Abstract
Raloxifene hydrochloride (RLX) is a selective estrogen-receptor modulator for treatment of osteoporosis and prevention of breast and endometrial cancer. By virtue of extensive presystemic clearance, RLX bioavailability is only 2%. The current study aimed to tailor and characterize RLX-loaded self-nanoemulsifying drug-delivery systems (SNEDDS) using bioactive excipients affecting drug metabolism. The potential of oral nanocarriers to enhance RLX delivery to endocrine target organs was assessed in fasted and fed female Wistar rats using high-performance liquid chromatography. RLX was loaded in the dissolved and dispersed status in the alkalinized (A-SNEDDS) and nonalkalinized (NA-SNEDDS) systems, respectively. Optimization and assessment relied on solubility studies, emulsification efficiency, phase diagrams, dilution robustness, cloud point, particle size, zeta potential (ZP), polydispersity index (PDI), and transmission electron microscopy. In vitro release was assessed using dialysis bag versus dissolution cup methods. NA-SNEDDS were developed with suitable globule size (38.49 ± 4.30 nm), ZP (31.70 ± 3.58 mV), PDI (0.31 ± 0.02), and cloud point (85°C). A-SNEDDS exhibited good globule size (35 ± 2.80 nm), adequate PDI (0.28 ± 0.06), and lower ZP magnitude (−21.20 ± 3.46 mV). Transmission electron microscopy revealed spherical globules and contended data of size analysis. Release studies demonstrated a nonsignificant enhancement of RLX release from NA-SNEDDS compared to drug suspension with the lowest release shown by A-SNEDDS. A conflicting result was elucidated from in vivo trial. A significant enhancement in RLX uptake by endocrine organs was observed after nanocarrier administration compared to RLX suspension. In vivo studies reflected a poor in vitro/in vivo correlation, recommended nanocarrier administration before meals, and did not reveal any advantage for drug loading in the solubilized form (A-SNEDDS). To conclude, NA-SNEDDS possessed superior in vitro characteristics to A-SNEDDS, with equal in vivo potential. NA-SNEDDS elaborated in this work could successfully double RLX delivery to endocrine target organs, with promising consequences of lower dose and side effects of the drug.
Introduction
Both osteoporosis and breast cancer are important health issues for postmenopausal women. It is well known that estrogen and estrogen receptors play an important role in the pathogenesis of both diseases. The key factor of the menopause is estrogen deficiency, which is the main cause of postmenopausal osteoporosis.Citation1 In past decades, hormone therapy, estrogen therapy, or a combination of estrogen and progesterone has been frequently used for treatment. However, there is an association between the risk of breast cancer and persistently elevated blood levels of estrogen.Citation2 Recently, selective estrogen-receptor modulators (SERMs) have represented a major therapeutic advance for clinical practice. SERMs work by virtue of their interaction with the estrogen receptors. Tamoxifen, a SERM, emerged as the first antiestrogenic agent that is clinically applicable to breast cancer. However, the long-term use of tamoxifen is associated with high risk of endometrial cancer, as it is an agonist on the endocrine system.Citation3
Raloxifene hydrochloride (RLX) is a second-generation SERM approved by the FDA in 1997 for the treatment of osteoporosis. It is commercially available under the trade name of Evista (Eli Lilly, Indianapolis, IN) in 60-mg dose tablets. RLX is a bone and liver estrogen agonist, which increases bone mineral density and decreases low-density-lipoprotein cholesterol. In addition, RLX has been found to be a breast and uterus estrogen-receptor antagonist, and thereby may decrease the risk of invasive breast and endometrial cancer.Citation4 Studies have demonstrated the superiority of RLX to tamoxifen, based on equal efficacy for prevention of breast cancer with fewer serious adverse events.Citation1 Studies have also reported equal efficacy of RLX with alendronate in preventing osteoporosis-related fractures with better safety profile (lower side effects).Citation1
Although RLX is characterized by high permeability,Citation5 the drug bioavailability is only 2%. RLX pertains to class II of the Biopharmaceutics Drug Disposition Classification System (BDDCS), where the drug is characterized by high permeability, poor solubility, and high metabolism. Poor bioavailability of such drugs is not only attributed to low solubility and dissolution but also to high presystemic clearance. RLX is a dual substrate for Uridine 5′-diphospho-glucuronosyltransferases (UGTs) and permeability glycoprotein (P-gp) in the intestine. Furthermore, the drug is subjected to extensive phase II metabolism (extensive first-pass effect) in the liver. RLX glucuronides are about 100-fold weaker in potency than RLX. According to the BDDCS, enhancing bioavailability of RLX could be obtained by inhibiting the efflux transporters responsible for the excretion of RLX conjugates, inhibiting metabolism.Citation6 In addition, bypassing the portal circulation would avoid further metabolism in liver and enterohepatic circulation. Inhibition of RLX presystemic clearance would improve the transport of the intact compound to the target organs, enhancing oral bioavailability.Citation5–Citation8 Biodistribution of RLX has been previously investigated to designate target organs with the highest uptake of the drug. Among different target organs, the endocrine system – mainly uterus – exhibited the highest RLX uptake.Citation7
So far, formulation attempts to improve RLX characteristics and oral bioavailability have focused only on solubility or permeability enhancement of the drug, regardless of drug metabolism. Trials encompassed use of hydrophilic binders,Citation9 cogrinding technique,Citation10 cyclodextrin complexation,Citation8 and use of mucoadhesive microspheres of the complex to give a controlled release pattern.Citation11 Although the reported permeability of RLX as class II drug is high, one attempt has adopted enhancing in vitro intestinal permeability of drug via microemulsion technique and a water titration method.Citation12 Nevertheless, the authors recommended that their in vitro assumptions could only be confirmed after in vivo animal experimentation, which they did not perform.Citation12
Other investigational attempts have been adopted for metabolic inhibition of RLX. A study based on flavonoid inhibitory effect on phase II metabolic enzymes was performed. Coadministration of apigenin flavonoid with RLX could increase the bioavailability of RLX. Due to the absorption and disposition of flavonoids being similar to that of RLX in enterocytes, apigenin competitively inhibited the formation of RLX glucuronide and RLX sulfate in the gut; therefore, apigenin could improve the absorption fraction of intact RLX from intestine during transport across mono-layer enterocyte.Citation13 Metabolic inhibition and kinetic RLX by bioactive excipients have been investigated by Kim et al.Citation14 Their study aimed at modulating poorly bioavailable drugs via decreasing the first-pass metabolism in gastrointestinal tract and liver and inhibition of P-gp efflux transporters. Nonionic surfactants such as Cremophor EL and Tween 80 have demonstrated significant inhibition of RLX metabolism with different mechanisms of action.Citation14 Nevertheless, such bioactive excipients have not been so far employed to formulate a bioactive delivery system for RLX hydrochloride.
Self-nanoemulsifying drug delivery systems (SNEDDS) are lipid-based nanocarriers that have recently exhibited an intriguing role in oral delivery of drugs. SNEDDS are nanoemulsion liquid preconcentrates composed of anhydrous isotropic mixtures of oil, surfactant(s), and co-surfactant(s). When introduced into the aqueous phase, SNEDDS spontaneously form oil-in-water nanoemulsions by simple peristaltic movement. Among lipid-based delivery systems, SNEDDS are superior with regard to excellent thermodynamic and shelf stability, high solvent capacity, possibility for large-scale production, and amenability to be filled in soft or hard gelatin capsules.Citation15,Citation16 SNEDDS have been extensively used to enhance oral bioavailability of drugs, particularly those pertaining to class II and IV of the BDDCS.Citation5 Mechanisms of enhancement encompass improved solubility, changing intestinal permeability, and interfering with enzymes and transporter activity via bioactive lipid excipients and surfactants. Furthermore, bypassing hepatic first-pass metabolism was also reported as a result of oral lymphatic targeting of drugs.Citation16–Citation28
By virtue of poor solubility and extensive presystemic clearance of RLX parallel to reported advantages of SNEDDS in this context, the current study endeavored to optimize RLX-loaded SNEDDS utilizing bioactive excipients of reported inhibitory effect on RLX metabolism. Full in vitro characterization was carried out for the optimized nanocarrier. The in vivo potential of the optimized SNEDDS to enhance RLX delivery to its endocrine target organs in female Wistar rats was assessed. The influence of different factors on RLX uptake by endocrine organs (uterus, ovaries, and fallopian tubes) was investigated. Factors encompassed fed state versus fasted state of animals, nanoformulation versus conventional RLX suspension, and simple SNEDD formulation versus triethanolamine-modified nanocarriers.
Materials and methods
Materials
RLX was obtained from Hetero Drugs, (Hyderabad, India). Caproyl 90 (propylene glycol monocaprylate), Plurol Oleique (ployglyceryl-6 diolate), Labrafac PG (propylene glycol dicaprylocaprate); Peceol (glyceryl monooleate), Maisine 35-1 (glyceryl monolinoleate), Labrasol (polyethylene glycol- 8 caprylic/capric glycerides), Lauroglycol FCC (propylene glycol monolaurate), and Transcutol HP (diethylene glycol monoethyl ether) were kind gifts from Gattefosse (Lyon, France). Cremophor RH40 (polyoxyl 40 hydrogenated castor oil) and Cremophor EL (polyoxyl 35 castor oil) were obtained from BASF (Ludwigshafen, Germany). Propylene glycol and Tween 80 were obtained from El-Nasr Pharmaceutical (Cairo, Egypt). Thiopental sodium A solution (50 mg/mL) was prepared for rat anesthesia by dissolving thiopental sodium lyophilized powder (Biochemie, Vienna, Austria) in saline. All other chemicals and reagents used were of analytical grade.
Preliminary investigation
Solubility studies
The saturation solubility of RLX in different oily phases, surfactants, and cosurfactants under investigation was assessed. An excess drug amount was added into each vehicle followed by vortex mixing for 30 seconds. Mixtures were shaken for 24 hours at 30°C in a thermostatically controlled shaking water bath (type 3047; Kottermann, Hanigsen, Germany), followed by equilibrium for 24 hours. Mixtures were then filtered through a Millipore (Billerica, MA) membrane filter (0.45 μ). Filtrates were suitably diluted with methanol and quantified spectrophotometrically (UV-160A; Shimadzu, Kyoto, Japan) for dissolved RLX at 287 nm.Citation15 For RLX solubility in each ingredient, ultraviolet measurement was carried out against the corresponding placebo additive in methanol. The experiment was repeated in triplicate. Results were represented as mean value (mg/ml) ± standard error of mean.
Surfactant screening
Emulsification efficiency of various nonionic surfactants under study was assessed based on percentage transmittance and number of flask inversions.Citation27,Citation29,Citation30 Briefly, 300 mg of the surfactant (Labrasol, Cremophor RH40, Cremophor EL, and Tween 80) were added to 300 mg of the selected oily phase. The mixtures were gently heated at 50°C for homogenization of the components. Each mixture, 50 mg, was then diluted with distilled water to 50 mL in a Stoppard conical flask. Ease of emulsification was judged by the number of flask inversions (time of emulsification) required to yield homogeneous emulsion. Emulsions were allowed to stand for 2 hours and their percentage transmittance was evaluated at 638.2 nm using distilled water as a blank. Dispersions formed were visually observed for any turbidity or phase separation.
Cosurfactant screening
Emulsification efficiency of cosurfactant of highest drug solubility was assessed using the oily phase(s) and surfactant(s) selected from preliminary screening. Surfactant mixture (2:1 surfactant to co-surfactant) and oily phase were blended in a ratio of 1:1. Blend preparation and assessment followed a similar pattern to that described in the surfactant screening section.
Ternary phase diagrams
Ternary phase diagrams representing oil:surfactant: cosurfactant ingredients of the selected SNEDD systems were constructed. The concentration of oil and surfactant varied from 30% to 70%, while that of cosurfactant varied from 0% to 30%. For any mixture, the total of component concentrations always added to 100%.Citation25 The oily phase was mixed with the surfactant/co-surfactant mixture and the dispersion was homogenized in a shaking water bath at 50°C for 10 minutes. From each mixture, 50 mg were diluted to 50 mL with distilled water. Only clear or slight bluish dispersions of globular size 200 nm or lower were considered in the nanoemulsion region of the diagram.Citation15,Citation27
Preparation of RLX-loaded SNEDDS
Preparation of nonalkalinized nanocarriers (NA-SNEDDS)
RLX was added to the selected formulations based on the aforementioned optimization steps. The drug was added in incremental amounts (2, 5, 10, 20, 30, 40, 50, 60 mg/g), and dispersion was gently shaken at 60°C for 30 minutes. Shaking was aided by sonication and vortex mixing. The prepared liquid preconcentrates were visually assessed for any physical changes (precipitation or phase separation) within 24 hours of preparation.Citation28,Citation31
Preparation of alkalinized nanocarriers (A-SNEDDS)
RLX in incremental amounts (2, 5, 10, 20, 30, 40, 50, 60 mg/g) was added to SNEDDS alkalinized with triethanolamine (TEA) (10% weight/weight [w/w]). Dispersion was gently shaken at 60°C for 30 minutes. Shaking was aided by sonication and vortex mixing till clear dispersion was obtained. The prepared liquid preconcentrates were visually assessed for any physical changes within 24 hours after preparation.
Influence of system alkalinization on chemical stability
The influence of TEA incorporation on RLX chemical stability in both concentrated and diluted forms of nanocarriers was investigated using pharmacopeial high-performance liquid chromatography (HPLC).Citation32 Regarding the concentrated form, preweighed alkalinized and nonalkalinized SNEDDS (2% w/w) were suitably diluted with the mobile phase and measured immediately and after 2 weeks of preparation. For the diluted form, alkalinized and nonalkalinized preconcentrates (2% w/w) were diluted with distilled water (1:500). The resultant nanoemuslions were analyzed immediately and after 6 hours of preparation. All samples were filtered through a Millipore membrane filter (0.45 μ) before being injected into the HPLC system. The pharmacopeial method of RLX separation was utilized using HPLC (series 200; PerkinElmer, Norwalk, CT) equipped with C8 column (4.6 mm × 15 cm, particle size = 3.5 μm).Citation32 The mobile phase consisted of a mixture of phosphate buffer (pH 2.5 ± 0.1) and acetonitrile (67:33). The mobile phase was delivered isocratically with a flow rate of 1.5 mL/minute. Chromatograms were monitored by a UV-detector set at 280 nm. The system was also equipped with a chromatography interface 600 series link, operated by TotalChrom chromatography data system software version 6.2 (PerkinElmer). The calibration curve of peak area against concentration was y = 3271.366 + 177646.3x, at a concentration range of 0.25–10 mg%. Retention time was 5.4 ± 0.3 minutes (r2 = 0.999, recovery = 98.02%).
Characterization of RLX-loaded nanocarriers
Physical robustness to dilution
Robustness of the selected formulations to dilution was assessed in the presence and absence of TEA (10%). Formulation was exposed to different folds (100, 500, and 1000) and media (distilled water, 0.1 N HCl, and phosphate buffer pH 7.4) of dilution. Percentage transmittance was then measured spectrophotometrically at 638.2 nm. The diluted nanoemulsions were stored for 6 hours and monitored for any physical changes (such as precipitation or phase separation).Citation15,Citation27
Cloud-point measurement
Selected formulations (both alkalinized and nonalkalinized) were assessed for cloud-point value. Formulations were diluted with distilled water (1:100) and placed in a water bath with gradual increase in temperature. At the cloud point, the drop in sample percentage transmittance from the zero point was measured spectrophotometrically at 638.2 nm.Citation27,Citation31
Emulsion droplet size and ζ-potential measurements
Selected formulations (alkalinized, nonalkalinized and placebo SNEDDS) were subjected to droplet size and ζ-potential (ZP) analysis by photon correlation spectroscopy using a Malvern Zetasizer Nano ZS (Malvern Instruments, Malvern, UK) with a dynamic light-scattering particle-size analyzer at a wavelength of 633 nm. The values of Z-average diameters were used. SNEDD formulations were diluted with distilled water (1:500) according to the method reported by Jeevana and SreelakshmiCitation33 and Shanmugam et al.Citation34 Samples for droplet- size measurements were placed in square glass cuvettes, while those for ZP were placed in clear disposable ζ-cells. Measurements were repeated in triplicate, each carried out for 13 times with good agreement being found between them.
Transmission electron microscopy
Transmission electron microscopy (TEM) was employed as a means for morphological examination and globular-size confirmation. Prior to the analysis, the SNEDDS sample was diluted in distilled water (1:1000), and a sample drop was placed on a copper grid. The excess was drawn off with filter paper. Samples were subsequently stained with saturated solution of uranyl acetate for 30 seconds.Citation22,Citation25
Drug release studies
Dialysis bag method
Drug release from the alkalinized and nonalkalinized preconcentrates compared to drug suspensions was assessed using the dialysis bag method (Visking 36/32, 28 mm, MWCO 12,000–14,000; Serva, Heidelberg, Germany).Citation15,Citation27,Citation28 In all release experiments, the dialysis bag was fixed to a 250-mL glass beaker containing 150 mL of 0.1% (w/v) Tween 80 in distilled water as a release medium.Citation32 The experiment was carried out in a thermostated shaking water bath at 37°C and 100 rpm. SNEDD formulations were dispersed in distilled water (5 mL) to yield drug strength of 2% (weight/volume [w/v]). Drug suspension in distilled water (2% w/v) was added to the dialysis bag in 5 mL volume to yield the same drug strength as in nanocarriers. Selected alkalinized and nonalkalinized formulations were added to the bags to omit a concentration gradient effect. Samples were taken at intervals of 0.1, 0.3, 0.5, 0.75, 1, 1.5, 2, 18, 20, and 24 hours. All samples were measured spectrophotometrically at 287 nm against 0.1% (w/v) Tween 80 in distilled water as blank.
Dissolution cup method
Another method utilized for assessment of RLX release was the dissolution cup method,Citation32 which was applied with some modifications. RLX release was assessed using a US Pharmacopeia dissolution tester, apparatus II at 37°C ± 0.5°C, and at a rotating speed of 50 rpm in 250 mL of 0.1% Tween 80 in distilled water.Citation32 Alkalinized and nonalkalinized preconcentrates (1 g) containing the same RLX concentration (2% w/w) were added into the dissolution cup. RLX suspension in water (0.4% w/v) was added into the dissolution cup in 5 mL volume to contain the same drug amount as nanocarriers (20 mg). At selected time intervals (10, 20, 30, 45, 60, 90, and 120 minutes), aliquots (5 mL) were withdrawn from the dissolution medium, filtered through a Millipore filter (0.45 μ) and replaced with an equivalent amount of the fresh dissolution medium. Concentrations of RLX were determined spectrophotometrically at 287 nm.Citation25
In vivo study
Animals and experimental protocol
In the current study, a novel method for in-vivo assessment of RLX-SNEDDS was introduced based on the reported mechanism of RLX action parallel to reported characteristics of SNEDDS and bioactive excipients to enhance bioavailability of similar drugs.Citation35–Citation42 In this novel method, the potential of the elaborated nanocarriers to increase RLX delivery to endocrine target organs (uterus, ovaries and fallopian tubes) was assessed in female Wistar rats weighing 190 ± 20 g. Rats were obtained from Vacsera (Cairo, Egypt). Experiments were performed in accordance with the European Community guidelines for the use of experimental animals and were approved by the institutional ethics committee. Rats were fed on a diet consisting of normal standard chow. Water was allowed ad libitum. Animals were housed in a room with controlled temperature and humidity.
Rats were divided into two main groups (n = 16 per group): fasted and fed state. In the first group, rats were fasted for 24 hours prior to experiment. The second group was fed on a high-fat meal of bread soaked in nutritional milk formula and cheese. Each main group was subdivided into four groups (n = 4) receiving placebo SNEDDS as a control, RLX aqueous suspension (2 mg/mL), NA-SNEDDS (2 mg/mL), and A-SNEDDS (2 mg/mL). Each group was administered a drug dose of 30 mg/kg.Citation41 RLX aqueous suspension and RLX-loaded SNEDDS (alkalinized or nonalkalinized) were accurately weighed (according to rat weight) and separately dispersed into distilled water (3 mL) by mixing homogeneously for 30 seconds prior to dosing. Each formulation was administered to rats by oral gavage using an animal-feeding needle.
Histological identification of the estrus-cycle phases
The criteria of animal selection depend on the rat estrous cycle regarding RLX role as SERM in postmenopausal conditions. Rats in estrus and proestrus phase were excluded. To identify the estrus-cycle phase, a sample of the vaginal fluid was collected by inserting the tip of a plastic pipette filled with 100 μL NaCl (0.9%) into the vaginal cavity. The vaginal fluid was placed on a glass slide and examined under a light microscope, with 10× and 40× objective lenses. The 40× objective lens helped characterize the cell type, whereas the 10× lens determined the proportions of individual cell types.Citation43
Sample preparation and extraction procedure
After oral administration of 30.0 mg/kg water solution of RLX in different formulations (suspension, alkalinized and non alkalinized) and under thiopental anesthesia, endocrine system samples (ovaries, fallopian tubes, and uterus) were isolated at 30 minutes.Citation35 Tissue samples were put into normal saline solution to remove the blood content, blotted on filter paper, and then weighed for wet weight. Samples were then homogenized using a Brinkmann homogenizer (Polytron; Kinematica, Kriens, Switzerland) at variable speed in saline solution at room temperature. The obtained tissue homogenates were stored at −20°C until analysis. Prior to HPLC analysis, 100 μL of methanol and 400 μL of acetonitrile were added to 200 μL of the tissue homogenates for protein precipitation. After centrifugation at 4000 rpm for 15 minutes at room temperature, 10 μL of the supernatant was injected into the HPLC system for analysis.Citation35
HPLC analysis
The aforementioned pharmacopeial method of RLX separation in the chemical stability session was utilized.Citation32,Citation35 In order to be applied to biological samples, RLX standards in blank tissue were prepared. Standard solutions at a concentration range of 5–70 μg/mL were prepared by adding 5 mL of the standard solutions to 400 μL of blank tissue homogenates. The calibration curve of peak area against RLX concentration in tissue homogenate was y = 11938 + 161862x, at a concentration range of 5–70 μg/mL. Retention time was 2.9 ± 0.19 minutes (r2 = 0.999, % recovery = 98.09%).
Statistical analysis
Statistical analysis of the results was carried out using Student’s t-test with the level of significance set at P < 0.05 (GraphPad Prism version 3.02; GraphPad Software, San Diego, CA).
Results and discussion
Preliminary studies
Solubility and emulsification efficiency studies
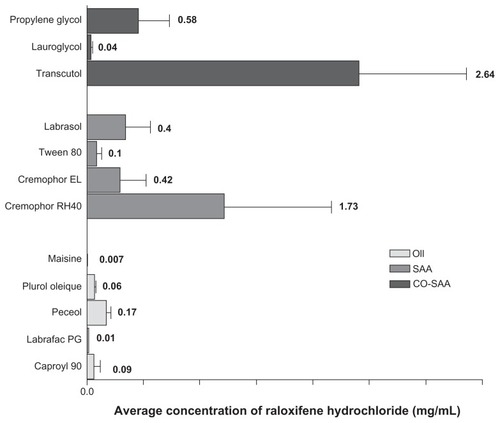
Self-emulsifying formulations consisting of oils, surfactants, cosurfactants, and drug should spontaneously form a clear and monophasic liquid at ambient temperature when introduced into aqueous phases.Citation25 For each drug, SNEDDS formulation should be “tailored” according to certain criteria, including drug solubility, emulsification efficiency, and bioactive effect. Oily phase is the most important component of SNEDDS formulations, as it can solubilize lipophilic drugs and increase the fraction of lipophilic drug transported via the intestinal lymphatic system, thereby increasing the absorption from the gastrointestinal tract, depending on the molecular nature of the triglycerides. Solubilizing capacity of an oily phase is crucial for development of SNEDDS, as it determines the drug-loading efficiency and soluble portion of drug during storage and in vivo dilution.Citation23 In the current study, solubility of RLX in five different oily phases was assessed. The oils were selected to possess various chemical composition and hydrophilic–lipophilic balance (HLB) characteristics. Oily phases screened included Caproyl 90 (propylene glycol monocaprylate, HLB = 6), Plurol Oleique (polyglyceryl-6 diolate, HLB = 6), Labrafac PG (propylene glycol dicaprylocaprate, HLB = 2), Peceol (glyceryl monooleate, HLB = 3), and Maisine 35–1 (glyceryl monolinoleate, HLB = 4). Results of solubility studies in oily phases are depicted in . Among all oils screened, the highest solubilization capacity was exhibited by Peceol (0.17 ± 0.089 mg/mL), followed by Caproyl 90 (0.09 ± 0.0065 mg/mL), with the lowest solubility obtained in Maisine (0.007 ± 0.0005 mg/mL). Peceol and Caproyl 90 were therefore selected for further investigation, while final selection would rely on emulsification properties with surfactant mixtures.
Figure 1 Solubility of raloxifene hydrochloride in various oil and surfactant mixtures.
Note: Data expressed as mean ± standard error of mean (n = 3).
Regarding surfactant and cosurfactant selection, drug solubility would come second to the main selection perspectives – emulsification efficiency and bioactivity.Citation15 Nonionic surfactants are usually utilized for fabrication of SNEDDS, as they are less toxic compared to their ionic counterparts.Citation23 Another important criterion in surfactant selection is bioactivity aspect. Some surfactants were categorized as “generally recognized as safe” bioactive excipients due to lymphotropic character (Tween 80) or inhibitory effects on P-gp and metabolic enzymes (Cremophor RH40 and Cremophor EL).Citation36 In this context, a competitive-type inhibition between Cremophor EL and RLX was recognized at the level of the active site of the UGT enzyme, therefore, Cremophor EL competitively inhibited formation of RLX metabolites. In addition, Tween 80 was reported to inhibit RLX metabolism via a mixed-competition mechanism.Citation14
In the current investigation, four different nonionic surfactants with reported bioactive effects (Tween 80, Labrasol, Cremophor EL, and Cremophor RH40) were screened for their emulsification efficiencies using different oily phases. It has been reported that well-formulated SNEDDS are dispersed within seconds under gentle stirring conditions.Citation25 High emulsification efficiency is therefore judged by a small number of flask inversions (lower than ten) in addition to high-percentage transmittance (higher than 90%).Citation15,Citation27,Citation37 One flask inversion requires only 1 second. Transmittance values of different mixtures are demonstrated in . Results indicated that both Cremophor RH40 and Cremophor EL exhibited the highest emulsification efficiency among all surfactants screened. The oily phase Caproyl 90 exhibited the highest emulsification efficiency with all surfactants employed. On the other hand, Peceol showed poor emulsification properties with all the surfactants screened, requiring a minimum of 40 flask inversions (40 seconds) in Cremophor RH40 emulsion (73% transmittance). The aforementioned results suggested two combinations: Caproyl 90/Cremophor RH40 and Caproyl 90/Cremophor EL for cosurfactant addition.
Table 1 Emulsification efficiency of various surfactants using different oily phases (1:1)
It was reported that amphiphilic cosurfactants are often used in the SNEDDS to improve drug loading, decrease time required for self-nanoemulsification, and impart thermodynamic stability to the system.Citation16 In the current investigation, three different cosurfactants, namely propylene glycol, Lauroglycol FCC, and Transcutol, were assessed for their solubilizing capacity. Results of solubility studies in cosurfactants are depicted in . The figure demonstrates that Transcutol provided the highest solubilizing capacity of RLX. Transcutol exhibited good emulsification efficiency (98% transmittance) with Caproyl 90 and Cremophor EL (oil:SAA:Co-SAA, 3:2:1) with only one flask inversion. Transcutol also exhibited a similar emulsification efficiency with Caproyl 90 and Cremophor RH40 (99% transmittance) with only two flask inversions.
Qualitative preliminary screening has so far suggested two different systems: system A (Caproyl 90/Cremophor RH40/Transcutol) and system B (Caproyl 90/Cremophor EL/Transcutol). The next step in optimization is a quantitative one pending phase diagram studies. Both systems consisted of liquid dispersions of viscous consistency.
Phase diagram study
Construction of phase diagrams provides a picture of probable concentrations of components that might yield spontaneous emulsion and the nature of the resultant dispersions, such as separated dispersions, coarse emulsions, and self-nanoemulsifing systems. Phase diagram studies can hence assist in selection of optimum formulation components and ratios.Citation16 Based on the preliminary investigations, ternary phase diagrams of the selected systems A (Caproyl 90/Cremophor RH40/Transcutol) and B (Caproyl 90/Cremophor EL/Transcutol) were constructed. For each system, 14 mixtures were prepared, as depicted in (system A) and (system B). Constructed phase diagrams were shown in and . Only clear or slight bluish dispersions of particle size 100 nm or lower are considered in the nanoemulsion region of the diagrams. The shaded region indicates the nanoemulsion region. The wider this region is, the better the self-nanoemulsifying ability.Citation15,Citation38 As demonstrated in and , system A could be self-nanoemulsified up to 60% oil load compared to 50% in system B.
Table 2 Composition of self-nanoemulsifying nanocarriers constructing phase diagram A (Caproyl 90, Cremophor RH40, and Transcutol)
Table 3 Composition of self-nanoemulsifying nanocarriers constructing phase diagram B (Caproyl 90, Cremophor EL and Transcutol)
Figure 2 Ternary phase diagram of system A (Caproyl 90/Cremophor RH40/Transcutol).
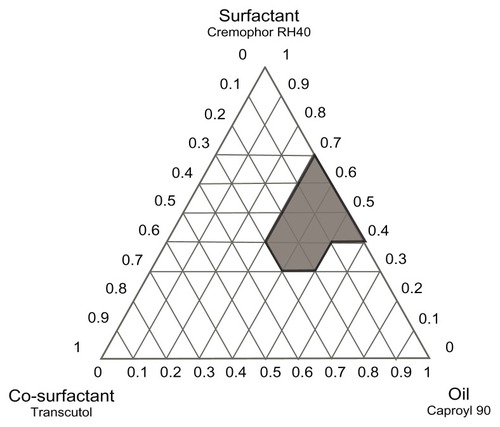
Figure 3 Ternary phase diagram of system B (Caproyl 90/Cremophor EL/Transcutol).
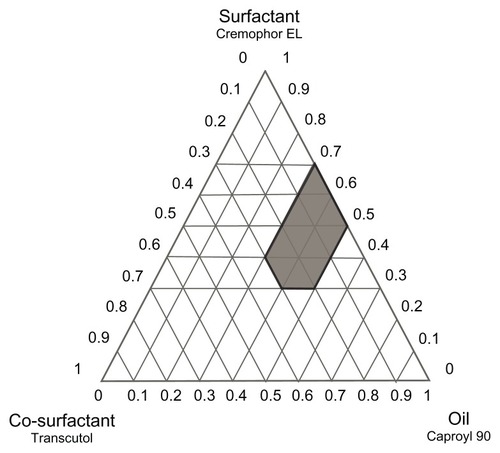
Results of preliminary investigation inferred an equal emulsification efficiency for both Cremophor RH40 and Cremophor EL, with higher drug solubilization in the former (). RLX solubility in Cremophor RH40 (1.73 mg/mL) was higher than in Cremophor EL (0.42 mg/mL). Furthermore, Cremophor RH40–based systems could self-emulsify a 10% higher oil content compared to the Cremophor EL–based system. Nevertheless, in view of low drug solubility in system ingredients (), such differences would not be expected to affect drug content significantly. In addition, extensive RLX metabolism is a crucial obstacle in its poor bioavailability, not only poor solubility.Citation12 Realizing reported inhibition of RLX metabolism by Cremophor ELCitation14 system B was selected for the next stage. P10 () formulation consisting of Caproyl 90:Cremophor EL:Transcutol in a ration of 5:4:1 was chosen for drug loading and characterization.
Preparation of drug loaded SNEDDS
Preparation of nonalkalinized SNEDDS (NA-SNEDDS)
Different concentrations of RLX were loaded to the selected SNEDD formulation (P10). Results demonstrated that formulations loaded with 2 mg/g and 5 mg/g of RLX were clear, while those with 10 mg/g and 20 mg/g RLX load showed good drug dispersibility in the system. On the other hand, formulations with RLX concentration higher than 20 mg/g exhibited poor dispersion and precipitation after 24 hours. These results came in accordance with summation of RLX solubility values in system ingredients. Formulation with drug load of 20 mg/g was selected for further investigation.
Preparation of alkalinized SNEDDS (A-SNEDDS)
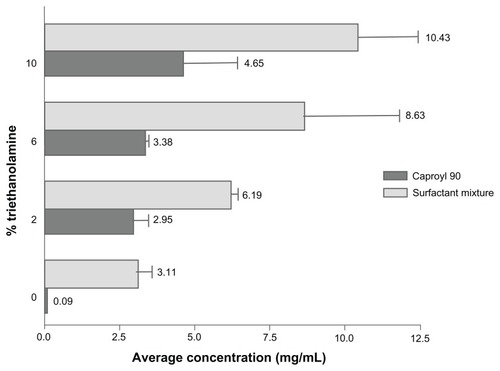
Results of solubility studies revealed poor RLX solubility in both water (0.25 ± 0.024 mg/mL) and oily phases screened (), constituting a challenge to drug-delivery formulation and drug loading. In an attempt to increase drug solubility and loading in SNEDDS ingredients, system alkalinization was performed by the addition of TEA. As demonstrated in , solubility in selected SNEDDS ingredients, represented as Caproyl 90 and surfactant mixtures (Cremophor EL/Transcutol), significantly increased with increasing TEA ratios. Furthermore, A-SNEDDS prepared with 10% TEA and different RLX concentrations exhibited more intense yellow color and better dispersions compared to NA-SNEDDS with the same RLX doses. A-SNEDDS prepared with RLX concentration up to 20 mg/g were clear. Lower drug dispersibility was demonstrated with higher RLX doses. Enhancement of drug solubility and system clarity by TEA modification might be ascribed to liberation of RLX base with high affinity to oily phase and surfactant mixture. It is noteworthy that the median lethal dose of TEA in humans is 5–15 g/kg, indicating the safety of the utilized doses of the excipient.Citation39 A maximum TEA concentration of 10% was selected in this study, as higher TEA concentration (20%) resulted in system precipitation after 24 hours, which could be attributed to surfactant instability. Clear alkalinized formulation with dissolved RLX (20 mg/g) was selected for comparison to NA-SNEDDS with dispersed drug loaded in the same concentration.
Figure 4 Solubility study of raloxifene in selected system ingredients with different triethanolamine ratios.
Note: Data expressed as mean value ± standard error of mean (n = 3).
Influence of system alkalinization on chemical stability
In order to detect any chemical degradation of RLX upon incorporation of TEA alkalinizer, RLX SNEDDS (2% w/w) was assessed in both concentrated and diluted forms using HPLC analysis. Fresh sample analysis showed no significant difference between nonalkalinized (2.05% w/w ± 0.045%) and alkalinized (2.03% w/w ± 0.01%) preconcentrates. The concentration remained unchanged after 2 weeks for both nonalkalinized (2.03% w/w ± 0.042%) and alkalinized (2.04% w/w ± 0.055%) formulations. No extra peaks were detected in the chromatogram of alkalinized nanocarriers in either case. These results confirmed the chemical stability of RLX in preconcentrate after TEA incorporation. On the other hand, the concentration of RLX in nanoemulsion dispersion after 6 hours remained significantly unchanged either before (1.9% ± 0.15% w/w) or after (1.84% ± 0.070%) addition of TEA relative to those at zero time both before (1.8 ± 0.07 % w/w) and after (1.72 ± 0.014%) addition of TEA. Results of HPLC analysis reflected good chemical compatibility of the system with the TEA alkalinizer. As a consequence, incorporation of TEA could be readily used for subsequent studies.
Characterization of selected nanocarriers
Physical robustness to dilution
Optimized NA-SNEDDS and A-SNEDDS with 20 mg/g drug and 10% TEA were exposed to different folds of dilution in different media and monitored for 6 hours. Percentage transmittance of the diluted nanoemulsions was measured spectrophotometrically at 638.2 nm. Results demonstrated that both NA-SNEDDS and A-SNEDDS () formulations were robust to dilution in different media and folds for 6 hours, with no signs of separation or precipitation. Lowest robustness of NA-SNEDDS to dilution was manifested in the most concentrated dispersion (100-fold) in phosphate buffer (82% T) rather than other media. This effect may be ascribed to an electrolyte effect on lyophobic colloidal dispersions rather than a dilution effect. On the other hand, A-SNEDDS exhibited lower robustness to dilution in both acidic (0.1 N HCl, 78%) and basic media (phosphate buffer, 79.7%) compared to the nonalkalinized form, which could be attributed to lower affinity of the liberated RLX base to aqueous media.
Table 4 Percentage transmittance of 2% RLX-loaded A-SNEDDS and NA-SNEDDS in different media with different folds of dilution at 638.2 nm
Cloud-point measurement
The cloud point is the temperature above which the formulation clarity turns into cloudiness. At the cloud point, dehydration of polyethylene oxide moiety of the nonionic surfactant will occur, resulting in phase separation and a drop in sample percentage transmittance. Since both drug solubilization and formulation stability will decline with this phase separation, the cloud point of the formulation should be higher enough than 37°C (body temperature).Citation27,Citation31 In this study, both nanocarrier formulations exhibited cloudiness at 85°C, with a drop in percentage transmittance from 95.5% ± 0.071% to 74.8% ± 2.899%. Results confirmed the stability of all SNEDDS formulations regarding separation at gastrointestinal tract temperature.Citation27,Citation31
Emulsion droplet size and ζ-potential measurements
The droplet size of SNEDDS is a crucial factor in selfemulsification performance because it determines the rate and extent of drug release, as well as the stability of the emulsion formed.Citation38 The polydispersity index (PDI) reflects the uniformity of particle diameter and can be used to depict the size distribution of the nanoemulsion population.Citation22 PDI varies from 0.0 to 1.0. The closer to zero the polydispersity value, the more homogeneous the particles are.Citation40 ZP measurement is used to identify the charge of the droplets. It has been suggested that ZP may serve as a partial indicator for the physical stability of the emulsion being formed. High absolute ZP values (±30 mV) should preferably be achieved in most of the emulsions prepared in order to ensure the creation of a high-energy barrier against coalescence of the dispersed droplets.Citation22 However, this suggested ZP cutoff point is only an experience-based value and cannot be reliably used to predict the stability of SNEDDS, because a wide range of absolute ZP values (ie, 1.5, 12.5, 45.5 mV) have been reported for SNEDDS in previous studies,Citation22,Citation26,Citation27,Citation37 most of which did not have any long-term stability assessment for verification purposes.
In the current study, globule size, ZP, and PDI of SNEDDS diluted in distilled water (500-fold) were measured by photon correlation spectroscopy using a Malvern Zetasizer ZS, and factors included drug addition to placebo formulation, system alkalinization, and drug dose. Selected samples encompassed placebo NA-SNEDDS, placebo A-SNEDDS, NA-SNEDDS (5 mg/g RLX load), A-SNEDDS (5 mg/g RLX load), NA-SNEDDS (20 mg/g RLX load), and A-SNEDDS (20 mg/g and 5 mg/g RLX). Results are depicted in . Results of globule-size analysis inferred that all formulations were in the nanometric range (less than 100 nm), with a maximum PDI of 0.3. The low negative value of ZP on the oil droplet of the nonalkalinized placebo formulation (−11.7 ± 1.25 mV) may be attributed to the presence of free fatty acids.Citation38 Upon loading with RLX (salt form), the positively ionized drug was anticipated to be adsorbed on the surface of the globules, neutralizing the free fatty acids and decreasing the negative charge. The higher the amount of drug loaded, the higher the magnitude of the positive charge obtained, as demonstrated from the higher ZP value of 20 mg/g drug load (31.7 ± 3.58 mV) compared to 5 mg/g drug load (6.76 ± 0.042 mV). Surface adsorption of RLX on NA-SNEDDS globules may be attributed to higher affinity of drug to surfactant than to oily phase, as inferred from the solubility studies.
Table 5 Particle size, zeta potential (ZP), polydispersity index (PDI), and pH of selected self-nanoemulsifying formulations
Regarding placebo A-SNEDDS, TEA is an amino alcohol anticipated to dissolve in oil globules. It is postulated to react with fatty acids leading to the formation of a surface active agent – TEA salt of fatty acids or ionizable fatty acid – which migrates to the globule surface, increasing the negative charge magnitude (−36.2 ± 2.79 mV). It may also lead to more stabilization (additional surfactant). Such negative charge is anticipated to be decreased upon RLX addition, where part of TEA will react with RLX salt, leading to the formation of RLX base and chloride salt of TEA, which is water-soluble salt lost after dilution. Such a decrease in negative charge upon drug loading is expected to increase with an increase in drug load, as demonstrated in the higher negative potential of 5 mg/g alkalinized formulation (−27.2 ± 1.56 mV) compared to 20 mg/g alkalinized formulation (−21.2 ± 3.46 mV). Additional decrease in the negative potential of 20 mg/g A-SNEDDS may be attributed to the remaining positively ionized drug, RLX, escaping from TEA alkalinization. Stability studies for the optimized formulations based on ZP and other parameters are currently under investigation.
Transmission electron microscopy
TEM of the selected samples (A-SNEDDS 20 mg/g; NA-SNEDDS 20 mg/g, and placebo SNEDDS) was performed for morphological examination and confirmation of particle-size analysis. TEM images of nanoformulations after dilution in distilled water (1:1000) are depicted in –. All figures reveal spherical nanoemulsion globules less than 100 nm in size, confirming the results obtained by the Malvern Zetasizer. Spherical, discrete, and nonaggregated globules in both nonalkalinized () and alkalinized () formulations inferred system stability in both alkalinized and nonalkalinized conditions. A thicker darker wall of nanoemulsion globules can be seen in the NA-SNEDDS image () compared to A-SNEDDS (), which may be ascribed to surface accumulation of RLX in the former. These results are in accordance with that obtained from ZP investigations. Nanocarriers developed showed drug molecules loaded inside the globules, as revealed in higher magnification images of drug-loaded A-SNEDDS () in comparison to faint surrounding membrane and empty core of its placebo formulation ().
Figure 5 Transmission electron microscopy photograph of nonalkalinized self-nanoemulsifying system with 1000-fold dilution in distilled water (×13,000).
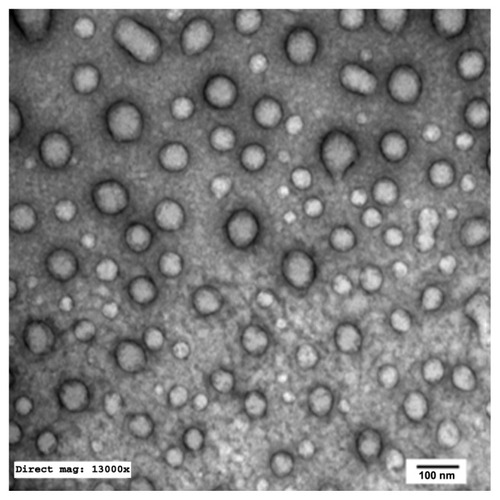
Figure 6 Transmission electron microscopy photograph of alkalinized self-nanoemulsifying system with 1000-folds dilution in distilled water (× 13,000).

Figure 7 Transmission electron microscopy photograph of drug-loaded alkalinized self-nanoemulsifying system with 1000-fold dilution in distilled water (×40,000).

Figure 8 Transmission electron microscopy photograph of placebo nanocarriers with 1000-fold dilution in distilled water (×40,000).

Drug-release studies
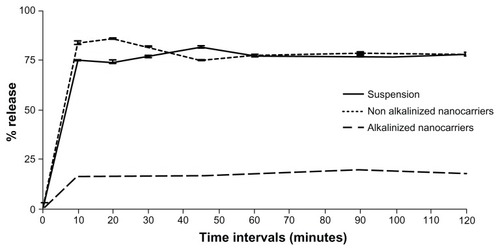
The in vitro release of RLX was assessed using two different techniques adopted for nanocarrier assessment: dialysis bag versus dissolution cup method. The former is usually performed for delivery systems where separation of system particles from drug molecules is necessary.Citation15,Citation27,Citation28 The latter is another attempt to circumvent problems of dialysis tubing, including slow initial release of drug from self-formulations, membrane saturation, and interaction.Citation15,Citation27,Citation28
Results of in vitro release studies by dialysis bag and dissolution cup method are depicted in and , respectively. In both methods, initial drug strength was unified among all formulations screened to omit the effect of concentration gradient. Both methods of release demonstrated a nonsignificant enhancement of RLX release from NA-SNEDDS compared to drug suspension. On the other hand, A-SNEDDS exhibited a significantly lower release profile compared to NA-SNEDDS and drug suspension. The in vitro release patterns of RLX from plain drug suspension and nonalkalinized preconcentrate revealed that almost 60% release could be attained after 10 minutes in the dissolution cup method in comparison to 4 hours in the dialysis bag method. Such a delay in dialysis method may be attributed to the presence of a diffusion membrane that constituted a hindrance against rapid drug release. An initial significant increase in RLX release was exhibited by NA-SNEDDS compared to suspension in dissolution cup but not in dialysis bag, which may be attributed to time required for self-emulsification. On the other hand, alkalinization of SNEDDS was anticipated to liberate RLX base. The latter would possess poor solubility in the aqueous dissolution medium and higher affinity to remain in the oily formulation, which was demonstrated as dramatically lower release profile in both methods.
Figure 9 In vitro release of raloxifene aqueous suspension compared to alkalinized and nonalkalinized nanocarriers in 0.1% Tween 80 using dialysis bag method.
Note: Data expressed as mean ± standard error of mean.
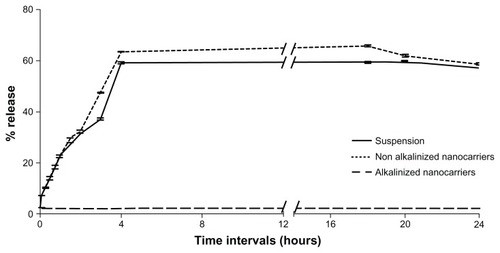
Figure 10 In-vitro release of raloxifene aqueous suspension compared to alkalinized (A-SNEDDS) and non-alkalinized (NA-SNEDDS) nanocarriers in 0.1% Tween 80 using dissolution cup method.
Note: Data expressed as mean ± standard error of mean.
Results confirm previous reports that RLX plain suspension has a good in vitro release profile (60% after 1 hour) that can be successfully enhanced by hydrophilic binders (polyvinylpyr rolidone) and cyclodextrin complexation.Citation8,Citation9 Despite marketed RLX tablets (Evista) containing polyvinylpyrrolidone as solubility enhancer, its bioavailability is as low as 2%. Solubility and dissolution rate are not therefore accused alone for such low bioavailability.Citation12
In vivo study
RLX was reported to be rapidly absorbed and eliminated, exhibiting a linear pharmacokinetic profile within the dose range of 4.0–30.0 mg/kg in rats after a single oral dose.Citation41 RLX target organs, being a SERM, include breast and endocrine organs, where the drug acts as an estrogen antagonist for prevention of breast and endometrial cancer.Citation42 Biodistribution of RLX in different body tissues was investigated by Bayrak et alCitation42 who reported the highest drug uptake in the uterus among the endocrine target organs. Consequently, measurement of RLX content in endocrine organs (uterus, ovaries, and fallopian tubes) was carried out in the current study to assess the potential of nanocarriers to increase the amount of the intact drug that could bypass gastric and liver metabolism and be delivered to its target organs. As investigated by Yang et al,Citation35 maximum RLX concentration in the endocrine system was observed 30 minutes after oral administration and the uptake decreased by time afterwards. Consequently, RLX concentration in endocrine tissue samples was measured by HPLC analysis for different formulations investigated. The influence of different factors on RLX uptake by endocrine organs was investigated. Factors encompassed food effect, nanocarrier formulation effect, and impact of formula alkalinization (dispersion status of the drug). To assess the influence of the aforementioned factors, eight different groups (n = 4) receiving four different formulations were investigated. The formulations included placebo SNEDDS as a control, aqueous drug suspension (2 mg/mL), A-SNEDDS (2 mg/mL), and NA-SNEDDS (2 mg/mL). To assess the food effect, each formulation was administered in two different groups of fasted and fed states (n = 4). Alkalinized SNEDDS were compared to nonalkalinized nanocarriers to detect any additional advantage to the dissolved drug state in the former to dispersed state in the latter.
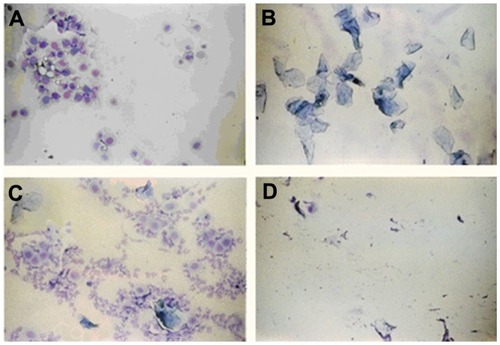
Previous studies have reported that RLX’s role as an SERM is manifested in postmenopausal conditions. The drug effect is insignificant with high estrogen levels, which were anticipated to decrease RLX binding to estrogen receptors.Citation1,Citation42 Consequently, selection of animals for the current investigation was based on histological identification of the estrus-cycle phases. Rats should be chosen in the estrus-cycle phases with reported lowest estrogen level in order to simulate the menopausal condition. The mean duration of the estrus cycle is 4 days for 60%–70% of female rats. However, some rats exhibit longer regular or irregular cycles. Vaginal smears revealed three different types of cells: (1) epithelial cells (round and nucleated), (2) cornified cells (irregular anucleated cells), and (3) leukocytes (little round cells). As shown in , the relative proportions of these three types of cells determine the phase of the estrus cycle.Citation43 It was reported that the proestrus phase exhibits the highest plasma estrogen levels, followed by the estrus phase.Citation44 Thereby, estrogen receptors will be occupied by estrogen, decreasing RLX uptake to receptor tissues (endocrine system). As a consequence, rats were chosen in diestrus and metaestrus phases with reported lowest estrogen level in order to simulate the menopausal condition.
Figure 11 Photomicrographs of vaginal smears from female rats at (A) proestrus (nucleated epithelial cells), (B) estrus (cornified cells), (C) metaestrus (leukocytes, cornified, and nucleated epithelial cells), and (D) diestrus phase (leukocytes).
illustrates the influence of all factors under investigation on drug uptake by target organs. Regarding food effect, the figure revealed that all formulations (drug suspension and nanocarriers) administered in the fasted state exhibited a significant enhancement in drug uptake compared to the fed state of rats. This effect may be attributed to delay in gastric emptying in the fed group.Citation5 High-fat meals were reported to produce a change in exposure time of drugs due to slowing of stomach emptying.Citation5 Lipid-based formulations were recognized to decrease or eliminate the high-fat-meal effect, therefore increasing the solubility of class II compounds.Citation45 These results are in accordance with those obtained by Wempe et alCitation8 and Jagadish et alCitation10 who reported bioavailability studies on RLX in the fasted state. It was reported that administration of RLX with a standardized, high-fat meal insignificantly increased the systemic exposure of RLX. Therefore, Evista can be administered without regard to meals.Citation46
Figure 12 Assessment of raloxifene concentration (μg/mL) in endocrine system (uterus, fallopian tubes, and ovaries) in fed and fasted states from different formulations (suspension, alkalinized, and nonalkalinized self-nanoemulsifying drug-delivery systems) using high-performance liquid chromatography analysis.
Note: Data expressed as mean ± standard error of mean.
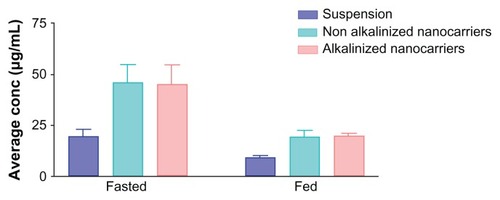
Another factor illustrated in is the nanoformulation effect. A significant enhancement in intact drug amount delivered from both alkalinized (44.9 ± 9.42 μg/mL) and nonalkalinized (45.9 ± 8.61 μg/mL) nanocarriers was observed compared with plain RLX suspension (19.5 ± 3.26 μg/mL). This improvement may be attributed to the ability of SNEDDS to decrease presystemic clearance of RLX owing to the presence of Cremophor EL as a bioactive excipient with reported inhibitory effect on RLX metabolism. Citation14 An additional postulated mechanism of improvement is bypassing the first-pass metabolism due to the oral lymphatic targeting of the drug provoked by the delivery system and its components.Citation26 Finally, the nanometric globule size of SNEDDS is reported to provide a large interfacial surface area for drug release and absorption.Citation28 The postulated aforementioned mechanisms are proposed to increase the part of unchanged RLX escaping metabolism into the systemic circulation and consequently to the target organs.
The last factor illustrated in is the impact of nanocarrier alkalinization. As discussed before, TEA-modified SNEDDS formulations were loaded with RLX in the dissolved state due to higher solubility of the liberated base form in system ingredients. Nevertheless, in vivo investigation indicated that drug loading in SNEDDS in the dissolved state does not confer additional advantage over a well-dispersed state of the drug (NA-SNEDDS). This effect was elucidated from an insignificant difference between RLX uptake from A-SNEDDS and NA-SNEDDS.
In vivo study indicated that in vitro release studies failed to reflect the in vivo conditions for a class II drug candidate like RLX. These results are in accordance with previous reports that solubility and dissolution are not the sole factors affecting the in vivo fate of drugs pertaining to the second category of the BDDCS.Citation5
Conclusion
In the current study, nanoemulsion liquid preconcentrates loaded with bioactive excipients were developed for enhanced oral-delivery characteristics of RLX. Full in vitro appraisal of SNEDDS formulated was performed. The potential of the optimized nanocarriers to enhance drug delivery to endocrine target organs was assessed. Formula alkalinization was performed to investigate the effect of drug loading in the solubilized form on in vitro and in vivo performance of the system. Optimized SNEDDS possessed promising in vitro characteristics, including robustness to dilution, particle size, ZP, PDI, morphological properties, and cloud point. A poor in vitro/in vivo correlation was reported for RLX SNEDDS. Despite in vitro release study not showing any significant difference, in vivo delivery to RLX endocrine organs from nanocarriers was twofold higher than plain drug suspension. In vivo studies supported administration of nanocarrier formulations before meals and showed no advantage for RLX loading in dissolved state (alkalinized nanocarriers) over dispersed state of the drug (nonalkalinized nanocarriers).
Disclosure
The authors report no personal or financial conflicts of interest.
References
- LeeWLChaoHTChengMHWangPHRationale for using raloxifene to prevent both osteoporosis and breast cancer in postmenopausal womenMaturitas2008609210718534794
- LeeWLChaoHTChengMHWangPHThe role of selective estrogen receptor modulators on breast cancer: from tamoxifen to raloxifeneTaiwan J Obstet Gynecol200847243118400579
- Williams-BrownMYSalihSMXuXThe effect of tamoxifen and raloxifene on estrogen metabolism and endometrial cancer riskJ Steroid Biochem Mol Biol2011126788621600284
- ChoCSShinTHLimJLMoonKYKimDKChoiYWStability- enhanced solid dispersion formulation of amorphous raloxifene hydrochlorideKorean J Chem Eng20102719061909
- CustodioJMWuCYBenetLZPredicting drug disposition, absorption/elimination/transporter interplay and the role of food on drug absorptionAdv Drug Deliv Rev20086071773318199522
- JeongEJLinHHuMDisposition mechanisms of raloxifene in the human intestinal Caco-2 modelJ Pharmacol Exp Ther200431037638515020665
- BayrakELambrechtFYDurkanKYilmazOIn vitro evaluation, biodistribution in rats of radiolabeled raloxifeneAppl Radiat Isot201068333619819155
- WempeMFWacherVJRubleKMPharmacokinetics of raloxifene in male Wistar–Hannover rats: influence of complexation with hydroxybutenyl-beta-cyclodextrinInt J Pharm2008346253717644287
- GargASinghSRaoVUBinduKBalasubramaniamJSolid state interaction of raloxifene HCl with different hydrophilic carriers during co-grinding and its effect on dissolution rateDrug Dev Ind Pharm20093545547019048425
- JagadishBYelchuriRBinduKTangiHMarojuSRaoVUEnhanced dissolution and bioavailability of raloxifene hydrochloride by co-grinding with different superdisintegrantsChem Pharm Bull20105829330020190431
- JhaRKTiwariSMishraBBioadhesive microspheres for bioavailability enhancement of raloxifene hydrochloride: formulation and pharmacokinetic evaluationAAPS Pharm Sci Tech201112650657
- ThakkarHNangeshJParmarMPatelDFormulation and characterization of lipid-based drug delivery system of raloxifene-microemulsion and self-microemulsifying drug delivery systemJ Pharm Bioallied Sci2011344244821966167
- ChenYJiaXChenJWangJHuMThe pharmacokinetics of raloxifene and its interaction with apigenin in ratMolecules201058478848721088662
- KimARLimSJLeeBJMetabolic inhibition and kinetics of raloxifene by pharmaceutical excipients in human liver microsomesInt J Pharm2009368374418977285
- ElnaggarYSEl-MassikMAAbdallahOYSelf-nanoemulsifying drug delivery systems of tamoxifen citrate: design and optimizationInt J Pharm200938013314119635537
- DateAADesaiNDixitRNagarsenkerMSelf-nanoemulsifying drug delivery systems: formulation insights, applications and advancesNanomedicine201051595161621143036
- NielsenFSPetersenKBMüllertzABioavailability of probucol from lipid and surfactant based formulations in minipigs: influence of droplet size and dietary stateEur J Pharm Biopharm20086955356218294829
- ChenMLLipid excipients and delivery systems for pharmaceutical development: a regulatory perspectiveAdv Drug Deliv Rev20086076877718077051
- PorterCJHPoutonCWCuineJFCharmanWNEnhancing intestinal drug solubilisation using lipid-based delivery systemsAdv Drug Deliv Rev20086067369118155801
- DixitRPNagarsenkerMSSelf-nanoemulsifying granules of ezetimibe: design, optimization and evaluationEur J Pharm Sci20083518319218652892
- WangLDongJChenJEastoebJLiXDesign and optimization of a new self-nanoemulsifying drug delivery systemJ Colloid Interface Sci200933044344819038395
- ZhaoYWangCChowAHLSelf-nanoemulsifying drug delivery system (SNEDDS) for oral delivery of Zedoary essential oil: formulation and bioavailability studiesInt J Pharm201038317017719732813
- NepalPRHanHKChoiHKPreparation and in vitro–in vivo evaluation of Witepsol H35 based self-nanoemulsifying drug delivery systems (SNEDDS) of coenzyme Q10Eur J Pharm Sci20103922423220035865
- RuanJLiuJZhuDPreparation and evaluation of self-nanoemulsified drug delivery systems (SNEDDSs) of matrine based on drug-phospholipid complex techniqueInt J Pharm201038628229019961910
- BasaliousEBShawkyNBadr-EldinSMSNEDDS containing bioenhancers for improvement of dissolution and oral absorption of lacidipine. I: Development and optimizationInt J Pharm201039120321120214965
- ParmarNSinglaNAminSKohliKStudy of cosurfactant effect on nanoemulsifying area and development of lercanidipine loaded (SNEDDS) self nanoemulsifying drug delivery systemColloids Surf B201186327338
- GuptaSChavhanSSawantKKSelf-nanoemulsifying drug delivery system for adefovir dipivoxil: design, characterization, in vitro and ex vivo evaluationColloids Surf A2011392145155
- GhaiDSinhaVRNanoemulsions as self-emulsified drug delivery carriers for enhanced permeability of the poorly water-soluble selective β1-adrenoreceptor blocker TalinololNanomedicine Epub September 14, 2011
- DateAANagarsenkerMSDesign and evaluation of self- nanoemulsifying drug delivery systems (SNEDDS) for cefpodoxime proxetilInt J Pharm200732916617217010543
- KaduPJKushareSSThackerDDGattaniSGEnhancement of oral bioavailability of atorvastatin calcium by self-emulsifying drug delivery systems (SEDDS)Pharm Dev Technol201116657420088679
- KallakuntaVRBandariSJukantiRVeerareddyPROral self emulsifying powder of lercanidipine hydrochloride: formulation and evaluationPowder Technol2012221375382
- US Pharmacopeial ConventionUSP 34RockvilleUSP201141134115
- JeevanaJBSreelakshmiKDesign and evaluation of self-nanoemulsifying drug delivery system of flutamideJ Young Pharm201134821607048
- ShanmugamSBaskaranRBalakrishnanPThapaPYongCSYooBKSolid self-nanoemulsifying drug delivery system (S-SNEDDS) containing phosphatidylcholine for enhanced bioavailability of highly lipophilic bioactive carotenoid luteinEur J Pharm Biopharm20117925025721550401
- YangZHeXZhangYThe determination of raloxifene in rat tissue using HPLCBiomed Chromatogr20072122923317236238
- WempeMFWrightCLittleJLInhibiting efflux with novel non-ionic surfactants: rational design based on vitamin E TPGSInt J Pharm20093709310219100824
- PatelJPatelARavalMShethNFormulation and development of a self-nanoemulsifying drug delivery system of irbesartanJ Adv Pharm Technol Res2011291622171286
- KohliKChopraSPharDAroraSKhanRSelf-emulsifying drug delivery system: an approach to enhance oral bioavailabilityDrug Discov Today20101595896520727418
- RowRCSheskeyPJWellerPJHandbook of Pharmaceutical Excipients4th edLondonPharmaceutical Press and American Pharmaceutical Association2003
- TangSYManickamSWeiTKNashiruBFormulation development and optimization of a novel Cremophore EL-based nanoemulsion using ultrasound cavitationUltrason Sonochem20121933034521835676
- YangZhYZhangZhFHeXBZhaoGYZhangYQValidation of a novel HPLC method for the determination of raloxifene and its pharmacokinetics in rat plasmaChromatographia200765197201
- KempDCFanPWStevensJCCharacterization of raloxifene glucuronidation in vitro: contribution of intestinal metabolism to presystemic clearanceDrug Metab Dispos20023069470012019197
- MarcondesFKBianchiFJTannoAPDetermination of the estrus cycle phases of rats: helpful considerationsBraz J Biol20026260961412659010
- El-MasMMEl-GowillySMGoharEYGhazalARAbdel-RahmanAAEstrogen dependence of the renal vasodilatory effect of nicotine in rats: role of α7 nicotinic cholinergic receptor/eNOS signalingLife Sci20118818719321092740
- FatourosDGKarpfDMNielsenFSMullertzAClinical studies with oral lipid based formulations of poorly soluble compoundsTher Clin Risk Manag2007359160418472981
- CelnikierDHPharmacokinetics of raloxifene and its clinical applicationEur J Obstet Gynecol Reprod Biol199985232910428318