 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Background
Integrins αvβ3 and αvβ5, both of which specifically recognize the Arg-Gly-Asp (RGD) motif, are overexpressed on many solid tumors and in tumor neovasculature. Thus, coupling the RGD motif to the liposomal surface for achieving active targeting can be a promising strategy for the treatment of tumors.
Methods
Cyclo(Arg-Gly-Asp-D-Phe-Cys) (cRGD) was covalently coupled with the liposomal membrane surface, followed by coating with poly(ethylene glycol) (PEG) using the post-insertion technique. The coupling efficiency of cRGD was determined. Doxorubicin as a model anticancer drug was loaded into liposomes using an ammonium sulfate gradient method to investigate the encapsulation efficiency, cellular uptake by the integrin-overexpressing human glioma cell line U87MG in vitro, and pharmacokinetic properties in Sprague-Dawley rats.
Results
cRGD was conjugated to the liposomal surface by a thiol-maleimide coupling reaction. The coupling efficiency reached 98%. The encapsulation efficiency of doxorubicin in liposomes was more than 98%. The flow cytometry test result showed that cRGD-modified liposomes (RGD-DXRL-PEG) had higher cell uptake by U87MG cells, compared with nontargeted liposomes (DXRL-PEG). The cellular uptake was significantly inhibited in the presence of excess free cRGD. Both the targeted (t1/2 = 24.10 hours) and non-targeted (t1/2 = 25.32 hours) liposomes showed long circulating properties in rat plasma. The area under the curve of the targeted and nontargeted liposomes was 6.4-fold and 8.3-fold higher than that of doxorubicin solution, respectively.
Conclusion
This study indicates preferential targeting and long circulating properties for cRGD-modified liposomes in vivo, which could be used as a potential targeted liposomal drug delivery system to treat human glioma.
Introduction
Liposomes have been extensively investigated as potential drug carriers for cancer chemotherapy. Drug-loaded liposomes typically have a prolonged circulation time in vivo, enhancing the efficacy of chemotherapy agents. However, their action can be limited by rapid clearance from the circulation via the reticuloendothelial system.Citation1–Citation3 Incorporation of a poly(ethylene glycol) (PEG) coating on liposomes can avoid this problem, in which case they are usually called “stealth liposomes” or “sterically stable liposomes” (SSL), with Doxil®/Caelyx® being the first stealth liposomal anticancer formulation on the market.Citation4 Stealth liposomes with a diameter of approximately 120 nm can accumulate passively in tumor tissues via the “leaky” vasculature surrounding tumors and the enhanced permeability and retention effect.Citation5,Citation6
Passive accumulation can be enhanced by active targeting of such PEGylated liposomes. Active targeting can be achieved by modification of sterically stable liposomes with various ligands, such as antibodies, folic acid, or transferrin.Citation7–Citation12 There are two main ways of modifying the liposomal membrane to display targeting ligands, ie, by direct coupling with the surface or with the distal ends of PEG-lipids. Citation7,Citation13,Citation14 Liposomes modified in such ways show targeting and long circulation times in vitro. However, MCC-465, a PEGylated liposomal formulation containing doxorubicin targeted with a F(ab’)2 fragment of human monoclonal antibody known as GAH, was the first targeted nanoparticle to enter clinical trials.Citation15
Among the various ligands, the integrin ligands have been widely studied.Citation16–Citation19 The most studied integrin ligands have been the Arg-Gly-Asp (RGD) motif and structurally related compounds.Citation20–Citation25 The RGD motif can be specifically recognized by integrins ανβ3 and ανβ5 which are overexpressed on many solid tumors and in tumor neovasculature, such as in human glioma.Citation26 Thus, modification of SSL with the RGD motif to achieve targeted drug delivery to tumors and the tumor neovasculature is a promising strategy in the treatment of glioma.
In the present study, by covalently coupling a cyclic RGD, cyclo(Arg-Gly-Asp-D-Phe-Cys) (cRGDfC, abbreviated as cRGD) to the liposomal surface, we constructed PEGylated liposomes with cRGD conjugated to the surface of the liposomal membrane and loaded with doxorubicin, a widely used anticancer drug. The cRGD-modified SSL as a liposomal drug delivery system was characterized as providing enhanced cellular delivery and an increased circulation time in vivo. Its targeted effect in U87MG cells in vitro along with its pharmacokinetic properties were evaluated in Sprague-Dawley rats.
Materials and methods
Materials
Hydrogenated soybean phosphatidylcholine (HSPC), N-(carbonyl-methoxypolyethylene glycol 2000)-1, 2-distearoyl-sn- glycero-3-phosphoethanolamine sodium salt (DSPE-PEG2000), and dipalmitoylphosphatidylethanolamine were purchased from Lipoid (Ludwigshafen, Germany). Cholesterol (CHOL) and Sephadex G-50/Sepharose CL-4B were obtained from Pharmacia Biotech (Piscataway, NJ). Maleimidobenzoylphosphatidylethanolamine (MBPE), the m-maleimidobenzoyl-N-hydroxysuccinimide derivative of dipalmitoylphosphatidylethanolamine, was prepared as previously described.Citation27,Citation28 Doxorubicin hydrochloride was gifted from Zhejiang Hisun Pharmaceutical Co, Ltd (Taizhou Zhejiang, China). The amino acids, resin, and reagents used in the synthesis of cRGD were purchased from GL Biochem (Shanghai, China). Trypsin, fetal bovine serum, and Dulbecco’s modified Eagle’s medium were purchased from Gibco-BRL Life Technologies (Carlsbad, CA). All the chemicals and solvents used in this study were of analytical or high-performance liquid chromatography (HPLC) grade.
Cell culture
A human glioma cell line, U87MG, was obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The cells were grown in Dulbecco’s modified Eagle’s medium in a fully humidified atmosphere supplemented with 10% fetal bovine serum, penicillin 100 U/mL, and streptomycin 100 U/mL at 37°C with 5% CO2. Based on cell density, the cells were subcultured regularly using trypsin-ethylenediamine tetra-acetic acid (EDTA, Gibco-BRL Life Technologies).
Synthesis of cRGD and MBPE
cRGD peptide (molecular weight 578.6) was synthesized by a Fmoc solid phase peptide synthesis method.Citation29,Citation30 The fully protected linear pentapeptide obtained, ie, HO-Gly-Asp(OtBu)-D-Phe-Cys(Trt)-Arg(Pbf)-H, was cyclized by diphenylphosphorylazide and deprotected by trifluoroacetic acid. The reaction product was then purified by preparative HPLC and lyophilized. cRGD was obtained with a purity > 95%, confirmed by analytical HPLC.
Preparation of liposomes
Liposomes were prepared by thin film hydration, followed by polycarbonate membrane extrusion, as described previously.Citation31,Citation32 Doxorubicin was remote-loaded into the liposomes by a transmembrane ammonium sulfate gradient with some modifications. The lipid composition of the nontargeted (DXRL-PEG) and cRGD-modified targeted liposomes (RGD-DXRL-PEG) were HSPC/CHOL/DSPE-PEG2000 (57:38:5 molar ratio) and HSPC/CHOL/MBPE/DSPE-PEG2000 (54.5:38:2.5:5 molar ratio), respectively. Briefly, a lipid membrane was prepared from a mixture of either HSPC/CHOL/DSPE-PEG2000 for DXRL-PEG or HSPC/CHOL/MBPE for RGD-DXRL-PEG. The lipid film was hydrated with 2 mL of 200 mM ammonium sulfate for 30 minutes at 60°C with occasional gentle mixing. The suspensions of lipids were then extruded 15 times through 400 nm, 200 nm, and 100 nm pore size polycarbonate membranes, using LF-1 Lipofast apparatus (Avestin, Ottawa, Canada) to form small unilamellar liposomes, respectively. The resulting liposome suspensions were chromatographed over a Sephadex G-50 packed column (1.6 × 20 cm) equilibrated with phosphate-buffered saline (pH 7.4) to establish a transmembrane NH4+ gradient. Liposomes, which were eluted in the void volume, were collected. Doxorubicin hydrochloride 4 mg dissolved in 1 mL of deionized water was mixed with the liposomes at a ratio of 1:15 (w/w) for doxorubicin-to-lipid, followed by incubation for 30 minutes at 60°C. The residual free doxorubicin in the liposomal suspensions was removed by size exclusion chromatography on a Sephadex G-50 column. For preparation of RGD-DXRL-PEG, 1 μmol of cRGD dissolved in phosphate-buffered saline (pH 7.4) was incubated with the liposome suspensions for 30 minutes at 37°C. Residual free cRGD in the liposomal suspensions was removed by size exclusion chromatography on a Sepharose CL-4B column. For conjugation of DSPE-PEG2000, the post-insertion approach for liposomes was adopted, as reported elsewhere.Citation33 In brief, following incubation with an aqueous micellar solution of DSPE-PEG2000 for 90 minutes at 60°C, the RGD-DXRL-PEG was prepared (). If necessary, the liposomes were further concentrated by ultrafiltration (molecular weight cutoff 10,000, Millipore, Billerica, MA). The mean diameters and zeta potentials of the two kinds of liposomes were determined by dynamic light scattering using a Nicomp 380 ZLS (Particle Sizing Systems, Santa Barbara, CA). The doxorubicin concentration in the liposomes was determined by measuring absorption at 480 nm on an ultraviolet-visible spectrometer (UV-1800, Shimadzu, Tokyo, Japan) following liposome lysis with 2% (v/v) Triton X-100. Doxorubicin encapsulation efficiency (EE), as assessed by running samples through a Sephadex G-50 column, was calculated by the equation:
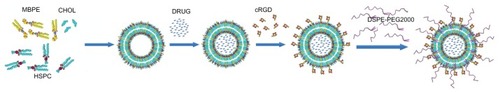
Figure 1 Schematic representation for preparation of RGD-DXRL-PEG.
Notes: The preformed liposomes were prepared by thin film hydration and polycarbonate membrane extrusion. Doxorubicin was loaded using a transmembrane ammonium sulfate gradient. cRGD was then covalently coupled to the liposomal surface via a chemical reaction between a thiol and maleimide group, followed by incubation with an aqueous micellar solution of DSPE-PEG2000.
Abbreviations: HSPC, hydrogenated soybean phosphatidylcholine; CHOL, cholesterol; MBPE, maleimidobenzoylphosphatidylethanolamine; DSPE-PEG2000, N-(carbonyl-methoxypolyethylene glycol 2000)-1, 2-distearoyl-sn-glycero-3-phosphoethanolamine sodium salt; DRUG, doxorubicin; DXRL-PEG, DXR-loaded PEGylated liposomes; RGD-DXRL-PEG, cRGD-modified DXRL-PEG.
HPLC analysis of cRGD coupling with liposomes
The extent of coupling of cRGD with the liposomes was determined by an HPLC-ultraviolet method previously described by Nallamothu et al, with some modification.Citation34 In brief, a C18 column (SinoChrom ODS-AP, 250 mm × 4.6 mm, Dalian Elite, Dalian, China) was used with a mobile phase consisting of acetonitrile/water/trifluoroacetic acid (20:80:0.1, v/v/v). The flow rate was 1 mL per minute and the column temperature was 30°C. The free cRGD was determined by measuring absorbance at 214 nm.
Flow cytometry analysis
The association of doxorubicin with U87MG cells was determined by flow cytometry. U87MG cells grown in a monolayer were suspended by brief treatment with trypsin-EDTA. Aliquots of U87MG cell suspensions were incubated with doxorubicin solution, DXRL-PEG, or RGD-DXRL-PEG at a doxorubicin concentration of 20 μg/mL diluted in culture medium for 2 hours at 37°C. In the competitive binding experiment, excess free cRGD (3 mM) in culture medium was preincubated with U87MG cells for 20 minutes, followed by continued coincubation with RGD-DXRL-PEG for another 2 hours at 37°C. The cells were then washed three times with cold phosphate-buffered saline (pH 7.4). The cells were then analyzed by flow cytometry using a FACSCalibur system (Becton Dickinson, San Jose, CA). Cell-associated doxorubicin was excited with an argon laser (488 nm) and fluorescence was detected at 550 nm. A minimum of 8000 gated events were collected and analyzed using the Cell Quest software supplied by the manufacturer.
Pharmacokinetic studies
Male Sprague-Dawley rats (weighting 200–250 g) were purchased from Shanghai SLAC Laboratory Animal Co, Ltd (Shanghai, China). The protocols for animal use and care were approved by the Institutional Animal Care and Use Committee of Shanghai Institute of Pharmaceutical Industry. The rats were housed at 25°C ± 2°C with free access to water and standard laboratory food. Rats (3–5 per group) were given intravenous injections of doxorubicin solution, RGD-DXRL-PEG, or DXRL-PEG at a doxorubicin dose of 5 mg/kg via the tail vein. Blood was sampled from the retro-orbital plexus at various predetermined time points, and blood samples (500 μL) were collected in heparinized tubes, followed by centrifugation at 3000 × g for 10 minutes to obtain plasma. For 300 μL plasma samples, 10 μL of daunorubicin (internal standard, 10 μg/mL) and 2 mL of chloroform/methanol (4:1, v/v) were added. After vortex mixing for 2 minutes, the mixture was centrifuged at 10,000 g for 5 minutes, and the subnatant was then transferred and evaporated to dryness under a stream of nitrogen at 40°C. The residue was dissolved in 200 μL of methanol, and 20 μL supernatant was injected into the HPLC system according to the method described by Shinozawa et al.Citation35 The pharmacokinetic parameters were calculated using Kinetica® software, version 5.0, including area under the curve (AUC0–t), plasma half-lives for the elimination phase (t1/2β), total body clearance (CL), and mean residence time.
Statistical analysis
Results were expressed as the mean ± standard deviation from at least three separate samples analyzed unless otherwise indicated. Graphing and statistical analysis was performed using SigmaPlot 12.0 (SPSS Inc, Chicago, IL). The cellular uptake efficiency and pharmacokinetic parameters for each liposomal formulation were compared using one-way analysis of variance, followed by Tukey’s test for post hoc comparison in order to ascertain differences between groups. The difference was considered to be statistically significant at a level of P < 0.05.
Results
Preparation and characterization of liposomal formulations
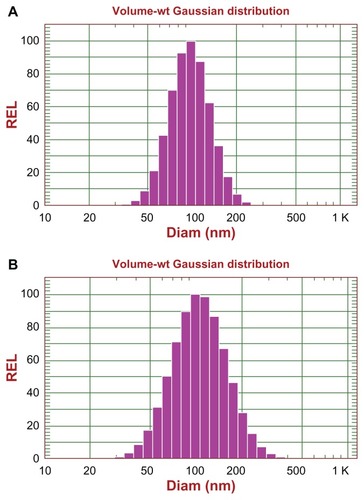
The RGD-DXRL-PEG was prepared by covalent coupling of cRGD onto the liposomal surface as described earlier. Nontargeted PEGylated liposomes, ie, DXRL-PEG, were prepared according to the procedure used for Doxil®.Citation36 For both kinds of liposomes, up to 2.0 mg/mL of liposomal doxorubicin was achieved after concentration by ultrafiltration, with more than 98% entrapment efficiency. The mean diameter of the two types of liposomes was 100.7 nm for DXRL-PEG and 114.1 nm for RGD-DXRL-PEG, as shown in . The zeta potentials for DXRL-PEG and RGD-DXRL-PEG were −20.06 ± 5.06 mV and −24.85 ± 8.55 mV, respectively.
Figure 2 Size distribution of DXR-encapsulating liposomes determined by dynamic light scattering using a NICOMP 380 ZLS: size distribution of DXRL-PEG (A), and RGD-DXRL-PEG (B).
Abbreviations: DXR, doxorubicin; DXRL-PEG, DXR-loaded PEGylated liposomes; RGD-DXRL-PEG, cRGD-modified DXRL-PEG.
HPLC determination of cRGD coupling to liposomes
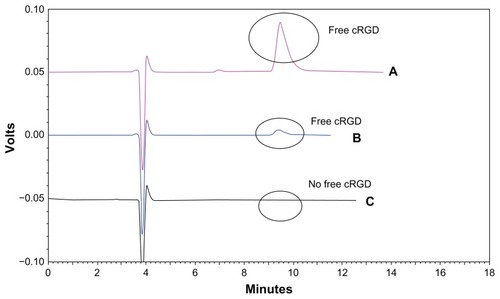
Coupling of cRGD to the liposomal surface was based on the chemical reaction between the maleimide and thiol groups. The coupling efficiency of the cRGD peptide to the maleimide groups on the liposomal surface was ascertained indirectly by determining the noncoupled cRGD fraction with an HPLC-ultraviolet method. cRGD dissolved in phosphate-buffered saline (pH 7.4) was eluted at about 10 minutes, as shown in . This peak was monitored for estimation of free cRGD in the final liposome formulations. The liposomal formulation sample was passed over a Sepharose CL-4B column following the coupling step, and then the free cRGD was collected and assayed. shows that there was still free cRGD unreacted with the maleimide group after excess free cRGD (1.25 μmol) was mixed with the liposome suspension. In , there was no significant peak around 10 minutes, indicating that there was hardly any free cRGD left unreacted in the formulation. Therefore, more than 99% of the cRGD peptide added to the formulation had been coupled with the liposomes. From the amount of cRGD used, it was calculated that about 2200 cRGD peptides might be present on the surface of each liposome, based on the assumption that 144,000 phospholipid molecules form one liposome vesicle of 120 nm.Citation37
Figure 3 High-performance liquid chromatography of cRGD coupling with the liposomes. (A) Free cRGD (500 μg/mL) eluted with a retention time of approximately 10 minutes. (B) Excess free cRGD after coupling with the liposomes gave the peak for free cRGD. (C) The liposome sample following the coupling step showed no significant peak for free cRGD at around 10 minutes.
Abbreviations: DXR, doxorubicin; cRGD, cyclo(Arg-Gly-Asp-D-Phe-Cys).
Cellular uptake of doxorubicin
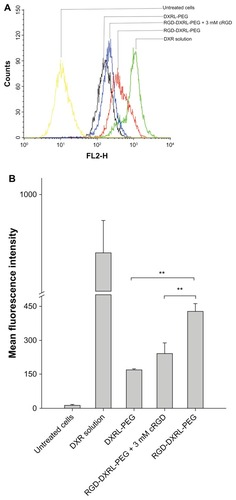
Flow cytometry was used to determine the total doxorubicin uptake by U87MG cells. and B show the cellular uptake of doxorubicin after U87MG cells were incubated with the different doxorubicin formulations for 2 hours at 37°C. A low level of background fluorescence was demonstrated. The cellular doxorubicin uptake for RGD-DXRL-PEG was about 2.5-fold higher than that for DXRL-PEG. The doxorubicin solution showed the highest cellular uptake of doxorubicin. The mean fluorescence intensities for the doxorubicin solution were approximately 5.8-fold and 2.3-fold higher than those for DXRL-PEG and RGD-DXRL-PEG, respectively. In addition, the mean fluorescence intensity of RGD-DXRL-PEG showed an intensity decrease of about 44% after incubation with excess free cRGD.
Figure 4 (A) Flow cytometry charts showing the cellular uptake of DXRL-PEG, RGD-DXRL-PEG, RGD-DXRL-PEG + 3 mM cRGD, and DXR solution by U87MG cells. (B) Mean fluorescence intensity as determined by flow cytometry experiments.
Notes: DXR solution served as a positive control while untreated cells were used as a negative control. U87MG cells were incubated with either free DXR, DXRL-PEG, or RGD-DXRL-PEG for 2 hours at 37°C. In the competitive binding experiment, excess free cRGD (3 mM) in culture medium was preincubated with U87MG cells for 20 minutes, followed by continued coincubation with RGD-DXRL-PEG for another 2 hours at 37°C. The cells were then washed with cold phosphate-buffered saline (pH 7.4) and cell-associated DXR was evaluated using flow cytometry. Data represent the mean ± standard deviation (n = 3). **P < 0.001.
Abbreviations: DXR, doxorubicin; DXRL-PEG, DXR-loaded PEGylated liposomes; RGD-DXRL-PEG, cRGD-modified DXRL-PEG.
Pharmacokinetic properties
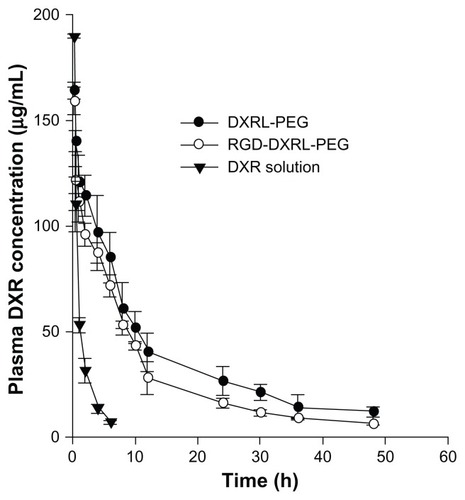
The pharmacokinetic profiles of doxorubicin in plasma after intravenous tail injection of a 5 mg/kg dose of doxorubicin, RGD-DXRL-PEG, and DXRL-PEG are shown in . The plasma concentration profiles of the three formulations were all best fitted by the two-compartment model using Kinetica software, and the pharmacokinetic parameters are summarized in . Both types of liposomal doxorubicin formulation showed a much longer systemic circulation time compared with the solution. The mean residence time of either type of liposomal doxorubicin formulation was significantly higher than that of the solution. Rats treated with RGD-DXRL-PEG showed a slightly lower plasma concentration than those treated with DXRL-PEG.
Table 1 Pharmacokinetic parameters of doxorubicin formulations in Sprague-Dawley rats following intravenous bolus administration
Figure 5 Plasma concentration versus time curves for DXR formulations in rats.
Notes: DXR formulations were administered via tail vein injection at a dose of 5 mg/kg. Each datum point is the average of 3–5 animals and the error bar is the standard deviation.
Abbreviations: DXR, doxorubicin; DXRL-PEG, DXR-loaded PEGylated liposomes; RGD-DXRL-PEG, cRGD-modified DXRL-PEG.
Discussion
In this study, we developed a simple and efficient way of attaching an cRGD peptide to a liposomal membrane. The method is different from that used in most recent studies, attaching peptide ligand to the distal ends of PEG-lipids.Citation9,Citation14 MCC-465, a PEGylated liposomal formulation containing doxorubicin targeted with an F(ab’)2 fragment of the human monoclonal antibody (GAH), was the first targeted nanoparticle to enter clinical trials.Citation15 For MCC-465, GAH was covalently coupled with the liposomal membrane surface. In consideration of feasibility, we attached cRGD to MBPE on the liposome surface.
Covalent coupling of thiolated cRGD to the maleimide terminus of MBPE was exploited to prepare RGD-DXRL-PEG. We chose that method because the reaction between the thiol and maleimide group has been reported to be one of the most efficient reactions in bioconjugate chemistry.Citation38
Due to the steric hindrance posed by PEGylation on the liposomes, the coupling reaction might be hindered if PEGylation was performed before the coupling of the cRGD to the liposomal membrane surface. Therefore, DSPE-PEG2000 was inserted into the prepared liposomes using the post-insertion procedure after coupling of cRGD, as previously reported.Citation39 In addition, in consideration of the high phase transition temperature of HSPC (55°C) in our study, loading of doxorubicin was performed before covalent coupling of cRGD with the preformed liposomes to avoid degradation of cRGD at high temperature (60°C). The procedure for loading doxorubicin in this study was the same as that used in most studies.Citation39–Citation41
In the cell uptake test, cRGD-modified liposomes (RGD-DXRL-PEG) showed significantly higher uptake by U87MG cells in vitro compared with nontargeted control liposomes (DXRL-PEG). The uptake could be blocked by excess free cRGD, suggesting that the observed uptake of RGD-DXRL-PEG was cRGD-mediated. In addition, the integrin receptors (αvβ3 and αvβ5) are overexpressed in U87 MG cells,Citation26 so might be closely involved in uptake by these cells, but this needs further investigation.
Pharmacokinetic studies of liposomal doxorubicin formulations demonstrated significantly longer systemic circulation times compared with doxorubicin solution. This result implies greater in vivo stability of both liposomal formulations following intravenous administration compared with doxorubicin solution. Further, it suggests that PEG chains were attached to the surface of liposomes by the post-insertion procedure. The area under the curve for either type of liposomal formulation was much greater than that for the doxorubicin solution. However, the cRGD-targeted formulation had faster clearance than the nontargeted liposomes. This is consistent with studies of RGD-SSL-DXR and SSL-DXR, as previous reported,Citation40 and this phenomenon is shared by other ligand-modified liposomes.Citation42,Citation43 As the published studies have explained, the liver and spleen might be responsible for the rapid clearance of ligand-modified liposomes.Citation44 Further studies are needed to define the impact of clearance on antitumor activity in vivo in the future.
Conclusion
In summary, cRGD was covalently coupled to the liposome surface via a chemical reaction between thiol and a maleimide functional group. Doxorubicin was loaded into the liposomes using an ammonium sulfate gradient method, followed by insertion of DSPE-PEG2000 into the membrane of the liposomes using the post-insertion procedure. The prepared RGD-DXRL-PEG liposomes had a negative charge (−24.85 ± 8.55 mV), a mean diameter of 114.1 nm, and an encapsulation efficiency > 98%. The flow cytometry test result showed a higher cell binding ability for RGD-DXRL-PEG than for DXRL-PEG. Furthermore, a competitive binding experiment indicated that integrin receptor-mediated endocytosis had been involved in the cell uptake. A pharmacokinetic study in Sprague-Dawley rats confirmed long circulating properties for the cRGD-modified liposomes. The results of the present study indicate that RGD-DXRL-PEG could be used as a potential targeted liposomal drug delivery system to treat human glioma.
Acknowledgments
The authors are grateful to the Shanghai Municipal Commission for Science and Technology, China (09ZR1430900) and the Shanghai Rising Star Program, China (10QB1404100) for funding this work. The authors also thank Lin Xiao and Mingyu Liu for assistance with the cell culture.
Disclosure
The authors report no conflicts of interest in this work.
References
- BanerjeeRLiposomes: applications in medicineJ Biomater Appl200116132111475357
- MamotCDrummondDCHongKKirpotinDBParkJWLiposome-based approaches to overcome anticancer drug resistanceDrug Resist Updat20036527127914643297
- MoghimiSPatelHSerum-mediated recognition of liposomes by phagocytic cells of the reticuloendothelial system – the concept of tissue specificityAdv Drug Deliv Rev1998321–2456010837635
- ImmordinoMLDosioFCattelLStealth liposomes: review of the basic science, rationale, and clinical applications, existing and potentialInt J Nanomedicine20061329731517717971
- YuanFLeunigMHuangSKBerkDAPapahadjopoulosDJainRKMicrovascular permeability and interstitial penetration of sterically stabilized (stealth) liposomes in a human tumor xenograftCancer Res19945413335233568012948
- MaedaHThe enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targetingAdv Enzyme Regul200141118920711384745
- SuzukiSWatanabeSMasukoTHashimotoYPreparation of long-circulating immunoliposomes containing adriamycin by a novel method to coat immunoliposomes with poly(ethylene glycol)Biochim Biophys Acta1995124519167654771
- MoreiraJNGasparRAllenTMTargeting stealth liposomes in a murine model of human small cell lung cancerBiochim Biophys Acta20011515216717611718672
- GabizonAShmeedaHHorowitzATZalipskySTumor cell targeting of liposome-entrapped drugs with phospholipid-anchored folic acid-PEG conjugatesAdv Drug Deliv Rev20045681177119215094214
- DeryckeASLKamuhabwaAGijsensATransferrin-conjugated liposome targeting of photosensitizer AlPcS4 to rat bladder carcinoma cellsJ Natl Cancer Inst200496211620163015523091
- LukyanovANElbayoumiTAChakilamARTorchilinVPTumor-targeted liposomes: doxorubicin-loaded long-circulating liposomes modified with anti-cancer antibodyJ Control Release2004100113514415491817
- HamaguchiTMatsumuraYNakanishiYAntitumor effect of MCC-465, pegylated liposomal doxorubicin tagged with newly developed monoclonal antibody GAH, in colorectal cancer xenograftsCancer Sci200495760861315245599
- HosokawaSTagawaTNikiHHirakawaYNohgaKNagaikeKEfficacy of immunoliposomes on cancer models in a cell-surface-antigen-density- dependent mannerBr J Cancer20038981545155114562030
- MaruyamaKTakizawaTYudaTKennelSJHuangLIwatsuruMTargetability of novel immunoliposomes modified with amphipathic poly (ethylene glycol)s conjugated at their distal terminals to monoclonal antibodiesBiochim Biophys Acta19951234174807880861
- MatsumuraYGotohMMuroKPhase I and pharmacokinetic study of MCC-465, a doxorubicin (DXR) encapsulated in PEG immunoliposome, in patients with metastatic stomach cancerAnn Oncol200415351752514998859
- EmsleyJKnightCGFarndaleRWBarnesMJLiddingtonRCStructural basis of collagen recognition by integrin α2β1Cell20001011475610778855
- XiongJPStehleTZhangRCrystal structure of the extracellular segment of integrin alpha Vbeta 3 in complex with an Arg-Gly-Asp ligandScience2002296556515115511884718
- HumphriesJDByronAHumphriesMJIntegrin ligands at a glanceJ Cell Sci2006119193901390316988024
- ShimaokaMXiaoTLiuJHStructures of the αL I domain and its complex with ICAM-1 reveal a shape-shifting pathway for integrin regulationCell200311219911112526797
- KeenanRMCallahanJFSamanenJMConformational preferences in a benzodiazepine series of potent nonpeptide fibrinogen receptor antagonistsJ Med Chem199942454555910052962
- FisherMJGunnBHarmsCSNon-peptide RGD surrogates which mimic a Gly-Asp β-turn: potent antagonists of platelet glycoprotein IIb-IIIaJ Med Chem19974013208521019207949
- ZablockiJAMiyanoMGarlandRBPotent in vitro and in vivo inhibitors of platelet aggregation based upon the Arg-Gly-Asp-Phe sequence of fibrinogen. A proposal on the nature of the binding interaction between the Arg-guanidine of RGDX mimetics and the platelet GP IIb-IIIa receptorJ Med Chem19933613181118198515420
- EgbertsonMSChangCTCDugganMENon-peptide fibrinogen receptor antagonists. 2. Optimization of a tyrosine template as a mimic for Arg-Gly-AspJ Med Chem19943716253725518057299
- ZablockiJARicoJGGarlandRBPotent in vitro and in vivo inhibitors of platelet aggregation based upon the Arg-Gly-Asp sequence of fibrinogen. (aminobenzamidino) succinyl (ABAS) series of orally active fibrinogen receptor antagonistsJ Med Chem19953813237823947608903
- HutchinsonJHCookJJBrashearKMNon-peptide glycoprotein IIb/IIIa antagonists. II. Design and in vivo evaluation of 3, 4-dihydro-1 (1H)-isoquinolinone-based antagonists and ethyl ester prodrugsJ Med Chem19963923458345918917647
- RuoslahtiERGD and other recognition sequences for integrinsAnnu Rev Cell Dev Biol19961216977158970741
- KimCKLimSJLiposome immunoassay (LIA) with antigen-coupled liposomes containing alkaline phosphataseJ Immunol Methods19931591–21011068445242
- KimCKChoiYJSoo-JeongLLeeMGLeeSHHwangSJLymph node targeting and pharmacokinetics of [3H] methotrexate-encapsulated neutral large unilamellar vesicles and immunoliposomesInt J Pharm1993981–3918
- RinkHSolid-phase synthesis of protected peptide fragments using a trialkoxy-diphenyl-methylester resinTetrahedron Lett1987283337873790
- BaranyGKneib-CordonierNMullenDGSolid-phase peptide synthesis: a silver anniversary reportInt J Pept Protein Res19873067057393326854
- HaranGCohenRBarLKBarenholzYTransmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak basesBiochim Biophys Acta1993115122012158373796
- BolotinEMCohenRBarLKAmmonium sulfate gradients for efficient and stable remote loading of amphipathic weak bases into liposomes and ligandoliposomesJ Liposome Res199441455479
- UsterPSAllenTMDanielBEMendezCJNewmanMSZhuGZInsertion of poly (ethylene glycol) derivatized phospholipid into preformed liposomes results in prolonged in vivo circulation timeFEBS Lett19963862–32432468647291
- NallamothuRWoodGCPattilloCBA tumor vasculature targeted liposome delivery system for combretastatin A4: design, characterization, and in vitro evaluationAAPS Pharm Sci Tech200672716
- ShinozawaSMimakiYArakiYOdaTDetermination of the concentration of adriamycin and its metabolites in the serum and tissues of Ehrlich carcinoma-bearing mice by high-performance liquid chromatographyJ Chromatogr198019634634697430305
- VaageJMayhewELasicDMartinFTherapy of primary and metastatic mouse mammary carcinomas with doxorubicin encapsulated in long circulating liposomesInt J Cancer19925169429481639542
- MaurerNFenskeDBCullisPRDevelopments in liposomal drug delivery systemsExpert Opin Biol Ther20011692394711728226
- HansenCBKaoGYMoaseEHZalipskySAllenTMAttachment of antibodies to sterically stabilized liposomes: evaluation, comparison and optimization of coupling proceduresBiochim Biophys Acta1995123921331447488618
- MoreiraJNIshidaTGasparRAllenTMUse of the post-insertion technique to insert peptide ligands into pre-formed stealth liposomes with retention of binding activity and cytotoxicityPharm Res200219326526911934232
- XiongXBHuangYLuWEnhanced intracellular delivery and improved antitumor efficacy of doxorubicin by sterically stabilized liposomes modified with a synthetic RGD mimeticJ Control Release2005107226227516125816
- XiangGWuJLuYLiuZLeeRJSynthesis and evaluation of a novel ligand for folate-mediated targeting liposomesInt J Pharm20083561293618258394
- IinumaHMaruyamaKOkinagaKIntracellular targeting therapy of cisplatin-encapsulated transferrin-polyethylene glycol liposome on peritoneal dissemination of gastric cancerInt J Cancer200299113013711948504
- TurkMJWatersDJLowPSFolate-conjugated liposomes preferentially target macrophages associated with ovarian carcinomaCancer Lett2004213216517215327831
- EmanuelNKedarEBolotinEMSmorodinskyNIBarenholzYTargeted delivery of doxorubicin via sterically stabilized immunoliposomes: pharmacokinetics and biodistribution in tumor-bearing micePharm Res19961368618688792423