
Abstract
Introduction
Carbon nanotubes (CNTs) have been widely employed as biomolecule carriers, but there is a need for further functionalization to broaden their therapeutic application in aqueous environments. A few reports have unraveled biomolecule–CNT interactions as a measure of response of the nanocarrier to drug-encapsulation dynamics.
Methods
Herein, the dynamics of encapsulation of the antimicrobial peptide HA-FD-13 (accession code 2L24) into CNTs and hydroxylated CNTs (HCNTs) is discussed.
Results
The van der Waals (vdW) interaction energy of CNT–peptide and HCNT–peptide complexes decreased, reaching −110.6 and −176.8 kcal.Mol−1, respectively, once encapsulation of the peptide inside the CNTs had been completed within 15 ns. The free energy of the two systems decreased to −43.91 and −69.2 kcal.Mol−1 in the same order.
Discussion
The peptide was encased in the HCNTs comparatively more rapidly, due to the presence of both electrostatic and vdW interactions between the peptide and HCNTs. However, the peptide remained encapsulated throughout the vdW interaction in both systems. The negative values of the free energy of the two systems showed that the encapsulation process had occurred spontaneously. Of note, the lower free energy in the HCNT system suggested more stable peptide encapsulation.
Introduction
Even after three decades of extensive use, carbon nanotubes (CNTs) still attract worldwide attention due to their exceptional characteristics. They provide the user with large aspect ratios,Citation1 low density,Citation1 excellent mechanical strength,Citation2,Citation3 and good electrical and thermal conductivityCitation4 and adsorption capacity.Citation5 CNTs have thus been considered for a wide range of industrial applications.Citation6,Citation7 For instance, today they are used in routine composite manufacturing and electronic devices.Citation8,Citation9 CNTs are also key nanocarriers for the delivery of drugs and therapeutic molecules.Citation10 However, to meet a specific and targeted mission, CNTs have to be functionalized with appropriate moieties to become highly efficient for targeting, imaging, and therapeutic purposes.Citation11,Citation12
Due to their tubular nanostructure with single- or multiwalled features, biomolecules, such as proteins and peptides, can be encased in CNTs for sustained and targeted drug release.Citation13,Citation14 Moreover, CNTs are promising nanostructures for drug protection against oxidization, destructive reactions, and environmental tensionCitation15 and delaying antimicrobial-drug resistanceCitation16,Citation17 with their unique structure that allows the penetration of cells, bacteria, and tissue.Citation18 However, the hydrophobic nature of CNTs can cause aggregation, preventing biomolecular encapsulation.Citation19 In this sense, there have been continued attempts to modify and functionalize CNTs with hydrophilic groups, such as hydroxyls,Citation20 carboxyls,Citation21 and a number of hydrophilic amino acids,Citation22 to make them water-soluble, nonimmunogenic, and less cytotoxic, which has broadened their therapeutic usage.Citation23–Citation25
A number of theoretical studies have been performed to evaluate the performance of functionalized CNTs as drug carriers. Maleki et alCitation26 simulated doxorubicin encapsulation in the single-walled carboxylated CNTs (SWCNT-COOH) using molecular dynamic (MD) simulation. They detected van der Waals (vdW) and electrostatic (Elec) energy values of −500 and −3,600 kJ.Mol−1 in neutral pH for SWCNT–doxorubicin and SWCNT-COOH–doxorubicin, respectively. However, the carboxyl groups showed no charge in an acidic environment, where vdW interaction was dominant in the adsorption process, facilitating release of the drug in cancerous tissue. Moradnia et alCitation27 reported adsorption of gemcitabine on the surface of polyethylene glycol (PEG)–modified SWCNTs using density functional theory. It was understood that OH hydrogen bonds between the PEG and gemcitabine dominated the intermolecular interactions in the drug–SWCNT complex. Kamel et alCitation28 demonstrated the role of hydrogen bonding between carboxyl groups of functionalized CNTs and the anticancer drug flutamide in stabilization of conformation of the drug–CNT complex. Moreover, the vdW interaction in carboxylated CNT–drug complex containing CNTs with chirality of (10,5) showed a more negative value (−350 kJ.Mol−1) than systems in which CNTs with chirality of (10,0) and (10,10) were considered (−100 and −150 kJ.Mol−1, respectively). Dehneshin et alCitation29 used valine and phenylalanine moieties to functionalize CNTs and simulate the adsorption of various drugs, including sunitinib, streptozotocin, and sorafenib using MD simulation. Although adsorption of sorafenib and sunitinib on functionalized CNTs occurred through π–π stacking and hydrogen bonding, streptozotocin absorbed on CNTs via hydrogen bonding. Hashemzadeh et alCitation30 simulated the adsorption of paclitaxel on the surface of CNTs and PEG-functionalized-CNTs, wherein functionalized CNTs enhanced the solubility of the CNTs in aqueous solution and remained stable in the tube (free energy of −2,159.35 kJ.Mol−1).
In light of these examples, it can be concluded that choosing an appropriate functional group is key to understanding and keeping stable the encapsulation of drugs in CNTs. MD simulation appears to be successful in capturing mechanical and thermal properties of nanostructures,Citation31–Citation38 and more importantly the interaction of drugs with nanostructures.Citation39–Citation42 CNT-based drug-delivery systems can be simulated in terms of variation in vdW and Elec energies. The possibility of encapsulation of the 13amino–acid peptide HA-FD-13 (ID 2L24) in the CNT and hydroxylated CNT (HCNTs) and stability of the drug–CNT complex was simulated in the present study. This drug is an α-helical antimicrobial peptide derived from the fusion domain (FD) of hemagglutinin (HA) of the influenza virus, and has antimicrobial characteristics, penetrating the anionic microbial membranes.Citation43 vdW and Elec interactions between drug and NTs, as well as the potential of mean force (PMF) of the encapsulated peptide, were calculated using MD simulation and compared for both unmodified and functionalized CNTs.
Simulation Method
In the present study, investigation of the insertion process of the α-helical antimicrobial peptide HA-FD-13 into the CNT and HCNT and subsequently the stability of the encapsulated HA-FD-13 in NTs was performed through MD calculation using LAMMPS software.Citation44 HA-FD-13 (containing amino acids Ile,Phe,Gly,Ala,Lys,Asn,Trp, and NH2) was chosen from the Protein Data Bank (https://www.rcsb.org/structure/2L24). This antimicrobial peptide has a positive charge so that it can penetrate anionic microbial membranes.
VMD software was used to visualize the simulated encapsulation process.Citation45 Considering the size of HA-FD-13, a CNT and HCNT with chirality of armchair (20,20) and length of 40 Å were selected as nanovehicles for this peptide. The HCNT contained hydroxyl groups on the edge with a degree of functionalization of 2.82%, which was calculated thus:
At the beginning of the simulation, HA-FD-13 was situated at an initial distance of 2 Å from the NT. The axial direction of the NT was parallel to the z-axis of the simulation box. CNT–HA-FD-13 and HCNT–HA-FD-13 complexes were immersed in simulation boxes containing TIP3P water molecules, as well as counterions to neutralize the simulated solution with periodic boundary conditions. All MD simulations were performed applying a CHARMM27 force field.Citation46 Tersoff potential was used to consider interactions among carbon atoms of NTs.Citation47 To verify the encapsulation process of the peptide, in the first step minimization of the system was achieved in the NVT ensemble at 300 K while the NT was fixed. In the next step, the MD runs were performed in the NPT ensemble for 15 ns with a time step of 1 fs. The inner and outer cutoff distances for the Lennard-Jones and coulombic potential were considered to be 8 and 12 Å, respectively. The parameters of Lennard-Jones potential for cross-vdW interactions between unbonded atoms was estimated using the Lorentz–Berthelot combination rule.Citation48 The vdW interaction between HA-FD-13 and the CNT was calculated:Citation45
where EvdW refers to vdW energy between HA-FD-13 and the NT and vdW interaction of HA-FD-13 combined with the CNT.
and
stand for vdW energies of HA-FD-13 and the CNT, respectively. To evaluate the stability of the peptide encapsulated inside the CNT, an external force was loaded on HA-FD-13 along the z-axis of the NT to pull it out in the direction opposite to the penetration process. The spring constant k and pulling velocity were chosen to be 15 kcal.Mol−1.Å−2 and 0.005 Å.ps−1, respectively.Citation49 The pulling process was simulated ten times to compute the PMF profile using Jarzynski’s equality:Citation46
where ΔG and W correspond to the free energy discrepancy between two states and the work performed on the system, respectively. β is equal to (T)−1, where
stands for the Boltzmann constant. The temperature and pressure of the system were adjusted to about 300 K and 101.3 kPa through applying Langevin dynamics and the Langevin piston Nosé–Hoover method, respectively.Citation50
Results and Discussion
Localization of HA-FD-13 Within the CNT–Peptide and HCNT–Peptide Complexes
The encapsulation process of HA-FD-13 inside the CNT and HCNT was investigated through MD simulation. Snapshots of these processes were taken by VMD to demonstrate the positions of the peptide at different times in the simulation box. and show these snapshots relating to the encapsulation of the peptide into the CNT and HCNT, respectively. As can be observed, HA-FD-13 was successfully encapsulated in the cavity of both the CNT and HCNT; however, the insertion process of the peptide confinement accelerated in the case of the HCNT, which means that the peptide overcame the energy barrier in the vicinity of the NT more rapidly. Moreover, the peptide remained stably encapsulated throughout the vdW interaction between the conjugated aromatic rings of the peptide and the interior walls of the NT to the end of the simulation (15 ns) in both systems. Conformational changes taking place inside the NTs are provided in Supplementary Information (Video 1: [CNT–peptide] and Video 2: [HCNT–peptide]). The center of mass (CoM) distance between the peptide and the CNT and HCNT are depicted in and , respectively. The sudden decrease in CoM distance at the beginning of the simulation in both systems demonstrated that the peptide was self-adjusting before entering the cavity of the NTs. The same downward trend was observed in work done by Kang et al.Citation49 They reported that the protein SmtA showed self-adjusted conformational changes before entering CNTs.
Figure 1 Representative snapshots of insertion of the HA-FD-13 peptide into an armchair (20,20) CNT at various times. For clarity, molecules of water have not been shown.
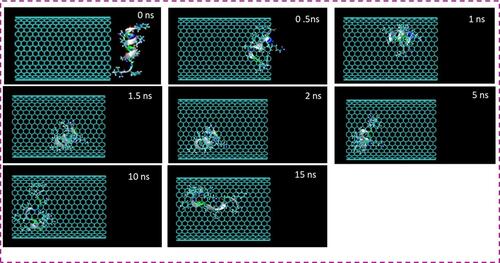
Figure 2 Representative snapshots of insertion of HA-FD-13 into an armchair (20,20) HCNT at various times. For clarity, molecules of water have not been shown.
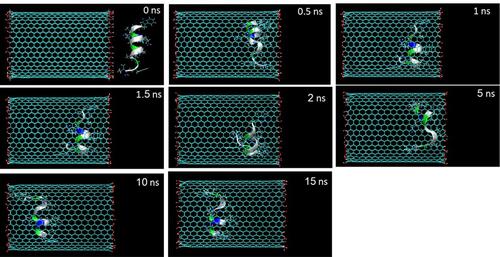
Figure 3 Center of mass (CoM) distance between (A) HA-FD-13 and the CNT; (B) HA-FD-13 and the HCNTs as a function of simulation time; vdW interaction between (C) HA-FD-13 and the CNT (20,20); (D) HA-FD-13 and the HCNT (20,20); (E) coulombic interaction between HA-FD-13 and the HCNT (20,20) as a function of simulation time.
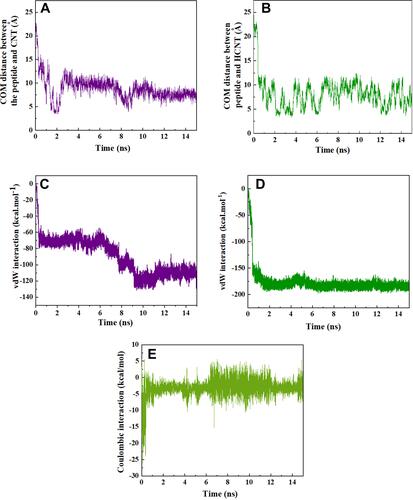
After the sudden drop had occurred in this curve for the CNT–peptide complex, it reached a minimum of 3.62 Å before fluctuating around the specified value at 2 ns, followed by the second minimum of 4.3 Å at 8 ns. On the other hand, for the HCNT–peptide complex, the CoM distance showed fluctuation around 7.5 Å after the initial drop, with no sign of a minimum. It can be posited that the presence of the hydroxyl groups in the entrance edge of the CNT created hydrogen bonding between the CNT and the peptide and subsequently a coulombic interaction (as shown in ) such that the peptide was rapidly adsorbed into the NT. This caused the sudden drop in CoM distance and the following fluctuation. In other words, the adsorption interaction between the OH groups on the edge and the peptide did not allow the peptide to reach the center of the NT and caused more intense fluctuation compared to the pristine CNT. For the CNT–peptide complex, the revealed two minima in the CoM-distance curve showed that after insertion of the peptide in the NT, it could more easily pass the edge of the NT and experience a sudden change in CoM distance with smooth fluctuating changes between these minima, due to the absence of the coulombic interaction between the CNT and peptide that caused that the vdW interactions being the dominant parameter in determining the encapsulation process.
Variations in vdW interaction energy between the peptide and NT in the CNT–peptide and HCNT–peptide complexes with simulation time during the encapsulation process are shown in and , respectively. As expected, vdW interaction energies of the CNT–peptide and HCNT–peptide complexes diminished with the drop in CoM distance between peptide and NT during the encapsulation process, reaching −110.6 and −176.8 kcal.Mol−1, respectively after the complete adsorption of the peptide into the NT cavity at 15 ns. The vdW interaction energy of the system containing the modified CNT decreased, which showed that the stronger vdW interaction between the HCNT and the peptide resulted in the development of a more favourable drug-delivery system with the ability to penetrate cells.Citation51 A similar decreasing trend in the vdW energy of insertion of DNA oligonucleotides and collagen-like peptides into CNTs has been observed in research performed by Gao et alCitation52 and Kang et al,Citation53 respectively. In another work done by Maleki et al,Citation54 Elec and vdW interactions between doxorubicin and CNTs were determined. According to their results, as a result of the zero value of Elec interactions, the absorbance of doxorubicin in the CNTs was due to vdW interactions with favorable negative values. Hashemzadeh et alCitation30 showed that vdW interactions between the anticancer drug paclitaxel and CNTs modified with PEG were stronger, with lower negative values than the vdW interaction of the pristine CNT–drug complex. As another observation from , the decreasing trend of vdW interaction energy of the CNT–peptide complex contained two steps: first at the beginning of the simulation, and second starting at 6 ns and ending at 9 ns of the simulation. This indicates that the peptide did not undergo conformational changes outside these periods. In the second decrease, the conformation of the peptide varied such that the vdW interaction energy reached the lowest negative value.
Free Energy Calculation by MD Simulation
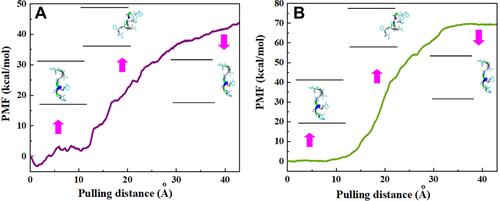
At the end of the simulation, when the peptide had completely adsorbed inside the NTs at 15 ns, its PMF profiles were calculated by pulling it out through the MD simulation at the speed of 0.005 Å.ps−1, which was selected based on the speed of the insertion process. This simulation was performed five times to obtain the average value of work (W) at each pulling distance, which is shown as PMF profiles in and for the CNT and HCNT systems, respectively. Peptide positions along the z-axis corresponding to the NTs are depicted also. As can be observed, the free energies of the simulated pristine-CNT and HCNT systems were elevated during the pulling process and reached 43.91 and 69.2 kcal.Mol−1, respectively, at a pulling distance of 43 Å. These results indicated that the encapsulation process of the peptide inside the CNT and HCNT occurred spontaneously, with free energy of −43.91 and −69.2 kcal.Mol−1, respectively. However, the lower free energy in the HCNT system showed more stability of the encapsulated peptide inside the cavity of the NT than the pristine CNT system. A similar spontaneous process was observed in work done by Veclani et al.Citation55 They obtained free energy value of −9.5 kcal.Mol−1 for the encapsulation process of ciprofloxacin on the surface of CNTs. Ghadri et alCitation56 reported that enhancement of active sites on CNTs caused an increase in hydrogen bonding between the drug fulvestrant and functional groups of CNTs such that the stronger affinity of the drug with the functionalized CNTs caused a lower free energy (−373.38 kcal.Mol−1) than the system with pristine CNTs.
Figure 4 Potential of mean force (PMF) for systems of (A) CNT–peptide and (B) HCNT–peptide complexes computed from ten pullings through the MD simulation. The images represent the positions of HA-FD-13 corresponding to the z-coordinate along the (A) CNT and (B) HCNT at key positions.
The energy contributions of the whole system in the spontaneous encapsulation of HA-FD-13 inside the CNT and HCNT are given in and , respectively. Considering the definition of free energy (ΔG = ΔH – TΔS, where ΔH and ΔS are changes in enthalpy and entropy, respectively), it can be concluded that the encapsulation process was an enthalpy-driven phenomenon, due to the greater contribution of enthalpy change to the free energy of the whole system and decrease in entropy during the encapsulation process in both systems.Citation57 As shown in and , the potential energy of the whole system (ΔP) as a part of enthalpy change consists in variations in vdW energy (ΔEvdW), Elec energy (ΔEele), and bonded-atom energy (ΔEbonded). For the pristine CNT system, the greater drop in vdW energy (310.5 kcal.Mol−1) compared to the decrease in Elec energy (198.28 kcal.Mol−1) proved that the vdW interaction played a more important role in the encapsulation process. In the HCNT system, the reduction in Elec energy (−677.48 kcal.Mol−1) was found to be the more important contributor to the process than the slight decrease in vdW energy (−7.176 kcal.Mol−1). The increase in Ebonded in both systems (80.52 and 43.766 kcal.Mol−1 for CNT and HCNT, respectively) corresponded to changes in energy of bonded atoms and subsequently conformational changes in the systems.
Table 1 Energy contributions (kcal.Mol−1) in the spontaneous encapsulation of HA-FD-13 inside CNTs (20,20). : Free energy variation of the whole system in the encapsulation process.
: Entropy variation of the whole system.
: Enthalpy variation of the whole system.
: Kinetic energy variation of the system in the encapsulation process.
: Multiplication of the volume of the system and pressure change of the whole system,
: Potential energy variation of the system, composed of vdW energy change (
), Elec energy change (
), and bonded-atom energy change (
)
Table 2 Energy contributions (kcal.Mol−1) in the spontaneous encapsulation of HA-FD-13 inside the HCNT (20,20). : Free energy variation of the whole system in the encapsulation process.
: Entropy variation of the whole system.
: Enthalpy variation of the whole system.
: Kinetic energy variation of the system in the encapsulation process.
: Multiplication of the volume of the system and the pressure change of the whole system.
: Potential energy variation of the system, composed of vdW energy change (
), Elec energy change (
), and bonded-atom energy change (
)
Variations in Conformation of HA-FD-13
Changes in the conformation of HA-FD-13 in the CNT and HCNT systems at 0 and 15 ns of the MD simulation are illustrated in and , respectively. The left images in these figures correspond to the peptide immersed in an aqueous solution in its natural structure at 0 ns. The right images show the state in which the conformation of the peptide was adjusted to the interior geometry of the NTs at 15 ns. Similar alterations in the arrangement and conformation of doxorubicin encapsulated within CNTs in accordance with NT chirality and diameter was reported by Zhang et al.Citation58 Root-mean-square deviations (RMSDs) revealing the variations in the peptide conformation in both systems as a function of simulation time are shown in and , respectively. As can be observed, for both systems, the RMSD showed fluctuation of 6–16 from the beginning of the simulation to 12 ns, which indicates the continuous conformational alteration of the peptide, or in other words self-adjustment of the peptide according to the interior geometry of the NT. However, the conformation of the peptide in the HCNT–peptide complex varied more intensely than the conformational changes in the CNT–peptide complex at the beginning of the simulation. This shows that the hydrogen bonding creating the Elec interaction between the hydroxyl groups on the edge of the CNT and the peptide caused not only the rapid adsorption of the peptide but also intense conformational change of the peptide.
Figure 5 (A) Axial views of HA-FD-13 at 0 and 15 ns in the MD simulation. For the sake of clarity, molecules of water have not been shown. (B) Root mean square deviation (RMSD) of HA-FD-13 as a function of simulation time. (C) Radius gyration of HA-FD-13 as a function of simulation time in the CNT– HA-FD-13 complex.
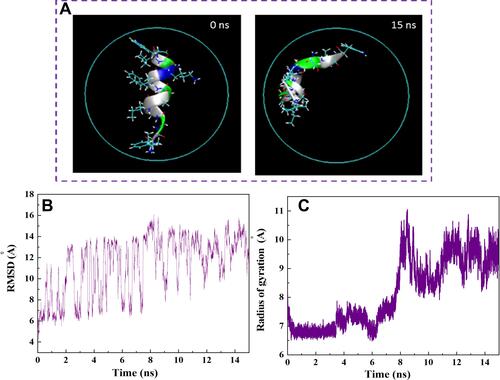
Figure 6 (A) Axial views of HA-FD-13 at 0 and 15 ns in the MD simulation. For the sake of clarity, molecules of water have not been shown. (B) Root mean square deviation (RMSD) of HA-FD-13 as a function of simulation time. (C) Radius gyration of HA-FD-13 as a function of simulation time in the HCNT–peptide complex.
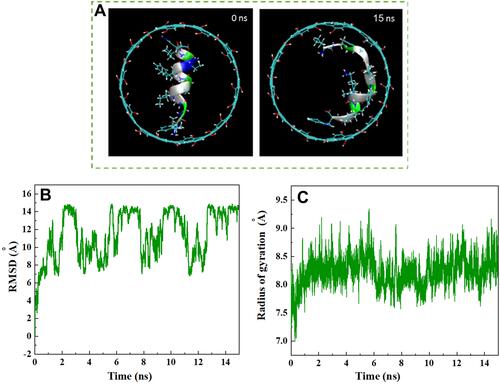
After 12 ns of the simulation, the peptide reached a more stable state, with less pronounced changes in conformation and subsequent RMSD so that the aromatic rings were situated parallel to the interior wall of the NT, causing the strong vdW interaction. This observation is in good agreement with the vdW interaction energies obtained between the peptide and both NTs. A similar structural variation during the encapsulation of the ssDNA oligomer inside the CNT was observed by Shen et al.Citation59 They showed that the RMSD of the biomolecule fluctuated slightly after complete encapsulation, and its conformation caused the creation of a π–π stacking interaction. In their other work,Citation60 it was reported that changes in RMSD for the encapsulated biomolecule were due to differences in the affinity of its hydrophobic and hydrophilic parts with the solvent and the NT, respectively. Besides the calculation of the RMSD, determining alterations in gyration radius of HA-FD-13 during the insertion process provided beneficial details about the conformational changes of the peptide. and show these variations in the CNT and HCNT systems, respectively, during the simulation time. In both systems, the structure of peptides stretched to adjust to NT geometry through peptide–NT vdW interactions. This conformational change was associated with the higher gyration radius of the confined peptide than the unconfined peptide.
Variations in distances between the CoM of HA-FD-13 and the central axis of the CNT and HCNT as a function of simulation time are demonstrated in and , respectively. It can be seen that the peptide was in a position in the simulation box such that that the CoM of the peptide was situated on the central axis of the CNT and HCNT at the beginning of the simulation. After the simulation had started, the peptide moved toward one side of the CNT and HCNT, causing an immediate increase in peptide–axis distance. These sudden movements correlated with the vdW interaction between the wall of the CNT and the peptide and both vdW and Elec interactions between the peptide and HCNT. However, these conformations were not stable and varied through the adsorption of the peptide to the other sidewall of the NTs, revealing a sudden drop in peptide–axis distance. After total insertion of the peptide in NTs, the distance between the CoM of the peptide and the central axis of the NTs fluctuated continuously, due to the completion of adsorption of the hydrophilic and hydrophobic groups of the protein by the water molecules andNTs, respectively. The slight fluctuation in the curve of peptide–axis distance in the HCNT system was related to the greater stability of the encapsulated peptide in HCNT than the pristine CNT.
Figure 7 Distance between the center of mass (CoM) of HA-FD-13 and the central axis of (A) the CNT and (B) HCNT as a function of simulation time. Potential energy of HA-FD-13 in the (C) CNT–peptide complex and (D) HCNT–peptide as a function of simulation time.
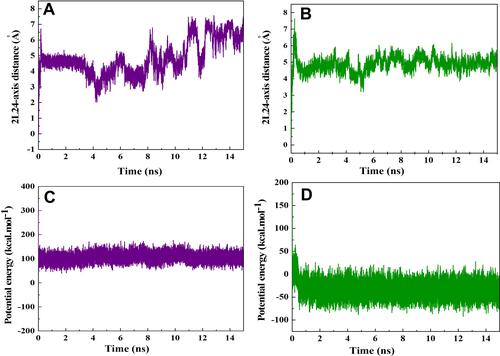
and present the changes in potential energy of HA-FD-13 during encapsulation into the CNT and HCNT, respectively. For the peptide in the HCNT, it was observed that its potential energy dwindled during the first 0.5 ns of the insertion procedure to −50 kcal.Mol−1, favoring the rapid adsorption and stability of the peptide in the cavity of the NT under the effect of both vdW and Elec interactions between the peptide and NT. However, for the peptide in the CNT–peptide complex, an unfavourable level of potential energy (100 kcal.Mol−1) correlated with less stability for the encapsulated peptide.Citation49
Conclusion
In this research, the encapsulation process of the antimicrobial peptide HA-FD-13 inside a CNT and HCNT in an aqueous solution was compared through MD simulation. The most important results of our research are:
The peptide was encapsulated in the cavity of HCNT more rapidly due to the presence of both Elec and vdW interactions between the peptide and NT.
The peptide remained encapsulated throughout the vdW interaction between the conjugated aromatic rings of the peptide and the interior walls of the NT until the end of the simulation (15 ns) in both systems.
The sudden decrease in CoM distance between the peptide and NTs at the beginning of the simulation in both systems demonstrated self-adjustment of the peptide before entering the cavity of NTs.
The vdW interaction energies of the CNT–peptide and HCNT–peptide complexes diminished, reaching −110.6 and −176.8 kcal.Mol−1, respectively, after complete adsorption of the peptide into the NT cavity at 15 ns.
Encapsulation of the peptide inside the CNT and HCNT occurred spontaneously, with free energy of −43.91 and −69.2 kcal.Mol−1, respectively. The lower free energy in the HCNT system showed more stability for the encapsulated peptide.
RMSD results showed that the conformation of the peptide in the HCNT–peptide complex varied more intensely at the beginning of the simulation. After 12 ns, in both systems the peptide had reached a more stable state, with lower changes in conformation and subsequent RMSD such that the aromatic rings of the peptide were placed parallel to the interior wall of the NTs, causing the strong vdW interaction.
The potential energy of the peptide reached −50 and 100 kcal.Mol−1 in the CNT and HCNT systems, respectively, revealing the greater stability of the HCNT complex.
Author Contributions
All authors made a significant contribution to the work reported, whether in conception, study design, execution, acquisition of data, analysis, and interpretation, or in all these areas, took part in drafting, revising, or critically reviewing the article, gave final approval to the version to be published, have agreed on the journal to which the article has been submitted, and agree to be accountable for all aspects of the work.
Acknowledgements
Ebrahim Mostafavi would like to acknowledge the support from the National Institute of Biomedical Imaging and Bioengineering (5T32EB009035).
Disclosure
The authors declare no conflicts of interest.
Additional information
Funding
References
- Ibrahim KS. Carbon nanotubes-properties and applications: a review. Carbon Lett. 2013;14:131–144.
- Mashhadzadeh AH, Fereidoon A, Ahangari MG. Surface modification of carbon nanotubes using 3-aminopropyltriethoxysilane to improve mechanical properties of nanocomposite based polymer matrix: experimental and density functional theory study. Appl Surf Sci. 2017;420:167–179. doi:10.1016/j.apsusc.2017.05.148
- Fereidoon A, Mashhadzadeh AH, Rostamiyan Y. Experimental, modeling and optimization study on the mechanical properties of epoxy/high-impact polystyrene/multi-walled carbon nanotube ternary nanocomposite using artificial neural network and genetic algorithm. Sci Eng Compos Mater. 2013;20(3):265–276. doi:10.1515/secm-2012-0178
- Rostamiyan Y, Mohammadi V, Mashhadzadeh AH. Mechanical, electronic and stability properties of multi-walled beryllium oxide nanotubes and nanopeapods: a density functional theory study. J Mol Model. 2020;26(4):1–13. doi:10.1007/s00894-020-4328-5
- Mashhadzadeh AH, Fathalian M, Ghorbanzadeh Ahangari M, et al. DFT study of Ni, Cu, Cd and Ag heavy metal atom adsorption onto the surface of the zinc-oxide nanotube and zinc-oxide graphene-like structure. Mater Chem Phys. 2018;220:366–373. doi:10.1016/j.matchemphys.2018.09.016
- Gupta N, Gupta SM, Sharma SK. Carbon nanotubes: synthesis, properties and engineering applications. Carbon Lett. 2019;29(5):419–447. doi:10.1007/s42823-019-00068-2
- Venkataraman A, Amadi EV, Chen Y, et al. Carbon nanotube assembly and integration for applications. Nanoscale Res Lett. 2019;14(1):220. doi:10.1186/s11671-019-3046-3
- Ferrer-Anglada N, Gomis V, El-Hachemi Z, et al. Carbon nanotube based composites for electronic applications: CNT–conducting polymers, CNT–Cu. Phys Status Solidi A. 2006;203(6):1082–1087. doi:10.1002/pssa.200566188
- Saeb MR, Najafi F, Bakhshandeh E, et al. Highly curable epoxy/MWCNTs nanocomposites: an effective approach to functionalization of carbon nanotubes. Chem Eng J. 2015;259:117–125. doi:10.1016/j.cej.2014.07.116
- Chandrasekhar P. CNT Applications in Drug and Biomolecule Delivery, in Conducting Polymers, Fundamentals and Applications. Springer; 2018:61–64.
- Pastorin G, Wu W, Wieckowski S, et al. Double functionalisation of carbon nanotubes for multimodal drug delivery. Chem Commun. 2006;11(11):1182–1184. doi:10.1039/b516309a
- Prato M, Kostarelos K, Bianco A. Functionalized carbon nanotubes in drug design and discovery. Acc Chem Res. 2008;41(1):60–68. doi:10.1021/ar700089b
- Skandani AA, Al-Haik M. Reciprocal effects of the chirality and the surface functionalization on the drug delivery permissibility of carbon nanotubes. Soft Matter. 2013;9(48):11645–11649. doi:10.1039/C3SM52126E
- Dehaghani MZ, Yousefi F, Seidi F, et al. Encapsulation of an anticancer drug Isatin inside a host nano-vehicle SWCNT: a molecular dynamics simulation. Sci Rep. 2021;11(1):1–10. doi:10.1038/s41598-021-98222-2
- Bernkop-Schnürch A, Bratengeyer I, Valenta C. Development and in vitro evaluation of a drug delivery system protecting from trypsinic degradation. Int J Pharm. 1997;157(1):17–25. doi:10.1016/S0378-5173(97)00198-1
- Smith AW. Biofilms and antibiotic therapy: is there a role for combating bacterial resistance by the use of novel drug delivery systems? Adv Drug Deliv Rev. 2005;57(10):1539–1550. doi:10.1016/j.addr.2005.04.007
- Zarrintaj P, Ramsey JD, Samadi A, et al. Poloxamer: a versatile tri-block copolymer for biomedical applications. Acta Biomater. 2020;110:37–67. doi:10.1016/j.actbio.2020.04.028
- Ramos MADS, Da Silva P, Spósito L, et al. Nanotechnology-based drug delivery systems for control of microbial biofilms: a review. Int J Nanomedicine. 2018;13:1179. doi:10.2147/IJN.S146195
- Ménard-Moyon C, Venturelli E, Fabbro C, et al. The alluring potential of functionalized carbon nanotubes in drug discovery. Expert Opin Drug Discov. 2010;5(7):691–707. doi:10.1517/17460441.2010.490552
- Sekar G, Kandiyil ST, Sivakumar A, et al. Binding studies of hydroxylated Multi-Walled Carbon Nanotubes to hemoglobin, gamma globulin and transferrin. J Photochem Photobiol B. 2015;153:222–232. doi:10.1016/j.jphotobiol.2015.09.023
- Chen Z, Pierre D, He H, et al. Adsorption behavior of epirubicin hydrochloride on carboxylated carbon nanotubes. Int J Pharm. 2011;405(1–2):153–161. doi:10.1016/j.ijpharm.2010.11.034
- Zardini HZ, Amiri A, Shanbedi M, et al. Enhanced antibacterial activity of amino acids-functionalized multi walled carbon nanotubes by a simple method. Colloids Surf B Biointerfaces. 2012;92:196–202. doi:10.1016/j.colsurfb.2011.11.045
- Liu P. Modification strategies for carbon nanotubes as a drug delivery system. Ind Eng Chem Res. 2013;52(38):13517–13527. doi:10.1021/ie402360f
- Tan JM, Arulselvan P, Fakurazi S, et al. A review on characterizations and biocompatibility of functionalized carbon nanotubes in drug delivery design. J Nanomater. 2014;2014:1–20. doi:10.1155/2014/917024
- Liu Z, Liu Y, Peng D. Hydroxylation of multi-walled carbon nanotubes reduces their cytotoxicity by limiting the activation of mitochondrial mediated apoptotic pathway. J Mater Sci Mater Med. 2014;25(4):1033–1044.
- Maleki R, Afrouzi HH, Hosseini M, et al. pH-sensitive loading/releasing of doxorubicin using single-walled carbon nanotube and multi-walled carbon nanotube: a molecular dynamics study. Comput Methods Programs Biomed. 2020;186:105210. doi:10.1016/j.cmpb.2019.105210
- Moradnia H, Raissi H, Shahabi M. The performance of the single-walled carbon nanotube covalently modified with polyethylene glycol to delivery of Gemcitabine anticancer drug in the aqueous environment. J Biomol Struct Dyn. 2020;39:881–888.
- Kamel M, Raissi H, Morsali A, et al. Assessment of the adsorption mechanism of Flutamide anticancer drug on the functionalized single-walled carbon nanotube surface as a drug delivery vehicle: an alternative theoretical approach based on DFT and MD. Appl Surf Sci. 2018;434:492–503. doi:10.1016/j.apsusc.2017.10.165
- Dehneshin N, Raissi H, Hasanzade Z, et al. Using molecular dynamics simulation to explore the binding of the three potent anticancer drugs sorafenib, streptozotocin, and sunitinib to functionalized carbon nanotubes. J Mol Model. 2019;25(6):159. doi:10.1007/s00894-019-4024-5
- Hashemzadeh H, Raissi H. The functionalization of carbon nanotubes to enhance the efficacy of the anticancer drug paclitaxel: a molecular dynamics simulation study. J Mol Model. 2017;23(8):1–10. doi:10.1007/s00894-017-3391-z
- Molaei F, Zarghami dehaghani M, Salmankhani A, et al. Applying molecular dynamics simulation to take the fracture fingerprint of polycrystalline SiC nanosheets. Comput Mater Sci. 2021;200:110770. doi:10.1016/j.commatsci.2021.110770
- Fooladpanjeh S, Yousefi F, Molaei F, et al. Thermal conductivity of random polycrystalline BC3 nanosheets: a step towards realistic simulation of 2D structures. J Mol Graph Model. 2021;107:107977. doi:10.1016/j.jmgm.2021.107977
- Dehaghani MZ, Esmaeili Safa M, Yousefi F, et al. Fracture behavior of SiGe nanosheets: mechanics of monocrystalline vs. polycrystalline structure. Eng Fract Mech. 2021;251:107782. doi:10.1016/j.engfracmech.2021.107782
- Bagheri B, Zarghami dehaghani M, Esmaeili Safa M, et al. Fracture fingerprint of polycrystalline C3N nanosheets: theoretical basis. J Mol Graph Model. 2021;106:107899. doi:10.1016/j.jmgm.2021.107899
- Salmankhani A, Karami Z, Hamed Mashhadzadeh A, et al. A theoretical scenario for the mechanical failure of boron carbide nanotubes. Comput Mater Sci. 2021;186:110022. doi:10.1016/j.commatsci.2020.110022
- Dehaghani MZ, Salmankhani A, Hamed Mashhadzadeh A, et al. Fracture mechanics of polycrystalline beryllium oxide nanosheets: a theoretical basis. Eng Fract Mech. 2021;244:107552. doi:10.1016/j.engfracmech.2021.107552
- Bagheri B, Zarghami Dehaghani M, Karami Z, et al. Correlation between surface topological defects and fracture mechanism of γ-graphyne-like boron nitride nanosheets. Comput Mater Sci. 2021;188:110152. doi:10.1016/j.commatsci.2020.110152
- Dehaghani MZ, Hamed Mashhadzadeh A, Salmankhani A, et al. Fracture toughness and crack propagation behavior of nanoscale beryllium oxide graphene-like structures: a molecular dynamics simulation analysis. Eng Fract Mech. 2020;235:107194. doi:10.1016/j.engfracmech.2020.107194
- Zarghami dehaghani M, Yousefi F, Sajadi SM, et al. Theoretical encapsulation of fluorouracil (5-FU) anti-cancer chemotherapy drug into carbon nanotubes (CNT) and boron nitride nanotubes (BNNT). Molecules. 2021;26(16):4920. doi:10.3390/molecules26164920
- Dehaghani MZ, Yousefi F, Bagheri B, et al. α-helical antimicrobial peptide encapsulation and release from boron nitride nanotubes: a computational study. Int J Nanomedicine. 2021;16:4277. doi:10.2147/IJN.S313855
- Dehaghani MZ, Bagheri B, Yousefi F, et al. Boron nitride nanotube as an antimicrobial peptide carrier: a theoretical insight. Int J Nanomedicine. 2021;16:1837. doi:10.2147/IJN.S298699
- Zarghami Dehaghani M, Bagheri B, Nasiriasayesh A, et al. Insight into the self-insertion of a protein inside the boron nitride nanotube. ACS Omega. 2020;5(49):32051–32058. doi:10.1021/acsomega.0c05080
- Zhu S, Aumelas A, Gao B. Convergent evolution-guided design of antimicrobial peptides derived from influenza A virus hemagglutinin. J Med Chem. 2011;54(4):1091–1095. doi:10.1021/jm1010463
- Plimpton S. Fast parallel algorithms for short-range molecular dynamics. J Comput Phys. 1995;117(1):1–19. doi:10.1006/jcph.1995.1039
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1):33–38. doi:10.1016/0263-7855(96)00018-5
- Park S, Schulten K. Calculating potentials of mean force from steered molecular dynamics simulations. J Chem Phys. 2004;120(13):5946–5961. doi:10.1063/1.1651473
- Tersoff J. New empirical approach for the structure and energy of covalent systems. Phys Rev B. 1988;37(12):6991–7000. doi:10.1103/PhysRevB.37.6991
- Hirschfelder JO, Curtiss CF, Bird RB. Molecular Theory of Gases and Liquids. Vol. 165. New York: Wiley; 1964.
- Kang Y, Liu Y-C, Wang Q, et al. On the spontaneous encapsulation of proteins in carbon nanotubes. Biomaterials. 2009;30(14):2807–2815. doi:10.1016/j.biomaterials.2009.01.024
- Feller SE, Zhang Y, Pastor RW, et al. Constant pressure molecular dynamics simulation: the Langevin piston method. J Chem Phys. 1995;103(11):4613–4621. doi:10.1063/1.470648
- Mousavi SZ, Amjad-Iranagh S, Nademi Y, et al. Carbon nanotube-encapsulated drug penetration through the cell membrane: an investigation based on steered molecular dynamics simulation. J Membr Biol. 2013;246(9):697–704. doi:10.1007/s00232-013-9587-y
- Gao H, Kong Y, Cui D, et al. Spontaneous insertion of DNA oligonucleotides into carbon nanotubes. Nano Lett. 2003;3(4):471–473. doi:10.1021/nl025967a
- Kang Y, Wang Q, Liu YC, et al. Dynamic mechanism of collagen-like peptide encapsulated into carbon nanotubes. J Phys Chem B. 2008;112(15):4801–4807. doi:10.1021/jp711392g
- Maleki R, Afrouzi HH, Hosseini M, et al. Molecular dynamics simulation of Doxorubicin loading with N-isopropyl acrylamide carbon nanotube in a drug delivery system. Comput Methods Programs Biomed. 2020;184:105303. doi:10.1016/j.cmpb.2019.105303
- Veclani D, Melchior A. Adsorption of ciprofloxacin on carbon nanotubes: insights from molecular dynamics simulations. J Mol Liq. 2020;298:111977. doi:10.1016/j.molliq.2019.111977
- Ghadri Z, Raissi H, Shahabi M, et al. Molecular dynamics simulation study of Glycine tip-functionalisation of single-walled carbon nanotubes as emerging nanovectors for the delivery of anticancer drugs. Mol Simul. 2020;46(2):111–120. doi:10.1080/08927022.2019.1679363
- Zhang D, Gullingsrud J, McCammon JA. Potentials of mean force for acetylcholine unbinding from the alpha7 nicotinic acetylcholine receptor ligand-binding domain. J Am Chem Soc. 2006;128(9):3019–3026. doi:10.1021/ja057292u
- Zhang L, Peng G, Li J, et al. Molecular dynamics study on the configuration and arrangement of doxorubicin in carbon nanotubes. J Mol Liq. 2018;262:295–301. doi:10.1016/j.molliq.2018.04.097
- Shen JW, Tang T, Wei XH, et al. On the loading mechanism of ssDNA into carbon nanotubes. RSC Adv. 2015;5(70):56896–56903. doi:10.1039/C5RA01941A
- Shen J-W, Wu T, Wang Q, et al. Induced stepwise conformational change of human serum albumin on carbon nanotube surfaces. Biomaterials. 2008;29(28):3847–3855. doi:10.1016/j.biomaterials.2008.06.013