 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Background and methods
Paclitaxel, a widely used antitumor agent, has limited clinical application due to its hydrophobicity and systemic toxicity. To achieve sustained and targeted delivery of paclitaxel to tumor sites, liposomes composed of egg phosphatidylcholine, cholesterol, and distearolyphosphatidyl ethanolamine-N-poly(ethylene glycol) (PEG2000) were prepared by a lipid film method. In addition, the liposomes also contained truncated fibroblast growth factor fragment-PEG-cholesterol as a ligand targeting the tumor marker fibroblast growth factor receptor. Physicochemical characteristics, such as particle size, zeta potential, entrapment efficiency, and release profiles were investigated. Pharmacokinetics and biodistribution were evaluated in C57BL/6 J mice bearing B16 melanoma after intravenous injection of paclitaxel formulated in Cremophor EL (free paclitaxel), conventional liposomes (CL-PTX), or in targeted PEGylated liposomes (TL-PTX).
Results
Compared with CL-PTX and free paclitaxel, TL-PTX prolonged the half-life of paclitaxel by 2.01-fold and 3.40-fold, respectively, in plasma and improved the AUC0→t values of paclitaxel by 1.56-fold and 2.31-fold, respectively, in blood. Biodistribution studies showed high accumulation of TL-PTX in tumor tissue and organs containing the mononuclear phagocyte system (liver and spleen), but a considerable decrease in other organs (heart, lung, and kidney) compared with CL-PTX and free paclitaxel.
Conclusion
The truncated fibroblast growth factor fragment-conjugated PEGylated liposome has promising potential as a long-circulating and tumor-targeting carrier system.
Introduction
Paclitaxel is a diterpenoid isolated from Taxus brevifolia, and is approved by the US Food and Drug Administration for the treatment of ovarian and breast cancers. Clinical trials have shown that paclitaxel has antineoplastic activity, particularly against primary ovarian epithelial carcinoma, breast, colon, head and neck cancers, and nonsmall cell lung cancer. Due to its low solubility in water, it is clinically administered dissolved in Cremophor® EL (polyethoxylated castor oil) and ethanol.Citation1,Citation2 These formulations are highly allergenic, require extensive premedication, and are responsible for most of the acute toxicity.Citation3–Citation10 To overcome these problems, new aqueous-based formulations of paclitaxel that do not require solubilization by Cremophor and ethanol and decrease the systemic toxicities of the drug are currently being developed.Citation11–Citation15 Liposomes consist of phospholipids that are highly biocompatible with low toxicity, and they are able to encapsulate both hydrophilic and hydrophobic drugs. Furthermore, one of the strong merits of liposomes is that they can be used for specific purposes, including long circulation and targeting, by alteration of their lipid composition or addition of functional agents.Citation16–Citation19 During recent decades, encapsulation of chemotherapeutics into liposomal formulations has had reasonable success as a pharmacological strategy in vitro. Sharma et al and Straubinger et al developed a liposomal formulation composed of phosphatidylcholine and phosphatidylglycerol, and showed that the growth-inhibitory activity of paclitaxel-loaded liposomes against a variety of tumor cell lines in vitro was similar to that of the free drug.Citation20–Citation22 However, in vivo, their success was limited due to rapid uptake by the reticuloendothelial system, thereby preventing them from reaching the target tissues.Citation20–Citation23 The advent of long-circulating liposomes prepared by incorporation of phospholipid-modified polyethyleneglycol (PEG) in the phospholipid bilayer allowed the limitations of conventional liposomes to be overcome and has proven to be a good approach to prolonging the circulation time of the drug and improving its antitumor efficacy. Long-circulating liposomes prepared by surface modification of a flexible hydrophilic polymer such as PEGCitation24–Citation28 are effective as drug carriers for oral, parenteral, and topical administration to avoid unwanted metabolism both in vivo and in vitro. PEGylated liposomal doxorubicin (Doxil®, Caelyx®; Alza Pharmaceuticals, San Bruno, CA) has shown an ability to inhibit interaction with plasma proteins and mononuclear phagocytes, and can markedly prolong the circulation time, with a terminal half-life of 55 hours in humans.Citation29 Sterically stabilized liposomes containing paclitaxel has also been reported.Citation30–Citation33
Figure 1 Synthetic scheme for preparation of targeted PEGylated liposomes.
Abbreviations: PEG, poly(ethylene glycol); EPC, egg phosphatidylcholine; Chol, cholesterol; DSPE, distearolyphosphatidyl ethanolamine; EDC, 1-ethyl-3-(3- dimethylaminopropyl)-carbodiimide hydrochloride; NHS, N-hydroxysuccinimide; tbFGF, truncated basic fibroblast growth factor.
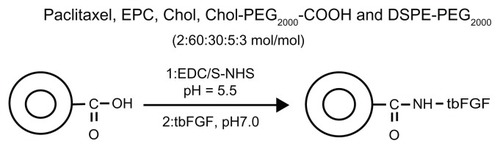
Various tumor cells, such as melanoma, breast cancer, prostate cancer, and nonsmall cell lung cancer are reported to overexpress basic fibroblast growth factor (FGF) and FGF receptors in comparison with normal tissues.Citation34–Citation36 In a previous study, we constructed a human truncated basic FGF peptide (30–115 amino acids) containing a basic FGF receptor binding site and part of a heparin-binding site. This truncated basic FGF peptide could effectively bind to FGF receptors on the cell surface, but did not stimulate cell proliferation.Citation37 Using this peptide, we have developed FGF receptor-mediated cationic liposomes to target doxorubicin or paclitaxel to tumor cells overexpressing the FGF receptor and to endothelial cells in the tumor neovasculature in vitro and in vivo. Such a delivery system not only improved the uptake of chemotherapeutic agents in tumor tissue and human umbilical vein endothelial cells, but also significantly inhibited tumor growth and improved survival in tumor-bearing mice.Citation38 We have also constructed a micellar drug delivery system formulated with cholesterol-block-PEG (COOH-PEG2000- cholesterol) copolymer and chemically surface-linked it with truncated basic FGF fragments. This micellar system has the advantages of delivering chemotherapeutic agents specifically to tumor cells, and increasing the efficacy of chemotherapeutic agents in vitro.Citation39 However, these cationic liposomes and the micellar drug delivery system do not have a noticeably prolonged circulation time in the bloodstream. Therefore, it has been necessary to develop a better tumor-targeted drug delivery system with a longer circulation time in the bloodstream.
In this study, liposomes composed of egg phosphatidylcholine, cholesterol, distearolyphosphatidyl ethanolamine-N-poly(ethylene glycol) (DSPE-PEG2000) and COOH-PEG2000-cholesterol were prepared using the lipid film method. In addition, truncated basic FGF fragments were conjugated to the distal terminals of PEG previously anchored on liposomes. The purpose of the DSPE-PEG2000 and COOH-PEG2000-cholesterol block copolymer was to keep the liposomes sterically stabilized, after which they were loaded with paclitaxel using the solvent evaporation method. Finally, the truncated basic FGF fragments were conjugated to the carboxyl terminals of COOH-PEG2000-cholesterol on the surface of the liposomes for targeted delivery to tumor cells. Ideally, such liposomes would substantially increase both passive and active accumulation of paclitaxel in tumor tissue. At the same time, we prepared conventional liposomes containing egg phosphatidylcholine, cholesterol, and paclitaxel (CL-PTX) and also prepared free paclitaxel. The physicochemical and pharmacokinetic properties of the different formulations were then investigated. The suitability of the liposomes as drug carriers to prolong the half-life of paclitaxel was evaluated. In addition, the effect of truncated basic FGF on the tumor-targeting characteristics and biodistribution of the liposomes was elucidated.
Materials and methods
Chemicals
Egg phosphatidylcholine, PEG (molecular weight 2000), cholesterol, Cremophor EL, Dulbecco’s modified Eagle’s medium, and RPMI 1640 medium were purchased from Sigma (St Louis, MO). Paclitaxel was purchased from Chengdu Mansite Pharmaceutical Co Ltd, Chengdu, China. Acetonitrile (high-performance liquid chromatography, HPLC, grade) was purchased from Fisher Chemical (Loughborough, Leicestershire, UK). Chloroform, methanol (analytical grade), 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC), and N- hydroxysuccinimide (NHS), were purchased from Chengdu Kelong Chemical Co Ltd, Chengdu, China. DSPE-PEG2000 was purchased from Lipoid (Ludwigshafen, Germany). COOH-PEG2000-cholesterol and truncated basic FGF peptides were prepared and purified in our own laboratory.Citation37,Citation39 All other reagents and solvents used were of analytical pure grade.
Animal models
Male and female C57BL/6 J mice (n = 90, 6–8 weeks old, 18–22 g) were obtained from the Laboratory Animal Center of Sichuan University in China. All the animals were raised under standard pathogen-free conditions in the Animal Center for Pharmaceutical Research. After a one-week acclimation period, the animals were used for the experiments. Murine B16F10 melanoma cells were purchased from the American Type Culture Collection (Rockville, MD) and cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% (v/v) heat-inactivated phosphate-buffered solution and 10 μL/mL penicillin-streptomycin. The cultures were sustained at 37°C in a humidified incubator containing 5% CO2. The cells were maintained in their exponential growth phase. To prepare tumor-bearing mice, 2 × 105 B16F10 cells in 50 μL were subcutaneously injected into the right flank of mice. All of the procedures involved in the animal experiments and the study protocols were approved by our institutional animal care and use committee (Sichuan University, Chengdu, China).
Preparation of paclitaxel liposomes
The long-circulating paclitaxel-encapsulated liposomes were prepared using a thin film evaporation-extrusion technique. Briefly, paclitaxel, egg phosphatidylcholine, cholesterol, COOH-PEG2000-cholesterol, and DSPE-PEG2000 (2:60:30:5:3 mol/mol) were dissolved in 4 mL of methanol and chloroform (1:3, v/v) as a mixed solvent at room temperature and dried to a thin film, firstly with nitrogen gas and then under vacuum for several hours. The lipid film was hydrated with 2 mL of 10 mM 2-(N-morpholino)ethanesulfonic acid (MES) buffer (pH 5.0) at 40°C for one hour. To obtain small and homogeneous vesicles, the liposome suspension was sonicated for 10 minutes in a bath-type sonicator (Bransonic 12) followed by three extrusion cycles through polycarbonate filters with 0.2 μm pores (Lipex™ Extruder, Northern Lipids Inc, Vancouver, BC).Citation40 For CL-PTX, paclitaxel, egg phosphatidylcholine, and cholesterol (molar ratio, 2:60:30) were dissolved in chloroform/methanol (3:1, v/v), and then prepared as for the above description of paclitaxel-loaded targeted PEGylated liposomes (TL-PTX) to obtain uniform CL-PTX. The resulting liposomes were purified on a Sephadex G-75 column to remove the nonencapsulated drug content.
Development of truncated basic FGF-conjugated liposomes
To activate the terminal carboxyl group of COOH-PEG2000- cholesterol, the paclitaxel-loaded liposomes (with 20 mg/mL COOH-PEG2000-cholesterol in 10 mM MES buffer) were incubated with 400 mM EDC and 100 mM NHS for 15 minutes at room temperature with gentle stirring and then neutralized with NaOH (0.01%) to pH 7.0. The resulting NHS-activated particles were covalently linked to the −NH2 groups of truncated basic FGF (1% weight compared with lipid concentration). The reaction was continued at 4°C overnight. Unreacted truncated basic FGF, EDC, and NHS were removed by gel filtration.Citation41 The final concentration of paclitaxel encapsulated in the liposomes was determined by HPLC assay. The encapsulation efficiency of paclitaxel in CL-PTX and in TL-PTX was calculated according to the following equation:
where EE is encapsulation efficiency, Wtotal is the total amount of paclitaxel in the liposomes before purification and Wencapsulated is the amount of paclitaxel encapsulated into the liposomes after purification.
Characterization of paclitaxelencapsulated liposomes
Size and zeta-potential determination and atomic force microscopy
The particle size distribution and zeta potential of CL-PTX and TL-PTX were measured by dynamic light scattering (Zetasizer, Nano-ZS, Malvern, UK). At least three different batches were analyzed to achieve a mean value and standard deviation for particle diameter and zeta potential. The shape and structure of TL-PTX were observed by atomic force microscopy. The prepared suspension was diluted with deionized water and deposited onto freshly cleaved mica lamella. The sample was dried for 3 hours at room temperature. Atomic force microscopic images were taken by tapping mode in air on an atomic force microscope (SPA400, Seiko, Tokyo, Japan).
Quantification of liposome-bound peptide
The amount of liposome-bound peptide was determined using a bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL) following the protocol recommended by the manufacturer. Briefly, triplicates of a truncated basic FGF-modified liposome sample along with duplicates of known serial dilutions of BCA (to build a standard curve), were added to a 96-well plate in the same volumes. After mixing samples with 200 μL of the BCA working reagent (1:50 ratio of 4% CuSO4: BCA, Na2CO3, NaHCO3, Na tartrate in 0.1 M NaOH), the plate was incubated at 37°C for 30 minutes. After cooling to room temperature, absorbance of the purple-colored complex of BCA and Cu1+ (product of protein oxidation by Cu2+) was measured at 562 nm using a Labsystems Multiscan MCC/340 plate reader (Labsystems and Life Sciences International Ltd, London, UK).
In vitro drug release
Release of the paclitaxel from CL-PTX, TL-PTX, and Cremophor EL in vitro was monitored using a dialysis method. Dialysis was carried out at 37°C using Spectra/Por dialysis membranes with a molecular weight cutoff of 14 kDa and phosphate-buffered saline (pH 7.4) as the sink solution. The initial concentration of paclitaxel in the formulations was 2 mg/mL. At scheduled intervals, 1 mL of the dialysis medium was collected and the same volume of fresh medium was added immediately. The concentration of paclitaxel in dialysis medium was monitored by HPLC assay. Chromatographic separations were carried out on a C18 column (250 × 4.6 mm, 5 μm) and samples were eluted using acetonitrile and water (50:50, v/v) at a flow rate of 1.0 mL per minute with the ultraviolet detector at 227 nm. The release percentage was calculated according to the following equation:
where Wtotal is the total amount of paclitaxel in the liposomes and Wrelease is the amount of paclitaxel released from the liposomes or Cremophor EL into the dialysis medium at any time.
Pharmacokinetics and biodistribution
The study protocol for the animal experiments complied with the guidelines on animal experimentation for Sichuan University, China. When the B16F10 tumors had reached a volume of about 500 mm3, the C57BL/6 J tumor-bearing mice were randomly and equally divided into three groups, which received free paclitaxel solution, CL-PTX, or TL-PTX administered as a single dose of 10 mg paclitaxel/kg body weight via the tail vein. Free paclitaxel solution, as the control, was prepared by dissolving 200 mg of paclitaxel in a 10 mL mixture of Cremophor EL (1.5%) and ethanol (1.5%) followed by dilution with 5% glucose (w/v) to 200 mL to generate a final concentration of 1 mg/mL. All the free paclitaxel solution mentioned below was made up as the same preparation. The concentration of paclitaxel in the CL-PTX and TL-PTX formulations was also 1 mg/mL.
For the pharmacokinetic studies, the mice were anesthetized by halothane inhalation and bled by eye enucleation at predetermined time points (0.5, 1, 3, 5, 12, and 24 hours following administration of the experimental treatments). Plasma samples were obtained and centrifuged immediately at 4000 × g for 10 minutes, and were then stored at −20°C until analysis. For the biodistribution studies, the mice from each group (five mice per point) were sacrificed following collection of blood at predetermined time points. Tissues of interest (tumor, heart, liver, spleen, lung, kidney, and brain) were rapidly excised from each mouse and immediately washed twice with normal saline (0.9% NaCl), dried with filter paper, weighed, homogenized, and frozen at −20°C until analysis.
Ultraperformance liquid chromatography (Waters, Milford, MA) was used to determine the paclitaxel concentration in the plasma and tissues. Paclitaxel was extracted by methyl-tertbutyl ether and quantified by reverse-phase ultraperformance liquid chromatography with a C18 column (2.1 × 50 mm, 1.7 μm) using an ultraviolet wavelength of 227 nm, with water and acetonitrile (40:60, vol/vol) as the eluant solution. The paclitaxel concentrations were fitted by a Pharmacokinetic Software of Drug and Statistics (edited by the Mathematical Pharmacology Professional Committee of China, version 2.1.1). The data were normalized to tissue weights.
Statistics
The data were expressed as the mean ± the standard deviation. Statistical differences were estimated using the Student’s t-test. The data were considered to be statistically significant at P < 0.05. Pharmacokinetic parameters were obtained using the practical pharmacokinetic program version 87 (Committee of Mathematic Pharmacology of the Chinese Society of Pharmacology, China). All statistical tests were two-sided, and were performed using SPSS version 16 for Windows statistical software (SPSS Inc, Chicago, IL).
Results
Physicochemical properties of CL-PTX ± truncated basic FGF fragments
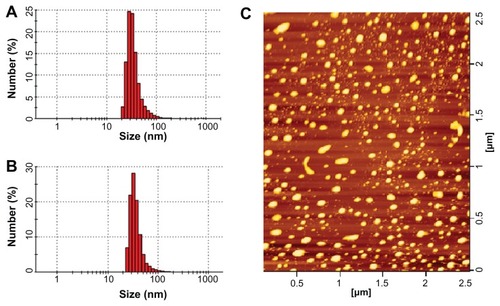
Both CL-PTX and TL-PTX were suitable as injectable formulations, and had a small particle size. The shape of the CL-PTX was round or oval, as observed by atomic force microscopy, and is shown in . The mean vesicle diameter of CL-PTX was 137.3 ± 23.1 nm (n = 3) which was determined by dynamic light scattering, and the polydispersity index was 0.217 ± 0.072 ( and ). The TL-PTX data showed that the mean liposome size increased slightly after attachment of the truncated basic FGF fragment (142.2 ± 46.4, n = 3), and the polydispersity index was 0.255 ± 0.086 ( and ). Zeta potential measurement indicated that the surface charge of CL-PTX and TL-PTX was −9.20 ± 0.31 mV (n = 3) and −5.05 ± 0.16 mV (n = 3), respectively (). Because the isoelectric point of the truncated basic FGF fragments was pH 8.77, the increase in zeta potential of TL-PTX (compared with CL-PTX, P < 0.05) might have contributed to the positive charge of the truncated basic FGF fragments in the buffer (pH 7.4). The average encapsulation efficiency of paclitaxel in CL-PTX and TL-PTX was 89.7% ± 3.6% and 89.1% ± 2.3%, respectively. The amount of truncated basic FGF conjugated to the liposomal surface was approximately 27.4 ± 1.8 μg truncated basic FGF/mg COOH-PEG2000-cholesterol (n = 3), as determined by BCA quantitative protein assay.
Table 1 Particle size and zeta potential (mean ± SD, n = 3) of CL-PTX and TL-PTX
Figure 2 Particle size distribution of paclitaxel-loaded conventional liposomes (A) and paclitaxel-loaded targeted PEGylated liposomes (B), and typical atomic force microscopy image of paclitaxel-loaded targeted PEGylated liposomes (C).
Stability of formulated liposomes
The stability of CL-PTX and TL-PTX was observed over a one-week time course with storage at 4°C. The results () show no significant differences in particle size distribution, zeta potential, and encapsulation efficiency for paclitaxel on any of the days in storage, indicating that CL-PTX and TL-PTX are stable for at least a week when stored at 4°C. The experiments were performed in triplicate.
Table 2 Stability of formulated liposomes (mean ± SD, n = 3)
Paclitaxel release from TL-PTX in vitro
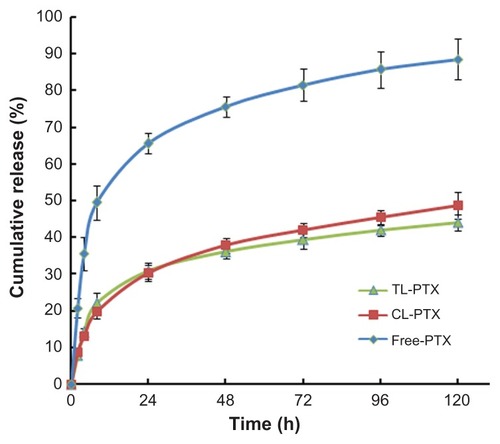
The paclitaxel release profile from TL-PTX in vitro is shown in . A typical two-phase release profile was observed for TL-PTX, ie, relatively rapid release in the first stage (22.1%, 0.442 mg versus 2 mg within 8 hours) followed by sustained and slow release over a prolonged period for up to several days. It was found that only 44.1% (0.882 mg versus 2 mg) was released from the liposomes within 120 hours, while about 88.5% of the free paclitaxel (1.770 mg versus 2 mg) was released into the outside medium. The drug release rate for free paclitaxel was about twice that of TL-PTX. The release profile for CL-PTX was similar to that of TL-PTX.
Figure 3 Time course of paclitaxel release from TL-PTX (▲), CL-PTX (■), and free PTX(◆) at 37°C and pH 7.0. The paclitaxel Released was separated by dialysis and quantified using high-performance liquid chromatography.
Notes: There were five mice per group per time point. Bars represent the mean ± standard deviation. Error bars correspond to 95% confidence intervals.
Abbreviations: CL-PTX, paclitaxel-loaded conventional liposomes; TL-PTX, paclitaxel-loaded targeted PEGylated liposomes; Free PTX, paclitaxel dissolved in Cremophor EL.
Pharmacokinetic parameters
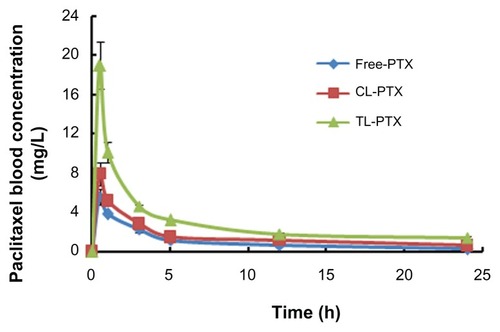
The plasma concentration-time curves and pharmacokinetic parameters after intravenous administration of the different paclitaxel formulations are shown in and . The highest plasma paclitaxel concentration was observed after injection of the TL-PTX formulation (maximum concentrations of TL-PTX, CL-PTX and free paclitaxel were 18.95 ± 1.03, 7.93 ± 0.47, and 5.58 ± 0.31, respectively, P < 0.05). This indicates that CL-PTX had a drug concentration-time curve similar to that of the free paclitaxel solution, meaning that paclitaxel was quickly removed from the circulation. In contrast, TL-PTX showed markedly delayed clearance in blood. TL-PTX prolonged the half-life of paclitaxel by 2.01-fold and 3.40-fold in plasma, and improved the AUC0→∞ values of paclitaxel by 1.56-fold and 2.31-fold compared with CL-PTX and free paclitaxel, respectively. The paclitaxel in TL-PTX was eliminated rather slowly, with a longer t1/2β (29.12 ± 1.81 hours) compared with CL-PTX and free paclitaxel solution. The t1/2β of CL-PTX (14.48 ± 1.07 hours) was a little longer than that of free paclitaxel (8.56 ± 0.91 hours). The AUC0→∞ of TL-PTX (62.17 ± 6.92 μg/mL*hour) was significantly larger than that of CL-PTX (39.88 ± 2.49 μg/mL*hour) and of free paclitaxel (26.89 ± 2.96 μg/mL*hour).
Table 3 Pharmacokinetic parameters for paclitaxel in plasma after intravenous administration of free PTX, CL-PTX, and TL-PTX (mean ± standard deviation)
Figure 4 Areas under the curve over 24 hours for paclitaxel in serum after treatment with free PTX (◆), CL-PTX (■), or TL-PTX (▲).
Notes: There were five mice per group per time point. Bars represent the mean ± standard deviation.
Abbreviations: CL-PTX, paclitaxel-loaded conventional liposomes; TL-PTX, paclitaxel-loaded targeted PEGylated liposomes; Free PTX, paclitaxel dissolved in Cremophor EL.
Biodistribution studies
In our study, the biodistribution of paclitaxel after intravenous injection in the three different preparations were evaluated in a B16 tumor-bearing mouse model. The three preparations consisted of TL-PTX, CL-PTX, and free paclitaxel. The paclitaxel distribution results are presented in and , and show that distribution of paclitaxel in the body could be markedly affected by the different formulations used for encapsulation. The paclitaxel concentration in tumor tissue achieved by TL-PTX was the highest of the three formulations at each time point. TL-PTX increased the concentration of paclitaxel at the tumor site by 1.89- fold (6.60 ± 0.57 μg/g versus 3.48 ± 0.38 μg/g, P < 0.01) and 4.66-fold (6.60 ± 0.57 μg/g versus 1.41 ± 0.12 μg/g, P < 0.01) at 0.5 hours, and 3.06-fold (0.86 ± 0.10 μg/g versus 0.28 ± 0.04 μg/g, P < 0.01) and 5.21-fold (0.86 ± 0.10 μg/g versus 0.17 ± 0.03 μg/g, P < 0.01) after 24 hours compared with CL-PTX and free paclitaxel, respectively (). Furthermore, the AUC0–∞ for TL-PTX in tumors was 4.61- fold higher than that for free paclitaxel (46.43 ± 8.08 mg/L/hour versus 10.07 ± 1.11 mg/L/hour, P < 0.01), 1.81- fold that for CL-PTX (46.43 ± 8.08 mg/L/hour versus 25.77 ± 0.95 mg/L/hour, P < 0.01), and the maximum concentration (Cmax) for TL-PTX in tumor tissue was 5.15- fold (6.60 ± 0.55 mg/L versus 1.28 ± 0.09 mg/L, P < 0.01) higher than that of free paclitaxel and 1.88-fold that of CL-PTX (6.60 ± 0.55 mg/L versus 3.51 ± 0.24 mg/L, P < 0.01). These data suggest that TL-PTX enabled drug accumulation specifically in tumors overexpressing the FGF receptor.
Table 4 Increase versus time of distribution of TL-PTX in the tumors compared with CL-PTX and free PTX, respectively
Figure 5 Mean paclitaxel concentration in tumor tissue at each time point after treatment with free PTX, CL-PTX, and TL-PTX. Paclitaxel was extracted from the tumors and measured by ultraperformance liquid chromatography.
Notes: There were five mice per group per time point. Bars represent the mean ± standard deviation.
Abbreviations: CL-PTX, paclitaxel-loaded conventional liposomes; TL-PTX, paclitaxel-loaded targeted PEGylated liposomes; Free PTX, paclitaxel dissolved in Cremophor EL.
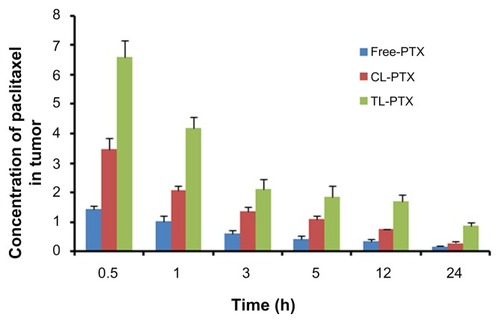
Figure 6 Mean paclitaxel concentration in tumor, lungs, heart, spleen, liver, and kidneys of mice treated with free PTX, CL-PTX, and TL-PTX. Mice were sacrificed at the indicated time points of (A) 0.5, (B) 1, (C) 3, (D) 5, (E) 12, and (F) 24 hours after administration, and paclitaxel was extracted from the tissues and measured by ultraperformance liquid chromatography.
Notes: There were five mice per group per time point. Bars represent the mean ± standard deviation.
Abbreviations: CL-PTX, paclitaxel-loaded conventional liposomes; TL-PTX, paclitaxel-loaded targeted PEGylated liposomes; Free PTX, paclitaxel dissolved in Cremophor EL.
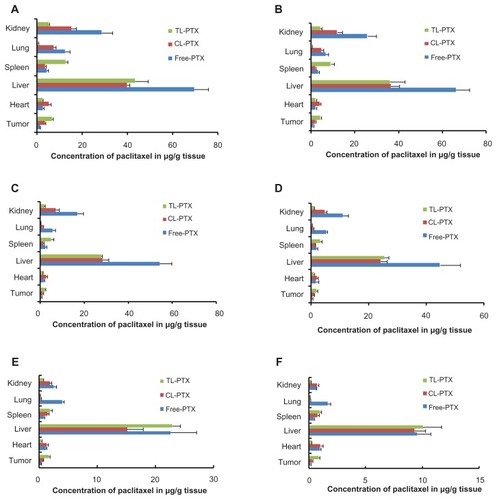
As shown in , paclitaxel concentrations from the TL-PTX formulation were significantly (P < 0.05) higher in the spleen, but lower (P < 0.05) in the lung, heart, and kidney compared with those from the CL-PTX or free paclitaxel formulations. Although the liposomes distributed mainly to the organs of the mononuclear phagocyte system, with most of the accumulation in the liver, paclitaxel concentrations in the liposomal formulation were significantly (P < 0.05) lower than those from free paclitaxel in the liver from 0.5 to 5 hours after injection.
Discussion
In chemotherapy for cancer, the crucial problem is how to avoid the side effects of drugs on healthy tissues.Citation42–Citation47 Only 2%–5% of a therapeutic drug dose is taken up by tumor tissue, with more than 90% taken up by normal tissues.Citation43,Citation46 As a result, side effects often result in dose reduction, frequent treatment, or discontinuation of therapy. Recent research in this area has focused on receptor-mediated drug delivery systems based on liposomes. Active targeting of tumor tissue by liposomes can be achieved by modifying the liposome with addition of a targeting ligand, such as an antibody and a peptide.Citation48–Citation51 However, the problem with these systems is that the majority of the liposomes are still cleared by the reticuloendothelial system, resulting in short half-lives, low accumulation in tumor tissue, and unwanted deposition in the liver and spleen. To achieve a specific therapeutic effect, both a prolonged blood circulation time and an active tumor-targeting effect are essential for an ideal drug delivery system. In our study, a novel formulation was designed for prolonged systemic circulation and selective targeting of tumor tissue showing amplified expression of the FGF receptor.
In this research, the particle size and zeta potential of CL-PTX and TL-PTX formulations were described. The liposomes were observed by atomic force microscopy to characterize their structures further. The particle size of TL-PTX (142.2 ± 46.4 nm) was slightly larger than that of CL-PTX (137.3 ± 23.1 nm) due to the fact that truncated basic FGF was attached to the surface of the liposomes so as to increase the diameter of TL-PTX. In contrast, part of the negative charge of TL-PTX was neutralized by truncated basic FGF so that the zeta potential of TL-PTX (−5.05 ± 0.16 mV) was slightly lower than that of CL-PTX (−9.20 ± 0.3 mV). There is some evidence that neutral or negatively charged liposomes could have a longer circulation time in the bloodstream because of their weaker interaction with serum proteins compared with positively charged liposomes.Citation52 An early study using liposomes demonstrated that small neutral unilamellar vesicles (≤100 nm) made from equimolar amounts of saturated phospholipids and cholesterol had a longer circulation time in rats (half-lives up to 20 hours) compared with their anionic counterparts (half-lives less than one hour).Citation53 These observations are in agreement with the process of complement opsonization of liposomes, whereby neutral vesicles were poor activators of the complement system when compared with positive or anionic liposomes.Citation54–Citation56 Therefore, small-sized neutral vesicles were not efficiently coated with the opsonizing complement proteins, so that they were poorly recognized by Kupffer cells. Truncated basic FGF fragments contain many basic amino residues, which might result in a positive charge on the liposomal surface. The positive charge might induce rapid uptake by the reticuloendothelial system and nonspecific binding with unintended cells, such as erythrocytes, after intravenous administration. To overcome this problem, we chose egg phosphatidylcholine, cholesterol, DSPE-PEG2000, and COOH-PEG2000-cholesterol as the primary elements of TL-PTX to achieve a surface charge of –5.05 ± 0.16 mV. The surface charge was close to neutral, and enabled paclitaxel to have prolonged circulation time in the bloodstream and improved its penetration and accumulation at tumor sites. Further, the PEG chains in COOH-PEG2000-cholesterol and DSPE-PEG2000 were expected to limit interaction with the reticuloendothelial system and other nontarget cells. The pharmacokinetics and biodistribution results for TL-PTX in vivo confirmed this hypothesis.
In vitro release of paclitaxel from the free paclitaxel, CL-PTX, and TL-PTX formulations was studied at 37°C in buffer (pH 7.4). The initial concentration of paclitaxel was selected as 2 mg/mL to enable a series of drug concentrations in dialysis medium to be appropriately detected after release from the three formulations. Relatively rapid release from free paclitaxel and slow release from CL-PTX and TL-PTX was observed. It was demonstrated that paclitaxel could pass through the dialysis membrane entirely into the dissolution medium and that paclitaxel could be released slowly from the liposomes. Therefore, CL-PTX and TL-PTX showed greatly increased stability compared with free paclitaxel.
Because the concentration of paclitaxel in the extracellular fluid (pH 7.4, 37°C) was relatively low, paclitaxel was released rapidly over 0–12 hours from TL-PTX and CL-PTX as a result of being driven by a concentration gradient. After 12 hours, the drug concentration in extracellular fluid reached a certain level and the effect of the concentration gradient became smaller, enabling slower release of paclitaxel from TL-PTX and CL-PTX, which ensured a long-circulating effect. This attenuated drug release might result in a more favorable pharmacokinetic profile in vivo.Citation57–Citation59
To evaluate the pharmacokinetics and tissue distribution of PEG-mediated and truncated basic FGF-mediated paclitaxel liposomes, we used C57BL/6J tumor-bearing mice and selected 10 mg paclitaxel/kg body weight as a single dose to be given via the tail vein consistent with a previous report.Citation60 Our present work provided an overall profile for circulation of the drug in the bloodstream, the main body tissues, and the tumor tissue. The pharmacokinetic parameters for paclitaxel changed significantly and were observed for TL-PTX, CL-PTX, and free paclitaxel. The higher plasma concentration of TL-PTX and CL-PTX compared with free paclitaxel in mice might be due to slower release of paclitaxel from the liposomes, such that paclitaxel in a liposomal form could circulate in the bloodstream for the required period of time. In addition, the slow clearance of paclitaxel from TL-PTX compared with CL-PTX might be due to the relatively better stability of TL-PTX in vivo. The reasons for its better stability might be associated with the physical barrier properties of the PEG coating, which might slow drug release from the liposomes and limit interaction with components of plasma. The slower biodegradability of TL-PTX in the bloodstream could enhance accumulation of paclitaxel in solid tumor vasculature.
Administration of TL-PTX and CL-PTX altered the distribution of paclitaxel in the tissues, with higher uptake in the tumor and spleen and lower uptake in other tissues compared with free paclitaxel. Importantly, TL-PTX showed significantly greater accumulation than CL-PTX and free paclitaxel in tumor tissue. The underlying mechanisms might be two-fold. First, long-circulating liposomes may slowly accumulate in tumor tissue and protect the encapsulated agent from enzymatic degradation, ultimately reaching high levels in tumor tissue due to the enhanced permeability and retention effect. Second, the truncated basic FGF might specifically target tumor cells by binding with FGF receptors, resulting in specific uptake of paclitaxel.
The lower concentration of paclitaxel released from TL-PTX than from CL-PTX and free paclitaxel in the lungs, heart, and kidneys at all time points might be due to the specific tumor targeting function of truncated basic FGF-modified liposomes, suggesting attenuated toxicity in healthy tissues.
During the first 5 hours following administration, liposomes distributed mainly to the bloodstream, tumor, and spleen. The concentration of free paclitaxel in the liver was higher than that of TL-PTX or CL-PTX, indicating that these liposomal formulations had a tendency to escape from phagocytic cells in the blood because of their small particle size and PEG coating over a certain time period. However, TL-PTX and CL-PTX accumulated in the liver to the same extent as free paclitaxel after 24 hours, suggesting that the liposomal formulations were still recognized as foreign material and cleared by phagocytic cells of the mononuclear phagocyte system, which are abundant in certain organs, such as the liver and spleen. Nevertheless, the higher level of paclitaxel released from TL-PTX reaching the blood and tumor tissue suggests that TL-PTX achieved targeting and had long-circulating effects.
The level of TL-PTX in the bloodstream was higher than for CL-PTX or free paclitaxel during the 5 hours following injection, possibly for two reasons, ie, TL-PTX might slow drug release from the liposomes and limit interaction with plasma components, and clearance of the drug from TL-PTX was slower. The slower biodegradability of TL-PTX did not enhance drug accumulation in the organs of the mononuclear phagocyte system, but did improve accumulation in the solid tumor vasculature. The results of our study also demonstrate that the additional benefit of less accumulation of TL-PTX in the organs of the mononuclear phagocyte system compared with CL-PTX and free PTX diminished over time, ie, at 12 hours and 24 hours after injection. At the same time, TL-PTX did not decrease accumulation of paclitaxel in tumor tissue. The higher paclitaxel concentration in the bloodstream and tumor tissue noted in the group treated with TL-PTX suggests that TL-PTX had targeted and long-circulating effects, which might enhance the therapeutic efficacy and reduce the systemic toxicity of antitumor drugs.
TL-PTX was targeted and long-circulating, which could maintain the concentration of the drug in the bloodstream and control any burst effect. It could also reduce the concentration of the drug in normal tissues, thereby reducing its toxicity. In this study, only paclitaxel was selected as the model drug. The main dose-limiting toxicity of paclitaxel was neutropenia, and TL-PTX accumulated in the bloodstream for a long time. Paclitaxel may not be very suitable for this new drug delivery system, but this does not mean that this new system cannot be used for other chemotherapeutic drugs. Furthermore, although TL-PTX accumulated to a higher degree than CL-PTX and free paclitaxel in the spleen, the paclitaxel concentration in the spleen was very low compared with levels in other organs. In other words, a small amount of paclitaxel in the spleen did not lead to important toxicity. In contrast, the concentration of free paclitaxel was relatively high in the heart, kidney and lung, which are the organs mainly at risk of toxicity, suggesting that the TL-PTX was safer compared to the free paclitaxel.
Conclusion
In conclusion, PEG-mediated and truncated basic FGF-mediated liposomes were developed in this study, with a prolonged circulation time in the bloodstream and enhanced accumulation of paclitaxel, a lipophilic anticancer drug, in tumor tissues. Longer persistence of the liposomes in the blood enabled better targeting of circulating tumor cells. These benefits, in turn, might improve the therapeutic efficacy of paclitaxel, enabling more specific tumor therapy.
Acknowledgments
This work was supported by the National Key Technologies R&D Program (2009ZX09501-015), the Natural Science Foundation of China (81071251), and the Open Research Fund of State Key Laboratory Breeding Base of Systematic Research, Development, and Utilization of Chinese Medicine. We acknowledge Minghai Tang for her help in the pharmacokinetic and biodistribution studies.
Disclosure
The authors report no conflicts of interest in this work.
References
- ChervinskyDBrecherMHoelcleMCremophor-EL enhances Taxol efficacy in a multi-drug resistant C1300 neuroblastoma cell lineAnticancer Res19931393968097392
- WebsterLLinsenmeyerMMillwardMMortonCBishopJWoodcockDMeasurement of Cremophor EL following Taxol: plasma levels sufficient to reverse drug exclusion mediated by the multidrug-resistant phenotypeJ Natl Cancer Inst199385168516908105099
- FjällskogMLFriiLBerghJIs Cremophor EL, solvent for paclitaxel, cytotoxic?Lancet1993342873
- RowinskyEEisenhauerEChaudhryVArbuckSDonehowerRClinical toxicities encountered with paclitaxel (Taxol)Semin Oncol1993204 Suppl 31158102012
- DorrRTPharmacology and toxicology of Cremophor EL diluentAnn Pharmacother1994285S11147915152
- GoldspielBRPharmaceutical issues: preparation, administration, stability, and compatibility with other medicationsAnn Pharmacother1994282326
- SongDHsuLFAuJLSBinding of Taxol to plastic and glass containers and protein under in vitro conditionsJ Pharm Sci19968529318926579
- KongshaugMChengLSMoanJRimingtonCInteraction of Cremophor EL with human plasmaInt J Biochem1991234734782015955
- SykesEWoodburnKDeckerDKesselDEffects of Cremophor EL on distribution of Taxol to serum lipoproteinsBr J Cancer1994704014047915910
- GaoJFGuanSDengWJClinical study of comparing LP and TP regimens in the treatment of advanced non-small cell lung cancerChinese-German J Clin Oncol20109198200
- ZhangQHuangXEGaoLLA clinical study on the premedication of paclitaxel liposome in the treatment of solid tumorsBiomed Pharmacother20096360360719019625
- HolvoetCVanderHYLoriesGPreparation and evaluation of paclitaxel-containing liposomesPharmazie20076212613217341033
- YangTCuiFDChaiMKLiposome formulation of paclitaxel with enhanced solubility and stabilityDrug Deliv20071430130817613018
- StinchcombeTENanoparticle albumin-bound paclitaxel: a novel Cremophor EL-free formulation of paclitaxelNanomedicine (Lond)2007241542317716129
- HureauxJLagarceFGagnadouxFLipid nanocapsules: ready-to- use nanovectors for the aerosol delivery of paclitaxelEur J Pharm Biopharm20097323924619560538
- YangTChoiMKCuiFDAntitumor effect of paclitaxel-loaded PEGylated immunoliposomes against human breast cancer cellsPharm Res2007242402241117828616
- WeinerALLiposomes as carriers for polypeptidesAdv Drug Deliv Rev19893307341
- ZhaoPQWangHJYuMPaclitaxel-loaded, folic-acid-targeted and TAT-peptide-conjugated polymeric liposomes: in vitro and in vivo evaluationPharm Res2010271914192620582454
- MengSYSuBLiWIntegrin-targeted paclitaxel nanoliposomes for tumor therapyMed Oncol2011281180118720645030
- SharmaAMayhewEStraubingerRMAntitumor effect of Taxol-containing liposomes in a Taxol-resistant murine tumor modelCancer Res199353587758817903197
- BalasubramanianSVStraubingerRMTaxol-lipid interactions: Taxol-dependent effects on the physical properties of model membranesBiochemistry199433894189477913831
- SharmaASharmaUSStraubingerRMPaclitaxel-liposomes for intracavitary therapy of intraperitoneal P388 leukemiaCancer Lett19961072652728947523
- LinYBXieRLinYZTherapeutic effect of paclitaxel liposome combined with cisplatin for III–IV degrees of cervical cancerChin J Woman Child Health Res (Chinese)200819571573
- ImmordinoMLDosioFCattelLStealth liposomes: review of the basic science, rationale, and clinical applications, existing and potentialInt J Nanomedicine2006129731517717971
- TorchilinVPTrubetskoyVSWhich polymers can make nanoparticulate drug carriers long-circulating?Adv Drug Deliv Rev199516141155
- KlibanovALMaruyamaKTorchilinVPHuangLAmphipathic polyethylene glycols effectively prolong the circulation time of liposomesFEBS Lett19902682352372384160
- BlumeGCevcGMolecular mechanism of the lipid vesicle longevity in vivoBiochim Biophys Acta199311461571688452853
- SeniorJDelgadoCFisherDTilcockCGregoriadisGInfluence of surface hydrophilicity of liposomes on their interaction with plasma protein and clearance from the circulation: studies with poly (ethylene glycol)-coated vesiclesBiochim Biophys Acta1991106277821998713
- ParkJLiposome-based drug delivery in breast cancer treatmentBreast Cancer Res20024959912052251
- YangTCuiFDChoiMKEnhanced solubility and stability of PEGylated liposomal paclitaxel: in vitro and in vivo evaluationInt J Pharm200733831732617368984
- YoshizawaYKonoYOgawaraKPEG liposomalization of paclitaxel improved its in vivo disposition and anti-tumor efficacyInt J Pharm201141213214121507344
- CrosassoPCerutiMBrusaPArpiccoSDosioFCattelLPreparation, characterization and properties of sterically stabilized paclitaxel-containing liposomesJ Control Release200063193010640577
- HuangYChenXMXiangBAntiangiogenic activity of sterically stabilized liposomes containing paclitaxel (SSL-PTX): in vitro and in vivoAAPS Pharm Sci Tech201011752759
- Penault-LlorcaFBertucciFAdélaïdeJExpression of FGF and FGF receptor genes in human breast cancerInt J Cancer1995611701767705943
- DanielsenTRofstadEKVEGF, bFGF and EGF in the angiogenesis of human melanoma xenograftsInt J Cancer1998768368419626350
- Kwabi-AddoBOzenMIttmannMThe role of fibroblast growth factors and their receptors in prostate cancerEndocr Relat Cancer20041170972415613447
- HeQWeiYTianLInhibition of tumor growth with a vaccine based on xenogeneic homologous fibroblast growth factor receptor-1 in miceJ Biol Chem2003278218312183612651849
- ChenXWangXWangYImproved tumor-targeting drug delivery and therapeutic efficacy by cationic liposome modified with truncated bFGF peptideJ Control Release2010145172520307599
- CaiLQiuNLiXA novel truncated basic fibroblast growth factor fragment-conjugated poly (ethylene glycol)-cholesterol amphiphilic polymeric drug delivery system for targeting to the FGFR- overexpressing tumor cellsInt J Pharm201140817318221277964
- SharmaAMayhewEBolcsakLActivity of paclitaxel liposome formulations against human ovarian tumor xenograftsInt J Cancer1997711031079096672
- SavvaMDudaEHuangLA genetically modified recombinant tumor necrosis factor-α conjugated to the distal terminals of liposomal surface grafted polyethyleneglycol chainsInt J Pharm1999184455110425350
- ReddWHMontgomeryGHDuHamelKNBehavioral intervention for cancer treatment side effectsJ Natl Cancer Inst20019381082311390531
- CheongIHuangXBettegowdaCA bacterial protein enhances the release and efficacy of liposomal cancer drugsScience20063141308131117124324
- Lotfi-JamKCareyMJeffordMSchofieldPCharlesonCArandaSNonpharmacologic strategies for managing common chemotherapy adverse effects: a systematic reviewJ Clin Oncol2008265618562918981466
- DharapSWangYChandnaPTumor-specific targeting of an anticancer drug delivery system by LHRH peptideProc Natl Acad Sci U S A2005102129621296716123131
- MoghimiSMHunterACMurrayJCNanomedicine: current status and future prospectsFASEB J20051931133015746175
- SuoTGeWZhangJZ17 cases of advanced non-small cell lung cancer treated with paclitaxel liposome plus nedaplatinChinese- German Journal of Clinical Oncology201211196198
- TorchilinVPLevchenkoTSRammohanRPapahadjopoulos-SternbergBD’SouzaGGCell transfection in vitro and in vivo with nontoxic TAT peptide-liposome-DNA complexesProc Natl Acad Sci U S A20031001972197712571356
- YuWPirolloKFYuBEnhanced transfection efficiency of a systemically delivered tumor-targeting immunolipoplex by inclusion of a pH-sensitive histidylated oligolysine peptideNucleic Acids Res2004325e4815026537
- KawakamiKKawakamiMPuriRKIL-13 receptor-targeted cytotoxin cancer therapy leads to complete eradication of tumors with the aid of phagocytic cells in nude mice model of human cancerJ Immunol20021697119712612471149
- NakaseMInuiMOkumuraKKameiTNakamuraSTagawaTp53 gene therapy of human osteosarcoma using a transferrin-modified cationic liposomeMol Cancer Ther2005462563115827336
- MoghimiSMHunterACMurrayJCLong-circulating and target-specific nanoparticles: theory to practicePharmacol Rev20015328331811356986
- SeniorJGregoriadisGIs half-life of circulating liposomes determined by changes in their permeability?FEBS Lett19821451091146897042
- VolanakisJEWirtzKWAInteraction of C-reactive protein with artificial phosphatidylcholine bilayersNature1979281155157471064
- ChonnACullisPDevineDThe role of surface charge in the activation of the classical and alternative pathways of complement by liposomesJ Immunol1991146423442412040798
- DevineDVBradleyAJThe complement system in liposome clearance: can complement deposition be inhibited?Adv Drug Deliv Rev199832192910837633
- LundbergBRisovicVRamaswamyMWasanKA lipophilic paclitaxel derivative incorporated in a lipid emulsion for parenteral administrationJ Control Release2003869310012490375
- GabizonAHorowitzATGorenDTargeting folate receptor with folate linked to extremities of poly (ethylene glycol)-grafted liposomes: in vitro studiesBioconjug Chem19991028929810077479
- LeeRJLowPSFolate-mediated tumor cell targeting of liposome- entrapped doxorubicin in vitroBiochim Biophys Acta199512331341447865538
- GustafsonDLMerzALLongMEPharmacokinetics of combined doxorubicin and paclitaxel in miceCancer Lett200522016116915766591