mL, 5 mg/mL, 10 mg/mL, 100 mg/mL, and 200 mg/mL (w/w). Immediately after treatment with LCNP, the medium was changed back to DMEM, and 0.01 mL of CCK-8 solution was added to each well. The cells were further incubated at 37°C for 2 hours, and color development was quantified photometrically at 450 nm using a multifunction microplate reader (Thermo Fisher Scientific, Pittsburg, PA). Caco-2 cell proliferation at the same concentration of Cremophor EL:ethanol (1:1, v/v) was also evaluated. Untreated cells were used as controls and a cell-free well with an equal volume of DMEM as a blank. Viability (%) was calculated as indicated below (n = 3),
Membrane fluidity
Cell membrane fluidity induced by LCNPs was evaluated by determining the fluorescence polarization of 1,6-diphenyl-1, 3,5-hexatriene (DPH), as previously described.Citation21 Briefly, one milliliter of the cell suspension in DMEM (3.5 × 105 cells/mL) was mixed with an equal volume of 0.2 mM cholesterol, 30 mM benzyl alcohol, and 20 mg/L, 200 mg/L, 2 mg/mL, and 20 mg/mL of empty LCNPs, respectively, followed by addition of DPH at a final concentration of 0.001 mM, and incubation for 30 minutes at 25°C. Fluorescence anisotropy values were measured using a FLS 920 fluorescence lifetime steady-state spectrometer (Edinburgh, UK) at an excitation wavelength of 362 nm and an emission wavelength of 462 nm. The membrane anisotropy value was given as a percentage of the blank control value.
Cellular association of Nile red-labeled LCNPs
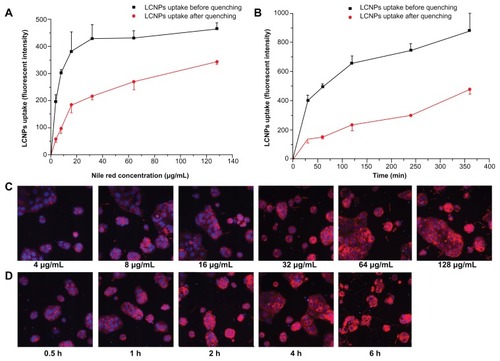
A quantitative analysis of the cellular association of Nile red-labeled LCNPs was performed using a high-content analysis system, as described elsewhere.Citation22 Briefly, Caco-2 cells were seeded into a 96-well plate at a density of 5000 cells/well and cultured for 24 hours. After that, the cells were incubated with Nile red-labeled LCNPs in DMEM at concentrations of 4, 8, 16, 32, 64, and 128 mg/L for 2 hours and 64 mg/L for 0.5, 1, 2, 4, and 6 hours. Immediately after treatment with LCNPs, the cells were washed three times with phosphate-buffered solution and fixed with 3.7% formaldehyde solution for 10 minutes. Following staining with 10 mg/mL Hoechst (bisbenzimide) 33258 at room temperature and away from light for 7 minutes, the cells were rinsed three times with phosphate-buffered solution and detected under a kinetic scan high-content analysis system reader (version 3.1; Cellomics Inc, Pittsburgh, PA) at a 20× objective. The cells were then treated with Trypan blue to quench the extracellular fluorescence and examined under the high-content analysis system once again. Cells treated with DMEM were used as controls. The assay protocol was configured to analyze 1000 cells per well, and all the experiments were performed in triplicate.
Endocytotic pathway of LCNPs
To study the endocytic pathway involved in LCNP internalization, cellular uptake experiments were performed in the presence of different inhibitors. Caco-2 cells were seeded in a 96-well plate at a density of 5000 cells/well and cultured for 24 hours. Cells were preincubated with 20 mM sodium azide and 100 mM 2-deoxyglucose (NaN3/DOG), 5 mM methyl-β-cyclodextrin, filipin 10 mg/L, cholesterol 40 mg/L, chlorpromazine 16 mg/L, and cytochalasin D 10 mg/L for 2 hours, respectively. After that, the cells were incubated with Nile red-labeled LCNPs (64 mg/L) in DMEM for 2 hours. Quantitative analysis of the cellular association of nanoparticles following the inhibitor treatments was performed as mentioned above and compared with that of the noninhibited controls.
Intracellular behavior of LCNPs
To understand further the intracellular behavior of LCNPs, the intracellular distribution of the fluorescence-labeled LCNPs was visualized by confocal laser scanning microscopy. Nile red was used as the model lipophilic agent and NBD-PE as the lipid component of the LCNPs. Caco-2 cells were seeded into multiple glass-bottomed tissue culture plates (10 mm; Shengyou Biotechnology Co, Ltd, Zhejiang, China) at an initial density of 1 × 105 cells/well and cultured for 24 hours before incubation with LCNPs that had been dual-labeled with Nile red (0.1%) and NBD-PE (1%) for 5, 15, 30, 90, and 240 minutes. Immediately after treatment with LCNPs, the cells were fixed, stained, and rinsed using the same method as mentioned above. Finally, the cells were examined under a Zeiss LSM 710 microscope.
In vivo pharmacokinetics
In order to evaluate the potency of LCNPs as nanocarriers for oral delivery of hydrophobic agents, the in vivo pharmacokinetics of paclitaxel-loaded LCNPs were investigated in Sprague Dawley rats after oral administration. The rats were fasted with water ad libitum 8 hours beforehand. Fifteen rats were randomly divided into three treatment groups, ie, for intravenous administration of Taxol, oral administration of Taxol, and oral administration of paclitaxel-loaded LCNPs, at a paclitaxel dose of 5 mg/kg. Blood was collected into heparinized tubes at 0.083, 0.25, 0.5, 1, 1.5, 2, 4, 6, 8, 12, and 24 hours after administration. Plasma was collected following centrifugation and stored at −20°C until analysis.
Plasma paclitaxel concentrations were determined by reverse-phase high performance liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS). To prepare the samples for analysis, 90 μL of methanol containing 60 ng/mL of docetaxel (internal standard) was added to 0.03 mL of plasma to precipitate the proteins. The mixture was vortexed and then centrifuged at 12,000 rpm for 10 minutes. The supernatant was mixed with an equal volume of deionized water, and 5 μL of the sample was subjected to analysis in the LC-MS/MS system.
An API 4000 triple quadrupole LC-MS/MS system (Applied Biosystems, Toronto, ON) was used, and was equipped with an electrospray ionization source, a G1312A quaternary pump, a G1379A vacuum degasser, a G1316A thermostatted column oven (Agilent, Waldbronn, Germany), and an HTS PAL autosampler (CTC Analytics, Zwingen, Switzerland). A Venusil XBP phenyl column (100 mm × 2.1 mm, 0.005 mm; Agela, Shanghai, China) with a mobile phase of 0.1% formic acid:methanol (3:7) used for chromatographic separations. The flow rate and column temperature were set at 0.3 mL per minute and 40°C, respectively. Quantitation was achieved with MS-MS detection in positive ion mode for all the analyses and internal standard. The ion spray voltage was set at 5000 V. The source temperature was maintained at 500°C. The ion source gas 1 (nitrogen), gas 2 (nitrogen), and curtain gas (nitrogen) pressures were set at 40, 55, and 20 psi, respectively. Nitrogen was used as the collision gas at a pressure of 4 L/minute for collision-activated dissociation. The parameters of the compound, including declustering potential, collision energy, entrance potential, and collision cell exit potential, were 160, 30, 10, and 15 V for paclitaxel, and 140, 32, 10, and 15 V for docetaxel, respectively. Detection of the ions was performed in the multiple-reaction monitoring mode, by monitoring the transition pairs of the m/z 876.6 precursor ion to the m/z 308.0 for paclitaxel (M + Na)+, and m/z 830.3 precursor ion to the m/z 549.1 for docetaxel (M + Na)+, respectively. Quadrupoles Q1 and Q3 were set on unit resolution. Data acquisition was performed using the Analyst 1.4.1 software package (Applied Biosystems). All the concentration data are plotted as plasma drug concentration-time curves.
The pharmacokinetic analysis was performed using a model-independent method. The terminal elimination rate constant (k) was determined by least-square regression analysis of the terminal log-linear portions of the plasma concentration-time profile (k = −2.303 × slope). The elimination half-life (T1/2) was calculated as 0.693/k. The area under the curve to the last measurable concentration (AUC0–t) was calculated using the linear trapezoidal rule. The area under the curve extrapolated to infinity (AUC0–∞) was calculated as AUC0–t + Ct/k, where Ct is the last measurable concentration. Absolute bioavailability was calculated from the dose-corrected areas under the curves for oral versus intravenous administration.
Statistical analysis
Comparison among multiple groups was performed by one-way analysis of variance followed by the Bonferroni test. Comparison between two groups was performed using the Student’s t-test. Statistical significance was defined as P < 0.05.
Results
Phase behavior of the bulk liquid crystalline matrix
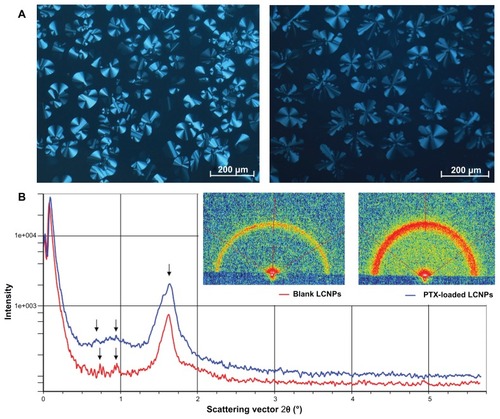
The phase behavior of the bulk liquid crystalline matrix was characterized using crossed polarized light microscopy and small-angle X-ray scattering. A birefringent fan-like texture was observed in the liquid crystalline matrix when in a dark environment. No significant change was found in the birefringent texture when 0.5% paclitaxel was incorporated into the bulk liquid crystalline phase (). As illustrated in , both the blank liquid crystalline matrix and the paclitaxel-loaded matrix showed three diffraction peaks in the small-angle X-ray scattering curves, with almost the same spacing ratio.
Figure 1 (A) Photomicrographs of bulk liquid crystalline matrices under crossed polarized light microscopy: left, blank liquid crystalline matrix; right, paclitaxel-loaded liquid crystalline matrix. (B) Small-angle X-ray diffraction pattern of liquid crystalline matrices: red line, blank liquid crystalline matrix; blue line, paclitaxel-loaded liquid crystalline matrix.
Abbreviations: LCNPs, liquid crystalline nanoparticles; PTX, paclitaxel.
Particle size and morphology of LCNPs
A homogeneous solution with blue opalescence was obtained after homogenizing. The particle size of the blank LCNPs was 76.33 ± 1.19 nm (polydispersity index 0.365 ± 0.014, ). A slight increase in diameter was observed in the paclitaxel-loaded LCNPs (84.70 ± 5.26 nm, polydispersity index 0.370 ± 0.016).
Figure 2 (A) Transmission electron microscopic images of liquid crystalline nanoparticles and (B) particle size of liquid crystalline nanoparticles when fit to Gaussian (up) and Nicomp (down) distributions, respectively.
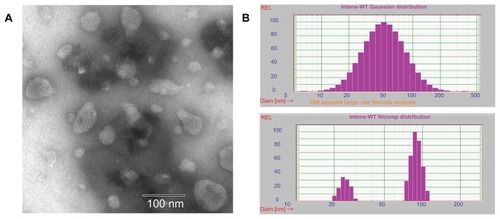
The morphology of the LCNPs was examined under a transmission electron microscope. As shown in , the nanoparticles formed had a spherical “flower-like” structure with an internal water channel. Albeit slightly smaller than that shown by dynamic light scattering, most LCNP diameters were in the range of 50–80 nm.
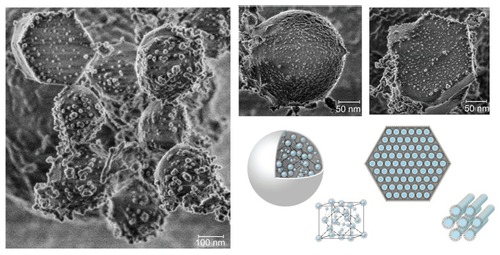
To study further the surface morphology of the LCNPs, the sample was examined under cryo-FESEM. The reversed micellar cubic particles exhibited a “ball-like” morphology, while the reversed hexagonal particles displayed clear hexagonal structures (). LCNPs were found to be comprised of an internal nanostructured phase with enclosed water channels, with a relatively smooth but rough nodule-like appearance, as reported previously (data not shown).Citation23
Figure 3 Cryo-field emission scanning electron microscopic images of liquid crystalline nanoparticles and schematic illustration of reversed micellar nanoparticles and reversed hexagonal nanoparticles.
Encapsulating efficiency, loading capacity, and in vitro drug release
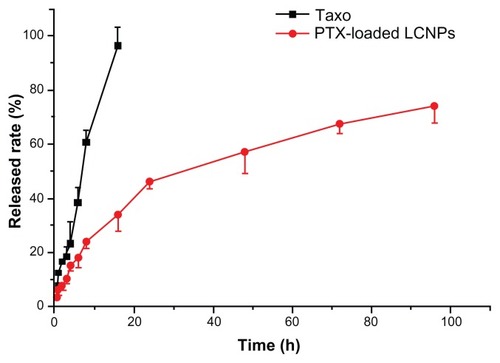
The EE% and LC% of paclitaxel-loaded LCNPs were 75% and 0.45%, respectively. In vitro paclitaxel release from the LCNPs was determined using an equilibrium dialysis method. As shown in , complete paclitaxel release from Taxol was obtained after 16 hours. LCNPs loaded with paclitaxel were shown to release their drug cargo in a sustained biphasic manner, with relatively fast drug release in the first 24 hours (50% release) and slower release in the following 72 hours (70% total release).
Figure 4 In vitro paclitaxel release from taxol and liquid crystalline nanoparticles in pH 7.4 HEPES buffer at 37°C (n = 3).
Note: The initial paclitaxel concentration was 30 mg/L.
Abbreviations: LCNPs, liquid crystalline nanoparticles; PTX, paclitaxel.
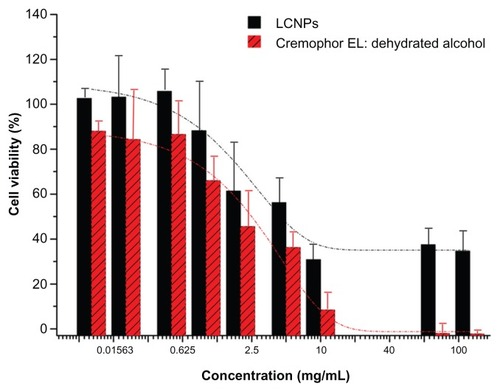
CCK-8 cell viability analysis
Cell viability following LCNP treatment was measured using the CCK-8 assay to evaluate the safety of LCNPs as a novel nanovehicle for delivery of paclitaxel. As shown in , Caco-2 cells were much less sensitive to LCNP-induced cytotoxicity. Compared with Cremophor EL:ethanol (1:1), a significant increase in cell viability was observed at a vehicle concentration of 1%–10%. The Taxol vehicle decreased Caco-2 viability, with an IC50 of 0.8433 mg/mL after 24 hours of treatment, while LCNPs had an IC50 value of 11.05 mg/mL.
Figure 5 Viability of Caco-2 cells following 24 hours of treatment with liquid crystalline nanoparticles and Cremophor EL:ethanol (1:1).
Note: The dotted lines presented the fitted dose-response curves (n = 3).
Abbreviation: LCNPs, liquid crystalline nanoparticles.
Membrane fluidity
Given that LCNPs prepared from glyceryl monooleate have been reported to have lipid-mixing and membrane-fusion properties,Citation24 soy phosphatidylcholine/glycerol dioleate-based LCNPs might act similarly and may result in changes in microviscosity of the cell membrane following oral administration. The change in membrane fluidity induced by LCNPs was investigated by determining the fluorescence polarization of DPH within the Caco-2 cell membrane. As noted in , LCNPs at concentrations of 10 mg/L and 100 mg/L had almost no effect on membrane fluidity, with anisotropy values of 99.5% and 97.9%, respectively. However, membrane fluidity was significantly increased after treatment with LCNPs at concentrations of 1 mg/mL and 10 mg/mL, achieving decreased anisotropy values of 55.5% and 41.1%, respectively.
Table 1 Anisotropy values for the DPH-labeled Caco-2 cell membrane after incubation with different concentrations of liquid crystalline nanoparticles at 25°C for 30 minutes
Cellular association of Nile red-labeled LCNPs
High-content system analysis demonstrated that the cellular association of Nile red-labeled LCNPs in Caco-2 cells increased in both a concentration-dependent and time-dependent manner (). The same difference in fluorescent intensity before and after quenching was obtained 2, 4, and 6 hours after Nile red-labeled LCNP treatment, indicating that LCNPs readily attach to Caco-2 cell surfaces and reach a plateau rapidly ().
Figure 6 Cellular association of Nile red-labeled liquid crystalline nanoparticles in Caco-2 cells (n = 3). (A and C) treated with Nile red-labeled liquid crystalline nanoparticles at concentrations of 4, 8, 16, 32, 64, and 128 mg/L for 2 hours. (B and D) treated with Nile red-labeled liquid crystalline nanoparticles at a concentration of 64 mg/L for 0.5, 1, 2, 4, and 6 hours. (C and D) Fluorescent images after quenching. Red, Nile red-labeled liquid crystalline nanoparticles; blue, cell nuclei stained with Hoechst.
Abbreviation: LCNPs, liquid crystalline nanoparticles.
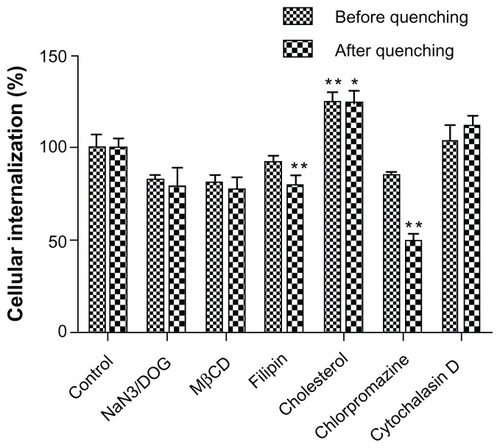
Endocytosis pathway for LCNPs
Endocytosis inhibition experiments showed that the cellular uptake of LCNPs was significantly increased by cholesterol but inhibited by chlorpromazine and filipin. NaN3/DOG and methyl-β-cyclodextrin were also associated with a reduction in cellular uptake of LCNPs, although the reduction was not statistically significant. No inhibition in uptake of LCNPs was found in cells pretreated with cytochalasin D (). The inhibition efficiencies obtained before and after quenching were similar for most of the inhibitors, except for chlorpromazine, which showed higher inhibition after quenching of the extracellular fluorescence.
Figure 7 Cellular association of Nile red-labeled liquid crystalline nanoparticles in the presence of different endocytosis inhibitors.
Notes: Data shown as the mean ± standard deviation (n = 3). *P < 0.05 and **P < 0.01, significantly different from controls.
Abbreviation: MβCD, methyl-β-cyclodextrin.
Intracellular behavior of LCNPs
To understand further the intracellular behavior of LCNPs, Caco-2 cells were treated with dual-labeled LCNPs (Nile red, red fluorescence; NBD-PE, green fluorescence) and examined under confocal laser scanning microscopy. As seen in , strong fluorescent signals were observed in the cells 5 minutes after treatment with LCNPs, indicating that internalization of LCNPs occurred rapidly. In addition, the agents internalized showed a yellow (colocalization of red and green) signal throughout the cytoplasm, together with some separated red and green signals. With an increase in incubation time, green NPD-PE was found to segregate from the red LCNPs and insert into the cell membrane (). Separation of the colocalized Nile red and NBD-PE was easily distinguished 30 minutes after LCNP treatment (). After 240 minutes of incubation, almost all the NBD-PE had separated from the LCNPs, leaving the Nile red-labeled LCNPs in the cytoplasm (). A three-dimensional image clearly shows that a considerable amount of NBD-PE escaped from the LCNPs, inserted into the membrane at 90 minutes, and exhibited a green “hat” covering the entire cell (shown by arrows, ).
Figure 8 Confocal image of internalization of dual-labeled liquid crystalline nanoparticles by Caco-2 cells at 5 minutes.
Notes: Liquid crystalline nanoparticles was prepared using NBD-PE (green) as a lipid component within the nanoparticles and Nile red (red) as a fluorescent probe for the nanoparticles. Blue, cell nuclei stained with Hoechst; yellow, colocalization of Nile red and NBD-PE.
Abbreviation: NBD-PE, N-(7-nitrobenz-2-oxa-1, 3-diazol-4-yl)-1, 2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine.
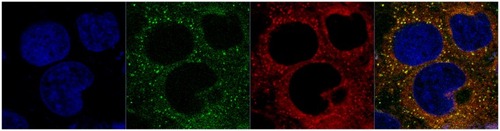
Figure 9 Confocal images of Nile red-labeled and NBD-labeled liquid crystalline nanoparticles in Caco-2 cells. (A) Most NBD colocalized with Nile red at 30 minutes, (B) NBD still colocalized with Nile red, and partly separated from Nile red at 90 minutes, and (C) most NBD separated with Nile red and inserted into the cell membrane at 240 minutes. Red, Nile red; green, NBD-PE.
Abbreviations: NBD, N-(7-nitrobenz-2-oxa-1, 3-diazol-4-yl)-1; NBD-PE, N-(7-nitrobenz-2-oxa-1, 3-diazol-4-yl)-1, 2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine.
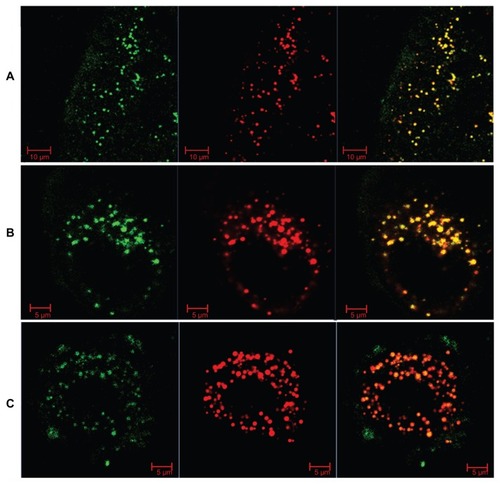
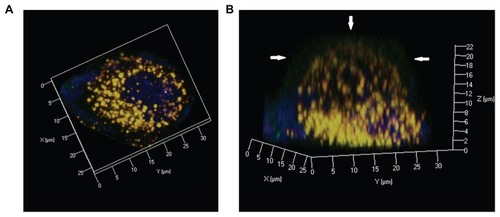
Figure 10 Confocal three dimensional images of Nile red-labeled and NBD-labeled liquid crystalline nanoparticles in Caco-2 cells at 90 minutes. (A) Top view and (B) side view. Red, Nile red; green, NBD-PE; blue, cell nuclei stained with Hoechst.
Note: Arrow, NBD-PE separated from liquid crystalline nanoparticles and inserted into the cell membrane.
Abbreviation: NBD-PE, N-(7-nitrobenz-2-oxa-1, 3-diazol-4-yl)-1, 2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine.
In vivo pharmacokinetics
The plasma concentration profiles of paclitaxel after intravenous administration of Taxol at 5 mg/kg paclitaxel are shown in . The mean AUC(0–24h) was 1333.30 ± 182.45 μg/L · h and the C5min was 1183.60 ± 156.02 μg/L. The T1/2 observed for paclitaxel was 5.10 ± 0.79 hours ().
Table 2 Pharmacokinetic parameters for paclitaxel after intravenous or oral administration of Taxol® and paclitaxel-loaded liquid crystalline nanoparticles to Sprague-Dawley rats at a paclitaxel dose of 5 mg/kg (mean ± standard deviation, n = 5)
Figure 11 Plasma paclitaxel concentration-time profiles after (A) intravenous administration and (B) oral administration of Taxol® and paclitaxel-loaded liquid crystalline nanoparticles to Sprague-Dawley rats at a paclitaxel dose of 5 mg/kg (n = 5).
Abbreviations: LCNPs, liquid crystalline nanoparticles; PTX, paclitaxel.
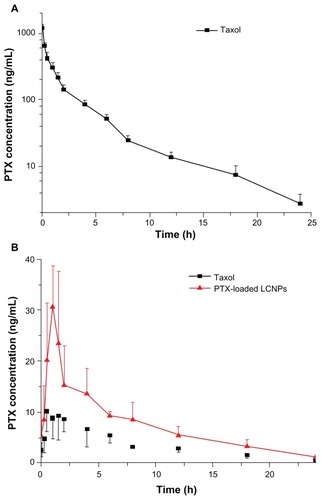
shows the pharmacokinetic profiles of paclitaxel obtained after oral administration. The AUC(0–24h) of paclitaxel-loaded LCNPs was 2.1 times that of Taxol. The Cmax of paclitaxel-loaded LCNPs and Taxol were 33.50 ± 5.46 μg/L and 12.65 ± 3.52 μg/L (P = 0.000095), respectively, with a similar Tmax. No significant difference was observed in the T1/2 for paclitaxel-loaded LCNPs and Taxol. The absolute bioavailability calculated for paclitaxel delivered in the LCNPs and Cremophor EL:ethanol formulations was 13.16% and 6.39%, respectively ().
Discussion
In this paper, LCNPs were developed as nanovehicles for oral delivery of poorly water-soluble agents. The discrete lipidic hydrophobic and aqueous hydrophilic domains within LCNPs have been used for diffusion-controlled release of hydrophobic, hydrophilic, and amphiphilic molecules.Citation14 However, the potential of LCNPs as oral drug nanovectors has not been fully explored, especially the internalization mechanism in intestinal epithelium cells. In this study, a soy phosphatidylcholine/glycerol dioleate-based LCNP system was prepared and characterized using crossed polarized light microscopy, small-angle X-ray scattering, transmission electron microscopy, and cryo-FESEM. In vitro release and in vivo pharmacokinetics of the paclitaxel-loaded LCNPs were evaluated. In vitro Caco-2 cell experiments were carried out to evaluate the mechanism of uptake of LCNPs by cells.
The phase behavior of the liquid crystalline matrix was studied using crossed polarized light microscopy and small-angle X-ray scattering. Crossed polarized light microscopy has a long history of providing information about the phase behavior of a liquid crystalline matrix. Small-angle X-ray scattering was used as a tool complementary to crossed polarized light microscopy for phase identification to provide a better understanding of the spacing ratio of the liquid crystalline phases. In this study, crossed polarized light microscopy data revealed the coexistence of reversed cubic and hexagonal phases (H2 and I2). Although it was hard to infer the space pattern of coexisting H2 and I2 phases from three diffraction peaks, our small-angle X-ray scattering data still at least partly reflect crystallization of the bulk phase. On the other hand, it has been reported that incorporation of additives in lipid cubic phase can interact with or modify the structure of the cubic phases.Citation25 However, in our study, negligible change in birefringent texture was observed in the presence of paclitaxel (0.5% in the total additives, ), indicating that no phase conversion occurred following incorporation of paclitaxel. Such speculation was justified by our small-angle X-ray scattering data, which showed the same spacing ratio in both the paclitaxel-loaded liquid crystalline matrix and the paclitaxel-free one ().
LCNPs were prepared using a solvent precursor method.Citation19 As shown in , the LCNPs constructed showed a bimodal polydispersed population with two distribution peaks (around 100 nm and 25 nm) and relative wide size distributions (polydispersity index 0.360–0.380). The particle distribution pattern was confirmed by our transmission electron microscopy data (), in which quite a few small particles (around 20 nm) were observed in the LCNP formulation. It has been pointed out that such extremely small particles might not originate from larger particles as a result of high shear forces during homogenization but form spontaneously without the need for a high energy input.Citation26
LCNPs showed winding water channels under transmission electron microscopy () and a “balllike”/“ hexagonal” morphology under cryo-FESEM (), which confirmed the nanostructure of LCNPs, as shown in . Nonlamellar liquid crystalline matrices (Q2 and H2) can be prepared from original lipid combinations without any energy input. In addition, dispersions of liquid crystalline matrices maintain their unique internal structure, with distinct two-dimensional and three-dimensional thermodynamic stability.Citation15 Thus, they might provide a promising sustained release drug nanocarrier system beyond traditional drug-delivery systems. On the other hand, cryo-FESEM seemed to yield somewhat larger diameters than dynamic light scattering and transmission electron microscopy, which we believe resulted from particle aggregation or water channel swelling at −100°C.
Although almost all liquid crystalline matrices have been reported to be sustained-release systems,Citation27 a large amount of literature has reported burst release from LCNPs.Citation28,Citation29 The major limitation of the application of LCNPs is thought to correlate with the increased surface area when dispersed into excess water.Citation30 In this study, LCNP nanocarriers showed sustained in vitro release of paclitaxel, with a relatively rapid release from LCNPs in the initial stage (over approximately 24 hours), and a slower release thereafter (during the following 72 hours, ). The initial rapid release is believed to derive from agents located at the outer layer of the particles. In contrast, the slower release is ascribed to those incorporated in the core of the nanocarrier and released in a sustained manner along with erosion or degradation of the matrix.
Cremophor EL, the excipient in Taxol, has been the subject of widespread safety concerns due to its side effect profile.Citation3 These concerns emphasize the need for development of a safe and effective alternative formulation of paclitaxel. Soy phosphatidylcholine and glycerol dioleate are generally accepted to be safe for oral administration, and polysorbate 80 is approved by the US Food and Drug Administration as an excipient with good biocompatibility and biodegradability. However, it has been suggested that LCNPs could fuse with cellular lipid membranes,Citation31,Citation32 which raises the question of potential toxicity. Thus, to evaluate the cytotoxicity of LCNPs, cell viability following LCNP treatment was assessed in comparison with Cremophor EL:ethanol. As shown in , compared with the Taxol vehicle, Caco-2 cells were much less sensitive to LCNP-induced cytotoxicity after 24 hours of treatment, showing a 12-fold higher IC50 than that of the Taxol vehicle. In addition, because LCNPs are formulated without Cremophor EL, their drug cargo is expected not to have the serious and potentially fatal hypersensitivity reactions that occur with Taxol. Accordingly, LCNPs can be considered to be promising alternative carriers for paclitaxel.
Although it has been pointed out that soy phosphatidylcholine/glycerol dioleate-based LCNPs show very limited lipid-mixing ability with model soy phosphatidylcholine membranes,Citation24 other researchers have claimed that mixtures of phosphatidylcholine and cholesterol could form stable inverted bicontinuous cubic phases under physiological conditions, thereby resulting in membrane “fusion”.Citation32 Therefore, it is necessary to determine the membrane fluidity of LCNPs in order to evaluate their impact on the cell membrane. In our steady-state fluorescence anisotropy study, positive controls (cholesterol) and negative controls (benzyl alcohol) increased and decreased the anisotropy values, respectively, suggesting that membrane fluidity was increased by cholesterol and decreased by benzyl alcohol. LCNPs had almost no effect on anisotropy values when their concentration was less than 100 mg/L, but significantly decreased anisotropy values when their concentration increased up to 10 mg/mL (), suggesting that only LCNPs over a certain concentration could affect fluidity of the cell membrane.
In order to profile the cellular internalization of LCNPs, quantitative analysis of the cellular association of Nile red-labeled LCNPs was done using a high content analysis system. It has been reported that Nile red is a convenient fluorescent marker for labeling lipid dispersionsCitation33,Citation34 and can be easily incorporated into lipid nanoparticles.Citation8,Citation35,Citation36 Poorly soluble Nile red was expected to mix with the lipophilic domains of the particles and show minimal leakage in our preliminary experiment. Therefore, it was used as the fluorescent marker for LCNPs to evaluate their cellular interactions and uptake. On the other hand, an earlier study has argued that transferring of lipophilic markers from nanoparticles to the cell membrane could lead to overestimation of cellular uptake of nanoparticles.Citation34 Therefore, Trypan blue was used to quench fluorescence on the cell membrane. From the data before and after quenching (), it was found that LCNPs readily attach to the plasma membrane, which could be as a result of the lipophilic properties of lipids and the bioadhesive properties of LCNPs. Internalization of LCNPs into the cytoplasm increased gradually, showing concentration-dependence and time-dependence.
Cellular endocytosis involves macropinocytosis, clathrin-mediated endocytosis, or a caveolae/lipid raft-mediated mechanism.Citation8 To characterize the endocytosis pathway involved in the cellular uptake of LCNPs, cellular uptake experiments were performed under ATP-depleted (NaN3/DOG), cholesterol-depleted (methyl-β-cyclodextrin), caveolae- inhibited (filipin), clathrin-inhibited (chlorpromazine), actin-disrupted (cytochalasin D), and cholesterolenriched (cholesterol) conditions, respectively.
The concentration-dependent, time-dependent, and energy-dependent cellular uptake of LCNPs () suggest that internalization of LCNPs was via active endocytosis. Incomplete inhibition of the LCNP interaction under biochemical ATP depletion (NaN3/DOG) might be attributed to mechanisms that mediate direct delivery, such as contact- mediated intermembrane lipid exchange/mixing and fusion of lipid particles.
Cellular uptake of LCNPs was also found to be inhibited by filipin and methyl-β-cyclodextrin (albeit not significantly so) but promoted by cholesterol (). Lipid rafts are cholesterol-enriched and sphingolipid-enriched microdomains found in cell membranes, which play a significant role in many biological processes.Citation35 It has been shown that both cholesterol and lipid raft involved in membrane trafficking are essential to the process of raft formation. As a specialized type of lipid raft, caveolae are flask-shaped invaginations in the plasma membrane, and are enriched with proteins as well as cholesterol and sphingolipids. Filipin, a sterol-binding pentaene macrolide antibiotic, which selectively inhibits caveolae invagination by formation of cholesterol precipitates,Citation36 is generally used to block caveolae-mediated endocytosis. Methyl-β-cyclodextrin, a cyclic oligomer of glucopyranoside that inhibits cholesterol-dependent endocytic processes by reversibly extracting steroids from the plasma membrane,Citation37 is often used to determine whether endocytosis is dependent on the integrity of the lipid raft. As shown in , the significant increase in LCNP internalization by cholesterol and decreased uptake by filipin and methyl-β-cyclodextrin indicates that caveolae/lipid raft-mediated endocytosis plays an important role in cellular uptake of LCNPs in Caco-2 cells.
Intracellular transport of LCNPs was inhibited significantly by chlorpromazine (), an inhibitor which causes clathrin to accumulate in late endosomes and thereby inhibits coated pit-mediated endocytosis,Citation38 indicating that clathrin-mediated uptake is also involved in the internalization of LCNPs. Furthermore, the fluorescence intensity in the presence of chlorpromazine showed a significant reduction after treatment with Trypan blue when compared with that before quenching, suggesting that clathrin-mediated endocytosis plays a major role in cellular internalization of LCNPs. These results suggest that at least two pathways exist in the cellular internalization of LCNPs, ie, clathrin-mediated and caveolae/lipid raft-mediated endocytosis.
As discussed above, LCNPs over a certain concentration may have an impact on the fluidity of the cell membrane. Due to susceptibility to membrane fusion,Citation32 we speculated that soy phosphatidylcholine might segregate from LCNPs when attached to the cell membrane and induce lipid mixing or membrane fusion, and thereby increase absorption of paclitaxel. To explore further the mechanism of cellular delivery of LCNPs, LCNPs were formulated with NBD-conjugated phosphatidylethanolamine and incorporated Nile red as the model agent. Both phosphatidylcholine and phosphatidylethanolamine are major constituent phospholipids in the cell membrane. In this study, NBD-PE was used as a fluorescent probe for soy phosphatidylcholine to show its behavior after cellular internalization.
Through observation of intracellular colocalization of Nile red and NBD-PE (), our confocal laser scanning microscopy results showed that endocytosis of intact LCNPs might be a major mechanism for their intracellular drug transport. As shown in , after 30–240 minutes of incubation, time-dependent lipid separation and membrane “fusion” of NBD-PE was observed, indicating that the lipid component could “stream” from LCNPs and distribute towards the plasma membrane, ie, exogenous LCNPs were trafficked into the cell via a particular endocytosis pathway, followed by intracellular divergence or degradation, enabling release of the lipid component and resulting in lipid particle fusion and/or lipid mixing. shows three-dimensional images of Caco-2 cells after incubation with dual-labeled LCNPs for 90 minutes, which (especially the reconstructed image along the z axis) clearly demonstrates that the fluorescent signals of NBD-PE are mixed with the cell membrane. These findings confirm our results showing that LCNPs can alter the membrane fluidity, but is in contrast with previous studies concluding that lipophilic materials located in nanoparticles can be delivered easily and directly to the plasma membrane.Citation39
The in vivo pharmacokinetics of paclitaxel formulated in Cremophor EL:ethanol or LCNPs after intravenous and oral administration are shown in and . Due to high expression of P-glycoprotein in intestinal epithelial cells and the poor solubility of paclitaxel, orally administered paclitaxel has little chance of reaching the bloodstream, thus has very low absolute oral bioavailability. Citation2 The reported oral bioavailability values for paclitaxel after intravenous administration of Taxol in rats were in the range of 2.5%–6.5%,Citation4,Citation40,Citation41 which is confirmed by our in vivo data (6.39%). When paclitaxel was loaded into LCNPs and administered orally, Cmax increased from 12.65 ± 3.52 μg/L (Taxol) to 33.50 ± 5.46 μg/L, and the AUC(0–24h) increased significantly from 82.01 ± 19.68 μg/L · h to 169.07 ± 52.69 μg/L · h. Enhancement of the oral bioavailability of paclitaxel (13.16% versus 6.39%) might be attributed to encapsulation of paclitaxel inside LCNPs, which would not only help paclitaxel to escape recognition by P-glycoprotein, but would also improve intestinal epithelial permeability by increasing membrane fluidity ().Citation42
Acknowledgment
This work was supported by the National Natural Science Foundation of China (81072592, 30801439), the National Key Basic Research Program (2010CB529800), the Innovation Program of Shanghai Municipal Education Commission (12ZZ107), and the Shanghai Science and Technology Committee (10QA1404100, 11430702200). The authors would also like to thank Tessa Nguyen for her help in editing the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
- GursoyRNBenitaSSelf-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugsBiomed Pharmacother20045817318215082340
- VarmaMVPanchagnulaREnhanced oral paclitaxel absorption with vitamin E-TPGS: effect on solubility and permeability in vitro, in situ and in vivoEur J Pharm Sci20052544545315890503
- MalingréMMMeerum TerwogtJMBeijnenJHPhase I and pharmacokinetic study of oral paclitaxelJ Clin Oncol2000182468247510856107
- GelderblomHVerweijJNooterKSparreboomACremophorELthe drawbacks and advantages of vehicle selection for drug formulationEur J Cancer2001371590159811527683
- YangSGursoyRNLambertGBenitaSEnhanced oral absorption of paclitaxel in a novel self-microemulsifying drug delivery system with or without concomitant use of P-glycoprotein inhibitorsPharm Res20042126127015032307
- MaXWangHJinSWuYLiangXJConstruction of paclitaxel-loaded poly (2-hydroxyethyl methacrylate)-g-poly (lactide)-1,2-dipalmitoylsn- glycero-3-phosphoethanolamine copolymer nanoparticle delivery system and evaluation of its anticancer activityInt J Nanomedicine201271313132822419875
- YangDVanSJiangXYuLNovel free paclitaxel-loaded poly(L-γ-glutamylglutamine)-paclitaxel nanoparticlesInt J Nanomedicine20116859121289985
- RogerELagarceFGarcionEBenoitJPLipid nanocarriers improve paclitaxel transport throughout human intestinal epithelial cells by using vesicle-mediated transcytosisJ Control Release200914017418119699246
- LeeSCKimCKwonICChungHJeongSYPolymeric micelles of poly(2-ethyl-2-oxazoline)-block-poly(ɛ-caprolactone) copolymer as a carrier for paclitaxelJ Control Release20038943744612737846
- YaghmurAMichaelRLiquid crystalline nanoparticles as drug nanocarriers Available at: http://www.ibr.oeaw.ac.at/people/Michael/Rappolt_Publ-Dateien/MR2010_5.pdfAccessed May 31, 2012
- YangDArmitageBMarderSRMinireviews cubic liquid-crystalline nanoparticlesAngew Chem Int Ed Engl2004434402440915317018
- YaghmurAGlatterOCharacterization and potential applications of nanostructured aqueous dispersionsAdv Colloid Interface Sci2009147–148333342
- EfratRAserinAKesselmanEDaninoDWachtelEGartiNLiquid micellar discontinuous cubic mesophase from ternary monoolein/ethanol/water mixturesColloids Surf A Physiochem Eng Asp2007299133145
- LeeKWYNguyenTHHanleyTBoydBJNanostructure of liquid crystalline matrix determines in vitro sustained release and in vivo oral absorption kinetics for hydrophilic model drugsInt J Pharm200936519019918790030
- KaasgaardTDrummondCJOrdered 2-D and 3-D nanostructured amphiphile self-assembly materials stable in excess solventPhys Chem Chem Phys200684957497517091149
- RosenbaumETavelinSJohanssonLBA characterisation study on the application of inverted lyotropic phases for subcutaneous drug releaseInt J Pharm2010388525720026201
- CervinCVandoolaeghePNistorCTibergFJohnssonMA combined in vitro and in vivo study on the interactions between somatostatin and lipid-based liquid crystalline drug carriers and bilayersEur J Pharm Sci20093637738519041716
- JohnssonMBarauskasJNorlinATibergFPhysicochemical and drug delivery aspects of lipid-based liquid crystalline nanoparticles: a case study of intravenously administered propofolJ Nanosci Nanotechnol200663017302417048513
- RizwanSBAssmusDBoehnkeAPreparation of phytantriol cubosomes by solvent precursor dilution for the delivery of protein vaccinesEur J Pharm Biopharm201179152221237267
- LaiJChenJLuYGlyceryl monooleate/poloxamer 407 cubic nanoparticles as oral drug delivery systems: I. in vitro evaluation and enhanced oral bioavailability of the poorly water-soluble drug simvastatinAAPS Pharm Sci Tech200910960966
- BatrakovaEVLiSVinogradovSVAlakhovVYMillerDWKabanovAVMechanism of Pluronic effect on P-glycoprotein efflux system in blood-brain barrier: contributions of energy depletion and membrane fluidizationJ Pharmacol Exp Ther20019948349311602658
- GaoXWangTWuBQuantum dots for tracking cellular transport of lectin-functionalized nanoparticlesBiochem Biophys Res Commun2008377354018823949
- RizwanSBDongYDBoydBJRadesTHookSCharacterisation of bicontinuous cubic liquid crystalline systems of phytantriol and water using cryo field emission scanning electron microscopy (cryo-FESEM)Micron20073847848517011783
- BarauskasJCervinCJankunecMInteractions of lipid-based liquid crystalline nanoparticles with model and cell membranesInt J Pharm201039128429120214966
- AngelovaAAngelovBMutafchievaRLesieurSCouvreurPSelf-assembled multicompartment liquid crystalline lipid carriers for protein, peptide, and nucleic acid drug deliveryAcc Chem Res20114414715621189042
- SiekmannBBunjesHKochMHJWestesenKPreparation and structural investigations of colloidal dispersions prepared from cubic monoglyceride-water phasesInt J Pharm2002244334312204563
- BoydBJKhooSMWhittakerDVDaveyGPorterCJHA lipid-based liquid crystalline matrix that provides sustained release and enhanced oral bioavailability for a model poorly water soluble drug in ratsInt J Pharm2007340526017467935
- BoydBJControlled release from cubic liquid-crystalline particles (cubosomes)LynchMLSpicerPTBicontinuous Liquid CrystalsBoca Raton, FLCRC Press Inc2005
- BoydBJCharacterisation of drug release from cubosomes using the pressure ultrafiltration methodInt J Pharm200326023924712842343
- GargGSarafSSarafSCubosomes: an overviewBiol Pharm Bull20073035035317268078
- ShenHCrowstonJGHuberFSaubernSMcLeanKMHartleyPGThe influence of dipalmitoyl phosphatidylserine on phase behaviour of and cellular response to lyotropic liquid crystalline dispersionsBiomaterials2010319473948120880581
- TenchovBGMacDonaldRCSiegelDPCubic phases in phosphatidylcholine-cholesterol mixtures: cholesterol as membrane “fusogen”Biophys J2006912508251616829556
- GreenspanPMayerEPFowlerSDNile red: a selective fluorescent stain for intracellular lipid dropletsJ Cell Biol1985109659733972906
- XiaoLXiongXXiongXRole of cellular uptake in the reversal of multidrug resistance by PEG-b-PLA polymeric micellesBiomaterials2011325148515721546083
- LamprechtABenoitJPEtoposide nanocarriers suppress glioma cell growth by intracellular drug delivery and simultaneous P-glycoprotein inhibitionJ Control Release200611220821316574265
- RogerELagarceFGarcionEBenoitJPReciprocal competition between lipid nanocapsules and P-gp for paclitaxel transport across Caco-2 cellsEur J Pharm Sci20104042242920438839
- XuPGullottiETongLIntracellular drug delivery by poly (lactic-co-glycolic acid) nanoparticlesMol Pharm2009619020119035785
- JacobsonKMouritsenOGAndersonRGWLipid rafts: at a crossroad between cell biology and physicsNat Cell Biol2007971417199125
- OrlandiPAFishmanPHFilipin-dependent inhibition of cholera toxin: evidence for toxin internalization and activation through caveolae-like domainsJ Cell Biol19981419059159585410
- GarmannDWarneckeAKalaydaGVKratzFJaehdeUCellular accumulation and cytotoxicity of macromolecular platinum complexes in cisplatin-resistant tumor cellsJ Control Release200813110010618691617
- ChiuYLHoYCChenYMThe characteristics, cellular uptake and intracellular trafficking of nanoparticles made of hydrophobically-modified chitosanJ Control Release201014615215920580915
- XiaoLXiongXXiongXRole of cellular uptake in the reversal of multidrug resistance by PEG-b-PLA polymeric micellesBiomaterials2011325148515721546083