 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Neuroinflammation, characterized by the accumulation of activated microglia and reactive astrocytes, is believed to modulate the development and/or progression of Alzheimer’s disease (AD). Epidemiological studies suggesting that nonsteroidal anti-inflammatory drugs decrease the risk of developing AD have encouraged further studies elucidating the role of inflammation in AD. Nanoparticles have become an important focus of neurotherapeutic research because they are an especially effective form of drug delivery. Here, we investigate the potential protective effect of indomethacin-loaded lipid-core nanocapsules (IndOH-LNCs) against cell damage and neuroinflammation induced by amyloid beta (Aβ)1-42 in AD models. Our results show that IndOH-LNCs attenuated Aβ-induced cell death and were able to block the neuroinflammation triggered by Aβ1-42 in organotypic hippocampal cultures. Additionally, IndOH-LNC treatment was able to increase interleukin-10 release and decrease glial activation and c-jun N-terminal kinase phosphorylation. As a model of Aβ-induced neurotoxicity in vivo, animals received a single intracerebroventricular injection of Aβ1-42 (1 nmol/site), and 1 day after Aβ1-42 infusion, they were administered either free IndOH or IndOH-LNCs (1 mg/kg, intraperitoneally) for 14 days. Only the treatment with IndOH-LNCs significantly attenuated the impairment of this behavior triggered by intracerebroventricular injection of Aβ1-42. Further, treatment with IndOH-LNCs was able to block the decreased synaptophysin levels induced by Aβ1-42 and suppress glial and microglial activation. These findings might be explained by the increase of IndOH concentration in brain tissue attained using drug-loaded lipid-core NCs. All these findings support the idea that blockage of neuroinflammation triggered by Aβ is involved in the neuroprotective effects of IndOH-LNCs. These data provide strong evidence that IndOH-LNC treatment may represent a promising approach for treating AD.
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder pathologically characterized by deposits of amyloid beta (Aβ) peptide in senile plaques, intracellular neurofibrillary tangles comprised of hyperphosphorylated tau, progressive synaptic dysfunction, and (much later) neuronal death, especially in the hippocampus. Extensive research in the last decade has revealed that most chronic illnesses, including neurological diseases, exhibit dysregulation of multiple cell-signaling pathways that have been linked to inflammation. It is now widely recognized that neuroinflammation is a prominent feature of AD brain tissue, with inflammatory responses playing a significant role in modulating disease progression.Citation1,Citation2
The rise of neuroinflammation-related hypotheses regarding AD began after initial epidemiological observations combined with direct observational evidence from postmortem brains suggested an association between inflammation and AD pathology. More recently, mechanisms underlying this link have been identified by experiments showing that brain tissue can flexibly alter paracrine signaling using autonomously produced and regulated inflammatory molecules.Citation3 The effects of inflammatory responses are multifaceted, with both detrimental actions that promote neurodegeneration as well as protective actions that promote neuronal survival and tissue repair. Prolonged and widespread activation of microglia and astrocytes are apparent in AD brain tissue. Importantly, the severity of glial activation correlates with the extent of brain atrophyCitation4 and cognitive decline.Citation5 The role of microglia in the development and/or progression of AD is somewhat controversial. Phagocytosis of Aβ by microglia is believed to be a protective mechanism.Citation6 However, neuronal loss also results from increased proinflammatory cytokine production by microglia.Citation7 In addition to the influence of microglia, astrocytes could potentially affect Aβ-induced neurotoxicity.Citation8 Both astrocytes and microglia release a myriad of pro- and anti-inflammatory cytokines that may result in neuronal damage, including interleukins (ILs), interferons, and tumor necrosis factors (TNFs), as well as chemokines, prostaglandins, leukotrienes, thromboxanes, coagulation factors, complement factors, proteases, and reactive oxygen species.Citation9,Citation10 Receptor binding of cytokines stimulate a variety of intracellular signaling pathways that have been implicated in AD, including activation of protein kinase C, c-jun N-terminal kinase (JNK), p38 mitogen-activated protein kinase, PI3 kinase, extracellular signaling-related kinase, caspase-1, and caspase-3.Citation11
Despite progress in symptomatic therapy for AD, effective therapeutic approaches that interfere with the neurodegenerative processes underlying AD are still unavailable. Epidemiological studies have documented a reduced prevalence of AD among users of nonsteroidal anti-inflammatory drugs (NSAIDs).Citation12,Citation13 It has been reported that NSAIDs blocking cyclooxygenase-2 ameliorate Aβ-induced neuronal dysfunction, and that this effect may be independent of an anti-inflammatory process or a reduction in the levels of Aβ1-42.Citation14 In addition, NSAID treatment in AD might also suppress cellular inflammatory reactions that are associated with Aβ.Citation1
Netland and coworkers showed that indomethacin (IndOH) was able to reduce activation of microglia surrounding the Aβ deposits after Aβ infusion into the lateral ventricle of rats.Citation15 However, the in vivo brain distribution of IndOH is severely limited by plasma protein binding, which reduces the plasma-free fraction in the blood circulation by >90%. This significantly reduces the amount of drug that can cross the blood–brain barrier (BBB).Citation16 Moreover, the BBB is the most serious obstacle limiting the development of new drugs for the central nervous system. During the past decade, numerous attempts have focused on this pivotal problem by designing different strategies to aid drug passage across the BBB. Among these, nanotechnology-based approaches have gained significant momentum, since some of them can effectively transport drug across the BBB. One such strategy is the use of nanoparticles for controlled drug delivery and release,Citation17,Citation18 such as the use of polymeric nanocapsules described previously by our group.Citation19–Citation21
The present study was designed to investigate the potential protective effect of IndOH-loaded lipid-core nanocapsules (IndOH-LNCs) against cell damage and neuroinflammation induced by an Aβ1-42 model of AD. Numerous animal models have been used to evaluate the role of inflammation in the course of AD. Here, we used organotypic hippocampal culture and intracerebroventricular (ICV) injection of Aβ1-42 in rats. Our results provide strong evidence that IndOH-LNC treatment reduces the neuroinflammation and the subsequent cell death triggered by Aβ1-42. Additionally, we show that IndOH-LNC treatment is able to counteract the behavioral and cell-signaling impairments triggered by ICV administration of Aβ1-42. One of the most powerful pieces of evidence from this report is the significant difference in the amount of IndOH in cerebral tissue achieved by using drug-loaded LNCs.
Materials and methods
Preparation of lipid-core nanocapsules
LNC suspensions were prepared by interfacial deposition of polymer.Citation22 At 40°C, IndOH (0.010 g), poly(ɛ-caprolactone) (0.100 g), capric/caprylic triglycerides (0.33 mL), and sorbitan monostearate (0.077 g) were dissolved in acetone (27 mL) at 40°C. In a separate flask, polysorbate 80 (0.077 g) was added to 53 mL of water. The organic solution was injected into the aqueous phase under magnetic stirring at room temperature. After 10 minutes, the acetone was eliminated and the suspensions were concentrated under reduced pressure. The final volume was adjusted to 10 mL for a drug concentration of 1 mg/mL. The control formulation (drug-unloaded NCs) was prepared, as described above without adding IndOH.
Characterization of lipid-core nanocapsules
After preparation, the pH values of the NC suspensions were determined using a potentiometer (B-474; Micronal, São Paulo, Brazil). The particle size, polydispersity, index, and zeta potential of the suspensions were determined using a Zetasizer nano-ZS ZEN 3600 model (Malvern Instruments, Malvern, UK). The samples were then diluted with prefiltered water (MilliQ; particle size and polydispersity index; Millipore, Billerica, MA) or 10 mM NaCl aqueous solution (zeta potential). The measurements were made in triplicate to assure accuracy. The total concentrations of IndOH in the formulations were measured by reverse phase high-performance liquid chromatography (HPLC; PerkinElmer [Waltham, MA] S-200, with injector S-200, detector UV-vis, a guard-column and a Lichrospher 100 RP-18 column of 250 mm, 4 mm, and 5 μm; Merck, Darmstadt, Germany). The mobile phase consisted of acetonitrile/water (70:30, v/v) adjusted to apparent pH 5.0 ± 0.5 with 10% (v/v) acetic acid. Each suspension (100 μL) was treated with acetonitrile (10 mL), and the solution was filtered (MilliQ 0.45 μm) and injected (20 μL). The HPLC method used here has been previously validated by other investigators.Citation23 Linear calibration curves for IndOH were obtained in the range of 1.00–25.00 μg/mL, presenting correlation coefficients higher than 0.9992. The encapsulation efficiency was determined by an ultrafiltration–centrifugation technique (Ultrafree-MC 10,000 MW; Millipore), at 15,300 × g for 10 minutes. The associated IndOH within the NCs was calculated from the difference between the total and the free drug concentrations determined in the NC suspension and in the ultrafiltrate, respectively.
Amyloid peptide preparation
The Aβ1-42 peptide or the nonamyloidogenic scramble of Aβ was dissolved in sterilized bidistilled water with 0.1% ammonium hydroxide at a concentration of 1 mg/mL and aliquots were stored at −20°C. The Aβ peptides were aggregated by incubation at 37°C for 72 hours before in vitro or in vivo use.
In vitro Aβ neurotoxicity
Organotypic hippocampal culture
Hippocampal slice cultures were prepared from 6- to 8-day-old male Wistar rats obtained from in-house breeding colonies at the Departamento de Bioquímica, Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil. All procedures used in the present study followed the Guide for the Care and Use of Laboratory Animals. NIH Publication No. 85–23. Revised 1985Citation24 and were approved by the local Ethics Committee on the Use of Animals (protocol number 2007977). All efforts were made to minimize the number of animals used and their suffering. Slice cultures were prepared as interphase cultures according to a protocol of Stoppini et alCitation25 with some modifications.Citation26,Citation27 Briefly, the animals were killed by decapitation, the brains were removed, the hippocampi were isolated, and transverse hippocampal slices (400 μm thickness) were prepared by using a McIlwain tissue chopper (Mickle Laboratory Engineering, Guildford, UK). The slices were placed on membrane inserts (0.4-μm Millicell-CM culture plate inserts; Millipore) in 6-well plates. Each well contained 1 mL of culture medium consisting of 50% minimum essential medium, 25% Hank’s balanced salt solution, 25% horse serum, supplemented with (mM, final concentration): glucose 36, HEPES 25, and NaHCO3 4; Fungizone (Life Technologies, Carlsbad, CA) 1%, and gentamicin 0.100 mg/mL, pH 7.3. Organotypic cultures were maintained in a humidified incubator gasified with a 5% CO2/95% O2 atmosphere at 37°C for 4 weeks. Culture medium was changed three times per week.
Indomethacin treatment and Aβ exposure of cultures
To induce Aβ neurotoxicity, on the 28th day in vitro, either Aβ1-42 or Aβ42-1 (2 μM – final concentration) were directly added to the medium and incubated for 48 hours. Control slices received no Aβ peptides. To evaluate whether the IndOH-LNCs would be protective against toxicity triggered by Aβ1-42, the cultures were treated with IndOH-LNCs (50 or 100 μM) for 48 hours simultaneously during Aβ exposure.
Quantification of cellular death
Cell damage was assessed by fluorescent-image analysis of propidium iodide (PI) uptake. Forty-six hours after the Aβ peptide exposure, organotypic cultures were stained with PI (5 μM) for 2 hours. PI fluorescence was observed by an inverted fluorescence microscope (Eclipse TE 300; Nikon, Tokyo, Japan). Images were captured using a charge-coupled-device camera (DXM1200C; Nikon Instruments, Melville, NY), stored and subsequently analyzed by using image-analysis software (Scion Image software; National Institutes of Health, Bethesda, MA). The amount of PI fluorescence was determined densitometrically after transforming the red values into gray values. For quantification of neural damage, the percentage of area exhibiting PI fluorescence above background level was calculated in relation to the total area of each slice. PI intensity (indicating cell death) was expressed as a percentage of cell damage:
where Fd is the PI uptake fluorescence of dead area of hippocampal slices and F0 is the total area of each hippocampal slice.
Determination of cytokine levels
After 48 hours of Aβ1-42 (2 μM) exposure, as well as IndOH-LNC treatment, the culture medium was collected, rapidly frozen, and stored at −20°C for later measurement of TNF-α, IL-6, and IL-10 levels using specific enzyme-linked immunosorbent assay kits (R&D Systems, Minneapolis, MN) in accordance with the manufacturer’s recommendations. Standard curves were obtained using recombinant rat TNF-α, IL-6, and IL-10. The values of cytokines were expressed as pg/mL medium.
In vivo Aβ neurotoxicity
Animals
Male adult Wistar rats (280–330 g) were obtained from inhouse breeding colonies at the Departamento de Bioquímica, Universidade Federal do Rio Grande do Sul. Animals were housed in cages under optimum light conditions (12-hour light–dark cycle), temperature (22°C ± 1°C), and humidity (50%–60%), with food and water provided ad libitum. All procedures used in the present study followed the “Principles of Laboratory Animal Care” from National Institutes of Health publication no 85-23 and were approved by the local Ethics Committee on the Use of Animals (protocol number 2007977). All efforts were made to minimize the number of animals and their suffering.
Surgical procedure and amyloid injection
Animals were anesthetized with Equithesin (3.5 mL/kg intraperitoneally [IP]) and placed in a stereotaxic frame. A middle sagittal incision was made in the scalp and was sterilized using standard procedures. Bilateral holes were drilled in the skull using a dental drill over the lateral ventricles. Injection coordinates were chosen according to the atlas of Paxinos and Watson:Citation28 0.8 mm posterior to bregma; 1.5 mm lateral to the sagittal suture; 3.5 mm beneath the surface of brain. Rats received a single infusion of 5 μL into each lateral ventricle of Aβ1-42 or Aβ42-1 (total of 2 nmol in 10 μL). Control animals received bilateral ICV injections of equal volumes of bidistilled water with 0.1% ammonium hydroxide. Microinjections were performed using a 10-μL Hamilton syringe fitted with a 26-gauge needle. All infusions were made at a rate of 1 μL/minute over a period of 5 minutes. At the end of infusion, the needle was left in place for an additional 3–5 minutes before being slowly withdrawn to allow diffusion from the tip and prevent reflux of the solution. After the injection, the scalp was sutured and the animals were allowed to recover from the anesthesia on a heating pad to maintain body temperature at 37.5°C ± 0.5°C. The animals were submitted to behavioral tasks 2 weeks after Aβ injection.
Drug administration and experimental design
One day after the surgical procedure, the animals were randomly divided into seven groups. Control animals infused ICV with water plus 0.1% ammonium hydroxide (Aβ-vehicle) were split into the following three groups: (1) untreated (control group), (2) treated with free IndOH (IndOH group), and (3) treated with IndOH-LNCs. Animals infused ICV with Aβ1-42 were divided into the following groups: (4) untreated (Aβ group), (5) treated with unloaded LNCs (Aβ LNC group) (6) treated with free IndOH (Aβ IndOH group), and (7) treated with IndOH-LNCs (Aβ IndOH-LNC group). Animals injected with scrambled Aβ received no treatment and completed behavioral tasks 2 weeks after Aβ injection.
IndOH was dissolved in calcium carbonate 3% (w/v) at a concentration of 1 mg/mL. This solution was freshly made up for each administration, and IndOH-LNCs were prepared as described above. Daily, 1 mg/kg of IndOH or IndOH-LNCs was administered IP to the animals for 14 consecutive days. Similarly, a vehicle-treated group (LNC) with identical volume to those treated with IndOH-LNCs was run in parallel in rats infused with Aβ. The behavior tests were started on day 14 after Aβ infusion and were carried out sequentially.
Behavioral analysis
Spontaneous alternation
Hippocampal-dependent memory performance was assessed by measuring spontaneous alternation performance during 8 minutes in the Y-maze test, which evaluates cognitive searching behavior but does not specifically isolate memory performance (reviewed by HughesCitation29). Spontaneous alternation behavior is considered to reflect spatial working memory, which is a form of short-term memory. The experimental apparatus used in the present study consisted of three arms (40 cm long, 25 cm high, and 10 cm wide, labeled A, B, and C) constructed of plywood and painted black with an equilateral triangular central area. This apparatus was used in a testing room with constant illumination. Each rat was placed at the end of one arm and allowed to move freely through the apparatus for 8 minutes. Behavior was recorded by a video camera mounted vertically above the test arena for later analysis using a video tracking program (ANY-Maze; Stoelting, Wood Dale, IL). The number of arm choices and pattern of choices were recorded for each animal. An arm entry was counted when the hind paws of the rat were completely within the arm. Spontaneous alternation behavior was defined as entry into all three arms on consecutive choices in overlapping triplet sets (ie, ABC, BCA, and CBA). The percentage of alternation was calculated as (total alternations/[total arm entries − 2] × 100).
Novel object-recognition task
The object-recognition task was set up following recently reviewed guidelines.Citation30 This task is based on the spontaneous tendency of rodents to explore novel objects.Citation31 The task was performed in an apparatus made of wood covered with impermeable Formica (dimensions 40 × 50 × 50 cm) that had black floor and walls. The apparatus was used in a testing room with constant illumination. The objects used in this test all had similar textures, colors, and sizes, but had different shapes. Objects were placed near the two corners at either end of one side of the apparatus. The objects chosen were two cuboid glass blocks, a cylindrical bottle filled with water, and a dodecahedronal block. These objects were heavy enough to prevent the rats from moving them.
A day before the tests, rats were submitted to a habituation session in which they were allowed to explore the apparatus for 5 minutes without objects. On the following day, rats were acclimated in the testing room for 2 hours before the beginning of the sessions. First, rats completed a training session (24 hours after habituation) that consisted of leaving the animals in the apparatus containing two identical objects (A and A1). After training, rats were placed in their home-cages for 3 hours. The testing session to evaluate short-term-recognition memory was performed 3 hours after the training session. Rats were once again allowed to explore the apparatus, but during this session, the apparatus contained two dissimilar objects: the familiar object from the training session, and a novel one (A and B, respectively). Long-term-recognition memory was evaluated 24 hours after the training session, and a different pair of dissimilar objects (a familiar and a novel one; A and C, respectively) were presented. In all sessions, each rat was always placed in the apparatus facing the wall and allowed to explore the objects for 5 minutes, after which the rat was returned to its home cage. Behavior was recorded by a video camera mounted vertically above the test arena and analyzed using a video-tracking program (ANY-Maze). Each animal underwent three trials, comprising the training and two test sessions.
The animals started to explore the objects 1 minute after they had been placed in the apparatus. The percentage of time spent exploring the novel object was calculated as a function of the total amount of time spent exploring both objects during testing: time spent with novel object/(time spent with novel object + time spent with familiar object). A higher percentage of time spent exploring the novel object was considered to be an index of enhanced cognitive performance (Recognition Index). Between trials, the objects were cleaned with 10% ethanol solution. Active exploration was defined by directing the nose to the object at a distance of no more than 2 cm and/or touching the object with the nose or forepaws. Sitting on the object was not considered exploratory behavior.
Quantification of indomethacin in the brain
In order to determine the levels of IndOH in the cerebral tissue, a reverse-phase HPLC analysis was performed, as has been described and validated previously.Citation19 Briefly, 1 hour after the last IP injection, animals were killed by decapitation. The brain was rapidly removed from the skull, weighed, washed in ice-cold 0.9% NaCl, and the hemispheres were separated. The right hemisphere was minced with scissors and placed in a homogenizer vessel. Five mL of acetonitrile was added in order to dissolve all components. The suspensions were centrifuged at 3000 rpm for 10 minutes and the supernatant was filtered (0.45 μM; Millipore) and injected (20 μL) for HPLC. The system consisted of a UV-Vis detector, pump and auto-injector S200 (PerkinElmer), a guard-column, and a LiChrospher 100 RP-18 column of 250 mm, 4 mm, and 5 μm (Merck). The mobile phase consisted of acetonitrile/water (70:30, v/v) adjusted to an apparent pH of 5.0 ± 0.5 with 10% (v/v) acetic acid. IndOH was detected at 267 nm with a retention time of 3.45 minutes. The HPLC method has been validated to consider the linearity, inter- and intraday variability, selectivity, accuracy, limit of quantification, and recovery. Citation23 Linear calibration curves for the IndOH dissolved in acetonitrile were obtained in the range of 1.00–25.00 μg/mL, presenting correlation coefficients of higher than 0.9992. The limit of quantification was 1.00 μg/mL. The area under the peak was calculated using numerical integration. The quantity of IndOH was calculated by comparing the peak area ratio from tissue samples of treated animals with those of the corresponding concentration standards of IndOH in acetonitrile injected directly into the HPLC system.
Western blotting assay
After obtaining the fluorescent images described above, the slices from the in vitro experiments were homogenized in lysis buffer (4% sodium dodecyl sulfate [SDS], 2 mM ethylenediaminetetraacetic acid [EDTA], 50 mM Tris) containing protease-inhibitor cocktail, and the protein concentration was measured.Citation32 In order to evaluate any cell-signaling disturbances triggered by ICV injection of Aβ1-42, animals were killed by a lethal dosage of anesthesia following completion of the behavioral tasks, and blood samples were collected by cardiac puncture. The brain was rapidly removed from the skull, and the hippocampus was dissected on dry ice. The hippocampus was then homogenized in ice-cold lysis buffer (4% SDS, 2 mM EDTA, 50 mM Tris) containing a protease-inhibitor cocktail. Both homogenates were denatured for 5 minutes at 100°C and then centrifuged at 10,000 × g for 30 minutes. The supernatant containing the cytosolic fraction was collected, the protein concentration was determined,Citation32 and β-mercaptoethanol was added to a final concentration of 5%. Equal amounts of proteins were resolved (50 μg per lane) on 10% SDS-PAGE. After electrophoresis, proteins were electrotransferred to nitrocellulose membranes using a semidry transfer apparatus (Trans-Blot SD; Bio-Rad, Hercules, CA). Membranes were incubated for 60 minutes at 4°C in blocking solution (Tris-buffered saline containing 5% nonfat milk and 0.1% Tween 20, pH 7.4) and further incubated with the appropriate primary antibody dissolved in the blocking solution overnight at 4°C. The primary antibodies against the following proteins were used: anti–glial fibrillary acidic protein (GFAP) (1:3000), anti-phospho-JNK1/2 (pTpY183/185) (1:1000), anti-JNK1/2 (1:1000), anti-synaptophysin (1:3000), anti–inducible nitric oxide synthase (iNOS) (1:500), and anti-β-actin (1:1000). After washing, the membranes were incubated with adjusted secondary antibodies coupled to horseradish peroxidase (1:1000) for 2 hours. The immunocomplexes were visualized by using the chemiluminescence detection system. Band-density measurements were performed using Optiquant software (Packard Instrument, Meriden, CT). For each experiment, the test groups were compared to control cultures not exposed to Aβ.
Isolectin B4 reactivity
In order to evaluate the activation of microglial cells after Aβ ICV injection, we analyzed isolectin B4 (IB4) reactivity. Proteins (25 μg per line) were resolved on 8% SDS-PAGE, and electrotransferred to nitrocellulose membranes, as described for the Western blotting assay. Membranes were incubated overnight at 4°C in albumin solution (5% albumin and 2% Tween 20 in phosphate-buffered saline, pH 7.4). After washing, IB4 peroxidase conjugate was incubated in phosphate-buffered saline containing 0.05% Tween 20 overnight in a final concentration of 0.250 μg/mL. Chemiluminescence was detected using X-ray films.
Measurement of hepatic enzymes in serum
In an attempt to evaluate whether treatments caused hepatic toxicity, the serum levels of hepatic enzymes were evaluated at the end of the treatments. The blood samples collected by cardiac puncture were analyzed by activities of hepatic enzymes γ-glutamyltransferase, alanine aminotransferase and aspartate aminotransferase, which were used as markers of metabolic and tissue toxicity. These experiments were performed in a LabMax 240 analyzer (Labtest Diagnostica, Lagoa Santa, Brazil).
Data analysis
All experiments were carried out at least in triplicate except for behavioral tests. The results are presented as the mean ± standard deviation of seven to 15 animals per group. The statistical comparisons of the data were performed by two-way analysis of variance followed by Bonferroni post hoc test using GraphPad Prism software version 5.01 (GraphPad Software, La Jolla, CA). P-values lower than 0.05 (P < 0.05) were considered significant.
Results
Physicochemical characterization of lipid-core nanocapsules
The LNC formulations were prepared by interfacial deposition of poly(ɛ-caprolactone) and did not require subsequent purification. IndOH-LNCs and LNCs were macroscopically homogeneous bluish-white opalescent liquids. After preparation, the mean particle diameters (Z-average) were 236 ± 5 nm (IndOH-LNCs) and 226 ± 7 nm (LNCs). The suspensions showed monomodal size distributions and a polydispersity index of 0.17 ± 0.02 nm (IndOH-LNCs) and 0.15 ± 0.03 (LNCs), indicating that the formulations were highly homogeneous with narrow size distributions. The pH values were 5.95 ± 0.2 (IndOH-LNCs) and 6.05 ± 0.3 (LNCs) and the zeta-potential values were −6.9 ± 1.5 mV and −7.3 mV ± 2 mV, respectively. The IndOH content was 0.997 ± 0.010 mg/mL and the encapsulation efficiency was close to 100% for all batches.
Indomethacin-loaded lipid-core nanocapsules protect from Aβ-induced cell death in rat organotypic hippocampal culture
These experiments were designed to reveal whether IndOH-LNC treatment can exert neuroprotective effects against the cell damage induced by Aβ. The exposure of cultures to Aβ1-42 peptide caused a marked fluorescence in the hippocampus, indicating a high incorporation of PI, as presented in the photomicrographs (). Quantification of PI fluorescence showed that Aβ1-42 caused damage to approximately 40% of the hippocampus (P < 0.001), a significant increase when compared to the control cultures (about 2%–4% of cellular damage; ). Cultures exposed to the same concentration of the scramble sequence of Aβ (Aβ42-1) showed no differences in cell survival compared to untreated control cultures (data not shown).
Figure 1 (A and B) Indomethacin-loaded lipid-core nanocapsules (IndOH-LNCs) attenuate cell damage after exposure of organotypic hippocampal slices cultures to Aβ1-42 peptide. (A) Representative photomicrographs of propidium iodide (PI) uptake in hippocampal slices after 48 hour exposure to 2 μM Aβ1-42 and treated with 50 or 100 μM IndOH-LNCs, as described in “Materials and methods”; (B) quantification of PI uptake in response to Aβ peptide and IndOH-LNCs. Values are expressed as percentage of cell death in hippocampus.
Notes: #Significantly different from control cultures; ***significantly different from Aβ1-42 2 μM group. Two-way analysis of variance followed by Bonferroni post hoc test, P < 0.001. Bars represent the mean ± standard deviation, n = 12.
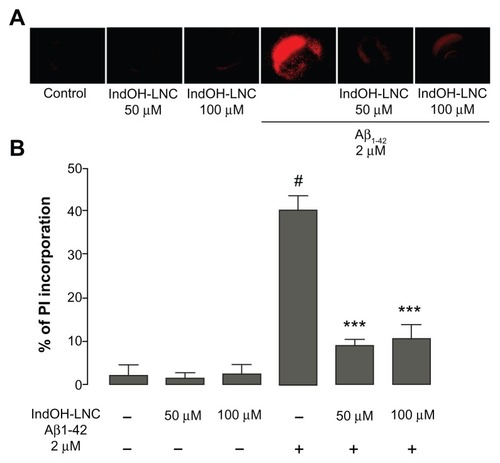
Treatment with IndOH-LNCs in the absence of Aβ (), as well as LNCs in the absence of Aβ (data not shown) were administered in order to test for possible intrinsic toxicity. However, these treatments incurred no significant differences in cell survival compared to untreated control cultures. When the cultures were exposed to Aβ1-42 and treated with 50 or 100 μM concentrations of IndOH-LNCs, cell death was significantly decreased to 9% ± 1% (P < 0.001) and 10% ± 3% (P < 0.001), respectively (). These results demonstrate that both concentrations of IndOH-LNCs exhibited similar neuroprotective effects against Aβ-induced cell death.
Indomethacin-loaded lipid-core nanocapsules modulate Aβ-induced cytokine release in rat organotypic hippocampal culture
Considering that inflammatory responses are associated with AD pathology, we investigated some anti- and proinflammatory cytokines thought to play a central role in the self-propagation of neuroinflammation, including TNF-α, IL-6, and IL-10. TNF-α and IL-6 levels were greatly increased in the culture medium after exposing the hippocampal cultures to Aβ1-42 for 48 hours (P < 0.001; ). IndOH-LNCs in both evaluated concentrations (50 and 100 μM) significantly attenuated the TNF-α and IL-6 increase induced by Aβ1-42 (P < 0.001; ). No significant differences were observed between the treatments using 50 and 100 μM concentrations of IndOH-LNCs ().
Figure 2 (A–C) Effects of indomethacin-loaded lipid-core nanocapsules (IndOH-LNCs) on the levels of cytokines in organotypic hippocampal cultures. (A) Tumor necrosis factor (TNF)-α, (B) interleukin (IL)-6, and (C) IL-10 levels in the culture medium after exposure of hippocampal cultures to Aβ1-42 for 48 hours and treatment with 50 or 100 μM IndOH-LNCs. IndOH-LNC treatment was administered simultaneously with the lesion and maintained during the recovery period for 48 hours.
Notes: #Significantly different from control cultures (P < 0.001); ***significantly different from Aβ1-42 2 μM group (P < 0.001). Two-way analysis of variance followed by Bonferroni post hoc test. Bars represent the mean ± standard deviation, n = 6.
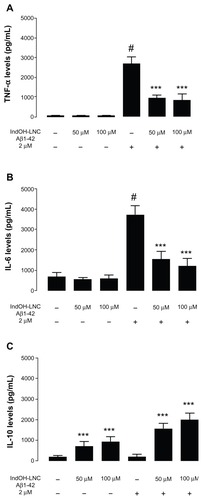
Since IndOH-LNCs decreased the proinflammatory cytokine release induced by Aβ, we reasoned that the anti-inflammatory effect of IndOH-LNCs could modulate the release of IL-10, a powerful anti-inflammatory cytokine. Our results showed that 50 or 100 μM concentrations of IndOH-LNCs elevated the amount of IL-10 present in the medium even in the control cultures (nonexposed to Aβ; P < 0.001, ). Interestingly, IndOH-LNC treatment had an overall effect in significantly increasing the amount of IL-10 in the medium of cultures (control and exposed to Aβ; P < 0.001; ). This effect was more pronounced after Aβ exposure.
Effects of indomethacin and indomethacin-loaded lipid-core nanocapsule treatment against Aβ1-42–induced memory impairment in rats
Considering that IndOH-LNC treatment had neuroprotective effects against the cell damage induced by Aβ in organotypic hippocampal culture, we were interested in the possibility of IndOH-LNCs being neuroprotective in vivo and potentially mitigating the deleterious cognitive symptoms associated with AD. Since AD is characterized clinically by a progressive decline in learning and memory processes, we evaluated the potential neuroprotective effects of IndOH and IndOH-LNCs against neurotoxicity using behavioral tests in Aβ1-42–treated rats. The spontaneous alternation and novel object–recognition tasks were used to investigate some of the mechanisms involved in Aβ-induced cognitive decline. Animals were treated daily with 1 mg/kg of IndOH or IndOH-LNCs for 14 days and the effects of IndOH were evaluated behaviorally. The results indicated that 2 weeks after a single ICV administration of Aβ1-42 (2 nmol), the rats displayed a decrease in spontaneous alternation in the Y-maze (P < 0.05; ). Treatment with IndOH failed to increase spontaneous alternation behavior in the Aβ-infused rat group. However, treatment with IndOH-LNCs at the same dosage significantly attenuated this impairment (P < 0.05).
Figure 3 Effect of indomethacin (IndOH) and indomethacin-loaded lipid-core nanocapsules (IndOH-LNCs) on spontaneous alternation behavior. Rats were injected (2 nmol, icv) with Aβ1-42 or Aβ-vehicle and daily administered with IndOH or IndOH-LNC (1 mg/kg, ip), starting 1 day after Aβ1-42 injection, and maintained for 14 days. Spontaneous alternation behavior during 8 minute sessions in the Y-maze task was measured after 14 days of treatment.
Notes: *Significant differences between the indicated columns (P < 0.05). Two-way analysis of variance followed by Bonferroni post hoc test. Columns indicate mean ± standard deviation; n = 7–10 animals in each experimental group.
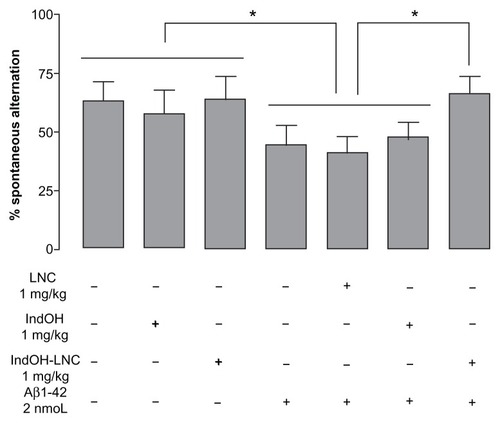
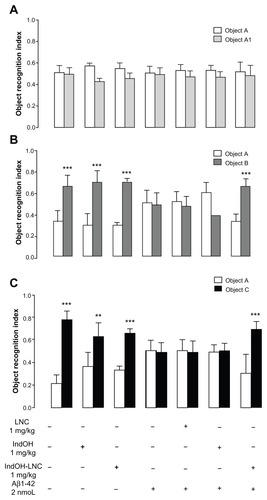
Next, we evaluated the effects of Aβ1-42 infusion, as well as IndOH and IndOH-LNC treatments on recognition memory by submitting the animals to a novel object–recognition task. As is seen in , when the animals were placed in the arena 3 hours after the first exploration period (training session), Aβ-infused rats were not able to discriminate between the familiar and novel objects, as indicated by similar exploration times for both objects (). Treatment with 1 mg/kg of IndOH did not improve short-term-recognition memory. However, discrimination was restored by treating with the same dose of IndOH-LNCs (P < 0.001; ). Similar results were found when long-term-recognition memory was evaluated 24 hours after the training session (P < 0.001; ). These results indicate that only rats treated with IndOH-LNC were able to distinguish between familiar and new objects following Aβ infusion. Animals ICV infused with Aβ vehicle were treated with IndOH or IndOH-LNCs on the same schedule in order to test for possible intrinsic toxicity. These animals showed no impairment in either the spontaneous alternation task or the recognition memory task when compared to control animals. Treatment with LNCs had no effects on the spontaneous alternation and recognition memory impairments triggered by Aβ ( and ). Animals ICV infused with Aβ42-1 had no differences in the spontaneous alternation and memory recognition tasks (data not shown).
Figure 4 (A–C) Effect of indomethacin (IndOH) and indomethacin-loaded lipid-core nanocapsules (IndOH-LNCs) on novel object–recognition memory. Rats were injected (2 nmol, intracerebroventricularly) with Aβ1-42 or Aβ vehicle and administered daily with IndOH or IndOH-LNC s (1 mg/kg, intraperitoneally), starting 1 day after Aβ1-42 injection, and maintained for 14 days. Graphics show object-recognition index during 5 minutes in the training session (A), short-term-memory test session performed 3 hours after training (B), and long-term memory-test session performed 24 hours after training session (C).
Notes: **Significant differences between familiar and new object for each group (P < 0.01); ***significant differences between familiar and new object for each group (P < 0.001). Two-way analysis of variance followed by Bonferroni post hoc test. Columns indicate mean ± standard deviation; n = 7–10 animals in each experimental group.
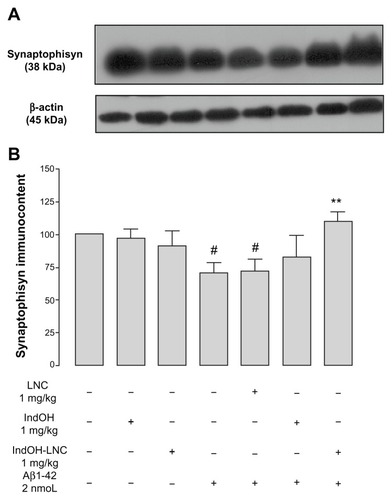
Indomethacin-loaded lipid-core nanocapsule treatment decreases synaptic dysfunction triggered by Aβ in rats
To investigate the synaptic integrity in our treatment groups, we quantified the prevalence of the protein synaptophysin, a specific presynaptic marker. A significant reduction in synaptophysin levels was found in Aβ1-42–infused rats 15 days after ICV injection (P < 0.01), suggesting that some form of synaptic dysfunction had been induced (). In accordance with the behavioral results, only treatment with IndOH-LNCs was able to block the decrease in synaptophysin levels following Aβ1-42 infusion (P < 0.01; ). Animals ICV infused with Aβ vehicle were treated with IndOH or IndOH-LNCs on the same schedule. Those animals showed no significant alteration in the synaptophysin levels when compared to control animals. Additionally, treatment with LNCs had no effect on the decreased synaptophysin levels triggered by Aβ ().
Figure 5 (A and B) Aβ1-42 injection causes synaptotoxicity, which is prevented by indomethacin-loaded lipid-core nanocapsule (IndOH-LNC) treatment. (A) Representative Western blotting analysis for synaptophysin and β-actin protein (loading control) was performed in the hippocampus of animals after injection with Aβ1-42 (2 nmol, intracerebroventricularly) and treated for 14 days with IndOH or IndOH-LNCs (1 mg/kg, intraperitoneally), starting 1 day after Aβ injection. (B) Graphic shows quantification of synaptophysin immunocontent normalized by β-actin protein (loading control).
Notes: #Significantly different from the respective control cultures (P < 0.01); **significantly different from Aβ1-42 2 μM and Aβ1-42 2 μM + LNC groups (P < 0.01). Two-way analysis of variance followed by Bonferroni post hoc test. The values represent synaptophysin levels, expressed as the average percentage increase (mean ± standard deviation) over basal levels; n = 7 animals in each experimental group.
Indomethacin-loaded lipid-core nanocapsule treatment suppresses glial and microglial activation triggered by Aβ in vitro and in vivo
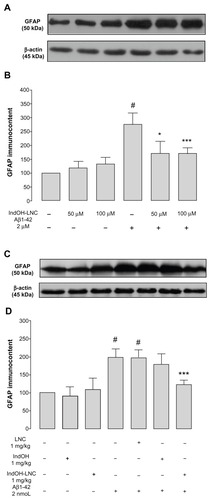
Considering the involvement of neuroinflammation in the physiopathology of AD, we tested the possible requirement of astrocytes and microglial activation in Aβ-induced toxicity. The results showed that exposure of the organotypic cultures to Aβ1-42 peptide caused a highly significant increase in GFAP immunocontent (P < 0.001; ). A significant reduction in the levels of GFAP immunocontent were observed in the cultures treated with 50 or 100 μM IndOH-LNCs (P < 0.05 and P < 0.001, respectively; ). Consistent with these results, a significant increase in GFAP immunocontent was found in the hippocampus of Aβ1-42–infused rats 15 days after ICV injection (P < 0.001; ) and only the treatment with IndOH-LNCs was able to reduce this astrocyte immunoreactivity (P < 0.001; ).
Figure 6 (A–D) Astrocytic activation induced by Aβ1-42 peptide can be reduced by indomethacin-loaded lipid-core nanocapsules (IndOH-LNCs). Representative Western blotting of glial fibrillary acidic protein (GFAP) immunoreactivity in (A) organotypic hippocampal cultures after 48 hours of exposure to Aβ1-42 and treatment with 50 or 100 μM IndOH-LNCs, and in (C) the hippocampus 15 days after intracerebroventricular injection of Aβ1-42 (2 nmol) and treatment with IndOH or IndOH-LNCs (1 mg/kg, intraperitoneally). Graphics show quantification of GFAP immunocontent normalized by β-actin protein (loading control). The values represent GFAP levels, expressed as the average percentage increase (mean ± standard deviation) over basal levels in (B) organotypic hippocampal cultures (n = 6) and (D) the hippocampus 15 days after intracerebroventricular injection of Aβ1-42 (n = 8).
Notes: #Significantly different from the respective control groups (P < 0.001); *significantly different from Aβ1-42 2 μM group (P < 0.05); ***significantly different from (B) Aβ1-42 2 μM group or (D) Aβ1-42 and Aβ1-42 treated with vehicle groups (P < 0.001). Two-way analysis of variance followed by Bonferroni post hoc test.
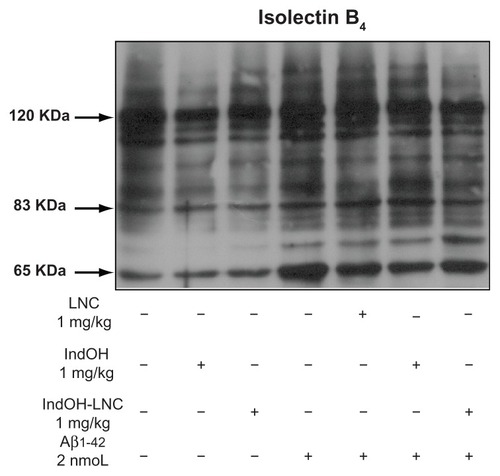
Similarly, immunodetection of iNOS in the hippocampus of Aβ1-42–infused rats was significantly increased, and only the IndOH-LNC treatment was able to significantly reduce this inflammatory reaction (P < 0.05; ). The hippocampi of Aβ1-42–infused rats also had increased reactivity of IB4, a hallmark of microglial activation (). In the same way, while IndOH was ineffective at blocking microglial activation, IndOH-LNC treatment decreased the reactivity of IB4 (). Animals ICV infused with Aβ-vehicle were treated with IndOH or IndOH-LNCs on the same schedule, and showed no alteration in the activation of astrocytes, microglia, or iNOS compared to control animals. Treatment with LNCs had no effect on the activation of astrocytes, microglia, and iNOS triggered by Aβ (–).
Figure 7 (A and B) Indomethacin-loaded lipid-core nanocapsule (IndOH-LNC) treatment reduces inducible nitric oxide synthase (iNOS) immunocontent in the hippocampus after icv injection of Aβ1-42. (A) Representative Western blotting of iNOS in the hippocampus 15 days after intracerebroventricular injection of Aβ1-42 (2 nmol) and treatment with IndOH or IndOH-LNCs (1 mg/kg, intraperitoneally). (B) Graphic shows quantification of iNOS immunocontent normalized by β-actin protein (loading control).
Notes: #Significantly different from all control groups (P < 0.05); *significantly different from Aβ1-42 and Aβ1-42 treated with vehicle groups (P < 0.05). The values represent iNOS levels, expressed as the average percentage increase (mean ± standard deviation) over basal levels; n = 5–8 animals in each experimental group. Two-way analysis of variance followed by Bonferroni post hoc test.
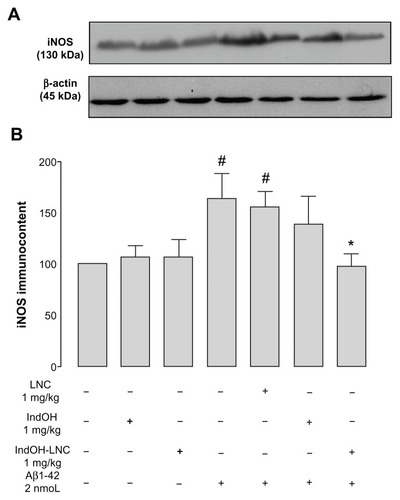
Figure 8 Indomethacin-loaded lipid-core nanocapsule (IndOH-LNC) treatment reduces microglial activation in the hippocampus after intracerebroventricular injection of Aβ1-42. Representative image showing isolectin B4 (IB4) reactivity (120 kDa) in the hippocampus 15 days after intracerebroventricular injection of Aβ1-42 (2 nmol) and treatment with IndOH or IndOH-LNCs (1 mg/kg, intraperitoneally).
Note: The images are representative of n = 6 animals in each experimental group.
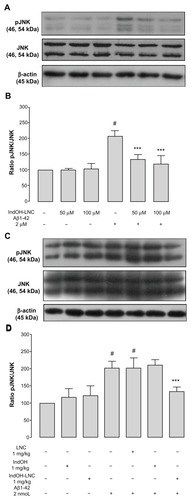
Indomethacin-loaded lipid-core nanocapsule treatment suppresses JNK activation triggered by Aβ in vitro and in vivo
To clarify the role of inflammation in Aβ toxicity, we investigated the possible requirement of JNK activation. As shown in , a significant increase in JNK phosphorylation was observed following Aβ1-42 peptide exposure in organotypic cultures (P < 0.001; ). Treatment with 50 or 100 μM IndOH-LNCs was able to significantly reduce the levels of JNK phosphorylation without modifying the total levels of JNK (P < 0.001; ). Consistent with these results, a significant increase in JNK phosphorylation was found in the hippocampi of Aβ1-42–infused rats 15 days after ICV injection (P < 0.001; ). It is important to note that only the LNC form of IndOH was capable of blocking JNK activation (P < 0.001; ). Animals ICV infused with Aβ vehicle were treated with IndOH or IndOH-LNCs on the same schedule, and showed no alterations in JNK signaling compared to control animals. Treatment with LNCs had no protective effects on the cell-signaling disturbances triggered by Aβ ().
Figure 9 (A–D) C-jun N-terminal kinase (JNK) phosphorylation induced by Aβ1-42 peptide can be reduced by indomethacin-loaded lipid-core nanocapsules (IndOH-LNCs). Representative Western blotting of phosphorylated JNK (pJNK), JNK, and β-actin immunocontent in (A) organotypic hippocampal cultures after 48 hours of exposure to Aβ1-42 and treatment with 50 or 100 μM IndOH-LNCs, and in (C) the hippocampus of animals after being injected with Aβ1- 42 (2 nmol, intracerebroventricularly) and treated by 14 days with IndOH or IndOH-LNCs (1 mg/kg, intraperitoneally). Histogram represents the quantitative Western blotting analysis of JNK phosphorylation state. The densitometric values obtained to phospho- and total JNK from treatments were normalized to their respective controls nonexposed to Aβ1-42 toxicity condition (control bar; 100%). Data are expressed as a ratio of the normalized percentages of pJNK and JNK. Bars represent the mean ± standard deviation for (B) organotypic hippocampal cultures (n = 6) and (D) hippocampus of animals after injection with Aβ1-42 (n = 7).
Notes: #Significantly different from the respective control groups (P < 0.001); ***significantly different from (B) Aβ1-42 2-μM group or (D) Aβ1-42 and Aβ1-42 treated with vehicles groups (P < 0.001). Two-way analysis of variance followed by Bonferroni post hoc test.
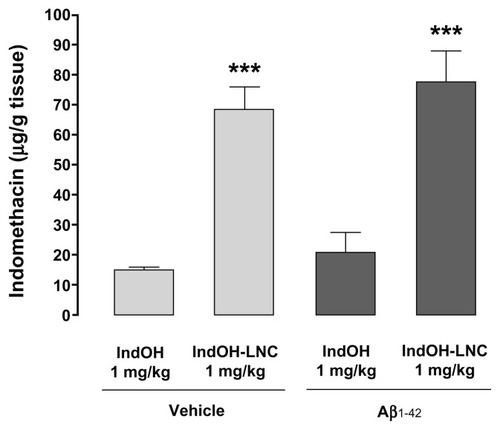
Lipid-core nanocapsules increase indomethacin concentration in brain tissue
Since drug-loaded LNC treatment was observed to enhance the effectiveness of IndOH against Aβ1-42–induced toxicity in rats, we hypothesized that the LNCs could facilitate these improvements by increasing the cerebral biodistribution of IndOH. Therefore, quantitative analyses were performed to assess the cerebral biodistribution of IndOH administered by polymeric NCs compared to IndOH after treatment using equal dosing (1 mg/kg/day). As can be seen in , a significantly higher quantity of IndOH was found in the brains of animals treated with IndOH-LNCs in comparison with animals treated with IndOH (P < 0.001). This result was observed in both animals ICV infused with Aβ vehicle and in Aβ1-42–infused animals (P < 0.001; ).
Figure 10 Lipid-core nanocapsules (LNCs) improve the brain biodistribution of indomethacin (IndOH). Brain amount of IndOH was analyzed by high-performance liquid chromatography in rats injected (2 nmol, intracerebroventricularly) with Aβ1-42 or Aβ vehicle and administered daily with IndOH or IndOH-LNCs (1 mg/kg, intraperitoneally) for 14 days, as described in “Materials and methods.”
Notes: ***Significantly different between IndOH-LNC group and the respective IndOH group (P < 0.001). Bars represent the mean ± standard deviation, n = 8. Two-way analysis of variance followed by Bonferroni post hoc test.
Investigation of possible side effects of treatments
Treatment with IndOH or IndOH-LNCs (1 mg/kg/day, IP), as well as the vehicles did not induce mortality or alter body weight within 14 days of treatment (data not shown). The activities of hepatic enzymes γ-glutamyltransferase, alanine aminotransferase, and aspartate aminotransferase were assessed in rat blood serum. None of the treated animals presented with significant alterations in the investigated enzymes, suggesting no hepatic alterations or metabolic toxicity in the animals in the tested conditions (data not shown). Further, none of the treated animals presented with alterations in hematological parameters (data not shown).
Discussion
The inflammatory reaction induced by Aβ involves the release of damaging factors such as cytokines, nitric oxide, and reactive oxygen species, which promote activation of intracellular pathways that contribute to the progression of AD.Citation33,Citation34 The development of therapies for this neurodegenerative disorder represents a major challenge to academic, biotechnology, and pharmaceutical researchers. In the present study, through use of organotypic hippocampal cultures and ICV injection of Aβ in rats, we show that neuroinflammation plays a significant role in the neurodegenerative events triggered by Aβ. We provide compelling evidence that IndOH-LNC treatment is neuroprotective against Aβ-induced toxicity.
Basic and clinical research advances over the past few decades have made significant advances in our understanding of AD. However, the mechanisms that govern Aβ production and control its conformational state remain to be determined. Although the amyloid fibrils found in plaques (as used in our model system) were originally believed to be responsible for AD pathogenesis, recent evidence indicates that the primary neurotoxic species in AD may actually be soluble oligomers of Aβ.Citation35,Citation36 Additionally, it appears unlikely that there are just one or two assembly forms of the peptide that induce neuronal dysfunction. Rather, various soluble oligomers of Aβ can likely bind to different components of neuronal and nonneuronal plasma membranes to induce complex patterns of synaptic dysfunction and network disorganization. These changes may in turn activate biochemical cascades, causing neuronal dysfunction and ultimately cell death.Citation37,Citation38 While oligomers of Aβ are often considered to be the main toxin involved in AD, we investigated the efficacy of IndOH-LNCs on aggregate assemblies of Aβ. However, the bioactivity of oligomers does not imply that plaques have no role in the progressive degeneration of neurons. The insoluble amyloid fibrils that make up the plaques might themselves be relatively inactive, but the presence of bioactive dimers and larger oligomers “trapped” within amyloid plaque cores strongly suggests that plaques serve as local reservoirs of diverse, small oligomers that can diffuse away to cause neurotoxicity.Citation37,Citation39
We first used organotypic hippocampal slice cultures to examine Aβ toxicity and evaluated the possible neuroprotective effects of IndOH by using LNCs as a nanocarrier system. Organotypic hippocampal cultures have several advantages over isolated cell cultures. In this preparation, neurons and glial cells survive long-term and physiologically mature in culture, allowing for extended survival studies.Citation40 For this reason, organotypic hippocampal culture provides an excellent model system for evaluating mechanisms of neurodegeneration and designing therapeutic agents.Citation24,Citation41 Our results showed a significant increase in cell death when cultures were exposed to Aβ, which may have been driven by the release of proinflammatory cytokines and glial activation. Inhibition of inflammatory responses with IndOH-LNCs decreased the toxicity induced by Aβ, suggesting that this treatment may be a promising candidate for treatment of the neuroinflammation processes closely associated with AD.
These results encouraged us to investigate the effects of IndOH-LNC treatment in an in vivo model of Aβ toxicity. Based on the ICV injection of Aβ1-42 model (1 nmol/site), we found that the hippocampi of rats receiving a single injection presented with a neuroinflammatory response, supporting the results observed in the organotypic culture. The hippocampal involvement of the neuroinflammatory response was particularly interesting to us, because this region is associated with the memory and synaptic dysfunction characteristic of early phases of AD. The Aβ disease model has been a useful complement to transgenic approaches to AD neuropathology in the development and evaluation of therapeutic approaches.Citation42
Notably, the memory dysfunction observed here appears to be related to synaptic loss. This finding supports the hypothesis that synaptic dysfunction induced by Aβ is the primary marker of AD and precedes neuronal death.Citation33 Our results also suggest that the physiological response to Aβ1-42 injection involves the activation of inflammatory brain cells (ie, astrocytes and microglia). Importantly, IndOH-LNC treatment was able to protect from behavioral impairments, glial and microglial activation, and cell signaling disturbances triggered by Aβ in vivo. By contrast, IndOH treatment failed to protect against neurotoxicity induced by Aβ. It is important to note that LNC delivery achieved dramatically higher intracerebral concentrations of IndOH, which may explain the neuroprotective effects observed here.
Epidemiological studies have suggested that long-term use of NSAIDs may protect against AD.Citation12,Citation13,Citation43 However, the hypothesis that IndOH slows the incidence and progression of AD remains controversial.Citation12,Citation13,Citation43 The presence of the BBB impedes effective treatment of many brain diseases, because large doses are required to reach the minimum effective concentration in the brain, thereby negatively affecting drug efficacy and tolerance.Citation44 For these reasons, exploitation of the therapeutic potential of IndOH has remained difficult because of the side effects that limit its use, particularly gastrointestinal complications.Citation45 In an attempt to overcome these limitations and increase intracerebral concentrations of IndOH, we used a nanocarrier system based on IndOH-LNCs. This method of drug delivery has been shown to decrease gastrointestinal side effects and increase the brain biodistribution of IndOH.Citation19,Citation46 In the present study, we reported that by using LNCs, even low concentrations of IndOH (such as 50 μM) were able to protect against cell death induced by Aβ1-42 exposure in organotypic culture. Possible mechanisms underlying the similar effects observed with both concentrations of IndOH-LNCs in the present study may include the NC uptake endocytosis by the cells achieved through endocytosis. Since this process is saturable, if the concentration of NCs rises beyond the cell endocytosis threshold, the drug efficiency will not increase beyond this threshold.Citation47 Although IndOH-treated cultures were not evaluated in the present study, we have previously demonstrated that organotypic cultures treated by the same concentration of IndOH-LNCs exhibited a strong neuroprotective effect against oxygen glucose–deprived lesions, whereas cultures receiving IndOH treatment were significantly damaged,Citation20 consistent with our initial hypothesis that LNCs improve IndOH bioavailability.
Aβ is acutely toxicCitation48 and can interfere with synaptic plasticity in the brain, suggesting that this peptide may be responsible for episodic memory deficits. Such memory deficits are an early symptom of AD linked to hippocampal injury.Citation33 Our experimental protocol (based on the ICV injection of Aβ1-42 in rats) caused a significant decrease in hippocampal levels of synaptophysin (a specific presynaptic marker), which might have led to impairment on the spontaneous alternation and object-recognition memory tasks. The synaptotoxic effects of Aβ1-42 may be crucial in causing the observed memory deficits. Our results are in accordance with previous work related to synaptic loss due to toxicity induced by ICV injection of Aβ toxicity.Citation49,Citation50 These data imply that one mechanism by which Aβ promotes neurotoxicity (at least in the hippocampus) is via a sustained increase of synaptic dysfunction. Additionally, the ICV infusion of Aβ42-1 did not affect either spontaneous alternation or the ability of animals in recognizing new objects, indicating that the observed behavioral impairments in Aβ1-42–exposed animals were dependent on the peptide sequence/structure. Remarkably, by using LNCs, subtherapeutic doses of IndOH (1 mg/kg) were able to reverse the decreased expression of synaptophysin triggered by Aβ1-42 and restore cognitive performance, whereas treatment with the same dose of IndOH failed to protect against the toxic damage induced by ICV Aβ injection. These data suggest additional research opportunities into the protective effects of IndOH, and for using subtherapeutic doses to overcome side effects. Our previous work has shown that treatment of rats with the same dose of IndOH-LNCs used in the present study induced a marked protective effect on the gastrointestinal mucosa compared with the ulcerative effect observed with the treatment with IndOH.Citation46 Additionally, neither free nor nanoencapsulated IndOH, altered hepatic function or hematological parameters in our experimental conditions.
Development of combined treatments using compounds with different bioavailability, pharmacokinetics, and metabolism is challenging. Nanotechnology represents a powerful tool that may allow clinicians to circumvent this problem by attaining increased efficacy associated with a marked reduction of adverse effects respective to each drug. We did not perform any experiments to verify directly that LNCs became lodged in the brain, but we have observed a larger amount of IndOH in the brain of animals treated with IndOH-LNCs in comparison with animals treated with IndOH. LNCs crossing the BBB remains to be determined. One proposed penetration mechanism is that the permeability of the BBB may be increased due to a concentration gradient established by the retention of nanoparticles in the brain blood capillaries, combined with adsorption to capillary walls. Other proposed mechanisms include solubilization of endothelial cell membrane lipids leading to membrane fluidization, endocytosis of the nanoparticles by endothelial cells followed by drug release into the brain, transcytosis of the nanoparticles with bound drug across the endothelial cell layer, and inhibition of the P-glycoprotein efflux system by coating the nanoparticles with polysorbate 80.Citation51,Citation52
Increasing evidence supports the possibility that neuroinflammation significantly contributes to the pathogenesis of AD. The presence of Aβ in the brain activates inflammatory cells, and tissue levels of pro- and anti-inflammatory mediators including cytokines and chemokines are altered.Citation53 In particular, the production and secretion of proinflammatory mediators may contribute to the initiation and progression of neurodegeneration by a variety of mechanisms. Proinflammatory cytokines, such as IL-1β, IL-6, and TNF-α, may play an important role in AD pathology and lead to increased production of nitric oxide through iNOS activation, neuronal stress, and further neuronal dysfunction and death.Citation54 Since inflammation can be damaging to host tissue, we hypothesized that NSAIDs such as IndOH might inhibit both the onset and the progression of AD. Our results clearly show that organotypic cultures treated with IndOH-LNCs exhibited decreased levels of TNF-α and IL-6 after Aβ exposure. In view of these results, we sought to evaluate whether IndOH-LNC treatments would be able to induce release of anti-inflammatory cytokine IL-10 to facilitate its anti-inflammatory effects. Our previous work has shown that this formulation increased the levels of IL-10 in peripheral inflammation models.Citation46 Here, we noticed that treatment with IndOH-LNCs increased levels of IL-10 in the presence or absence of Aβ. In this way, our data demonstrate that IndOH was able to break the inflammatory response induced by Aβ, culminating in the protective effect on cell death, as observed by PI incorporation.
The accumulation of cytokines triggered by Aβ does not appear to be a mere consequence of the degenerative process, but seems to play a role in the cascade of events inducing neuronal dysfunction by several stress-activated signal transduction pathways, such as JNK. The activation of JNK has been described in cultured neurons after Aβ exposure, and its inhibition attenuates Aβ toxicity.Citation55 Additionally, JNKs are reported to be involved in the enlargement of microglia, as well as in the induction of proinflammatory cytokine genes coding for TFN-α, IL-6, or MCP-1 in addition to cyclooxygenase-2,Citation56 suggesting that JNKs are relevant comediators of the activation of microglia. Further, activation of JNKs in the brain induced by Aβ leads to enhanced expression of iNOS, contributing to the neurodegenerative process and cognitive damage.Citation49 Taken together, our results suggest that Aβ-induced astrocyte and microglial activation, which in turn led to increased synthesis and the release of proinflammatory cytokines and sustained JNK and iNOS activation, culminating in synaptic dysfunction and cell death. Only treatment with IndOH-LNCs was able to decrease glial and microglial activation, as evidenced by GFAP and IB4 reduction, respectively, as well as JNK and iNOS activation. In this way, our results suggest that Aβ induces glial and microglial activation, leading to increased proinflammatory cytokine release, which in turn activates JNK and iNOS, driving a sustained proinflammatory and oxidative environment culminating in synaptic dysfunction and consequently behavioral impairments. IndOH, probably through inhibition of prostaglandin formation in astrocytes and microglia, was able to attenuate this vicious cycle. This may be correlated with increased cell survival and improvements in memory performance following Aβ-induced impairment.
Conclusion
In summary, the present study provides strong evidence that IndOH negatively modulates in vitro and in vivo neuroinflammation triggered by Aβ, culminating in the amelioration of synaptic integrity, cell survival, and cognitive performance. Furthermore, significantly higher efficiency was achieved by delivering IndOH with LNCs. Hence, the combination of IndOH and LNC-based delivery system may pave the path for future therapeutic interventions in Alzheimer’s disease.
Acknowledgments
This study was supported by the following Brazilian agencies: CAPES, FAPERGS, CNPq, and CNPq-FAPERGS (#10/0048-4). A Bernardi was the recipient of the CAPES and CNPq postdoctoral fellowship. RL Frozza and JB Hoppe were recipients of CNPq PhD fellowships.
Disclosure
The authors report no conflicts of interest in this work.
References
- LeeYJHanSBNamSYOhKWHongJTInflammation and Alzheimer’s diseaseArch Pharm Res201033101539155621052932
- McGeerEGMcGeerPLNeuroinflammation in Alzheimer’s disease and mild cognitive impairment: a field in its infancyJ Alzheimers Dis201019135536120061650
- HensleyKNeuroinflammation in Alzheimer’s disease: mechanisms, pathologic consequences, and potential for therapeutic manipulationJ Alzheimers Dis201021111420182045
- CagninABrooksDJKennedyAMIn-vivo measurement of activated microglia in dementiaLancet2001358928046146711513911
- ParachikovaAAgadjanyanMGCribbsDHInflammatory changes parallel the early stages of Alzheimer diseaseNeurobiol Aging200728121821183317052803
- SimardARSouletDGowingGJulienJPRivestSBone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s diseaseNeuron200649448950216476660
- HickmanSEAllisonEKEl KhouryJMicroglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease miceJ Neurosci200828338354836018701698
- ChenYVartiainenNEYingWChanPHKoistinahoJSwansonRAAstrocytes protect neurons from nitric oxide toxicity by a glutathione-dependent mechanismJ Neurochem20017761601161011413243
- JohnstoneMGearingAJMillerKMA central role for astrocytes in the inflammatory response to beta-amyloid; chemokines, cytokines and reactive oxygen species are producedJ Neuroimmunol1999931–218219310378882
- WhitneyNPEidemTMPengHHuangYZhengJCInflammation mediates varying effects in neurogenesis: relevance to the pathogenesis of brain injury and neurodegenerative disordersJ Neurochem200910861343135919154336
- AnismanHCascading effects of stressors and inflammatory immune system activation: implications for major depressive disorderJ Psychiatry Neurosci200934142019125209
- McGeerPLMcGeerEGAnti-inflammatory drugs in the fight against Alzheimer’s diseaseAnn N Y Acad Sci1996777172132208624086
- HoozemansJJRozemullerJMvan HaastertESVeerhuisREikelenboomPCyclooxygenase-1 and −2 in the different stages of Alzheimer’s disease pathologyCurr Pharm Des200814141419142718537664
- KotilinekLAWestermanMAWangQCyclooxygenase-2 inhibition improves amyloid-beta-mediated suppression of memory and synaptic plasticityBrain2008131Pt 365166418292081
- NetlandEENewtonJLMajochaRETateBAIndomethacin reverses the microglial response to amyloid beta-proteinNeurobiol Aging19981932012049661994
- ParepallyJMMandulaHSmithQRBrain uptake of nonsteroidal anti-inflammatory drugs: ibuprofen, flurbiprofen, and indomethacinPharm Res200623587388116715377
- SilvaGANanotechnology applications and approaches for neurore-generation and drug delivery to the central nervous systemAnn N Y Acad Sci2010119922123020633128
- BrambillaDLe DroumaguetBNicolasJNanotechnologies for Alzheimer’s disease: diagnosis, therapy, and safety issuesNanomedicine20117552154021477665
- BernardiABraganholEJägerEIndomethacin-loaded nano-capsules treatment reduces in vivo glioblastoma growth in a rat glioma modelCancer Lett20092811536319286307
- BernardiAFrozzaRLHornAPProtective effects of indomethacin-loaded nanocapsules against oxygen-glucose deprivation in organotypic hippocampal slice cultures: involvement of neuroinflammationNeurochem Int201057662963620691236
- FrozzaRLBernardiAPaeseKCharacterization of trans-resveratrol-loaded lipid-core nanocapsules and tissue distribution studies in ratsJ Biomed Nanotechnol20106669470321361135
- JägerEVenturiniCGPolettoFSSustained release from lipidcore nanocapsules by varying the core viscosity and the particle surface areaJ Biomed Nanotechnol20095113014020055116
- PohlmannARSoaresLUCruzLda SilveiraNPGuterresSSAlkaline hydrolysis as a tool to determine the association form of indomethacin in nanocapsules prepared with poly(eta-caprolactone)Curr Drug Deliv20041210311016305375
- NIHGuide for the Care and Use of Laboratory AnimalsNIH Publication Vol. No. 85-23 Revised 1985. Available from: http://oacu.od.nih.gov/regs/guide/guide.pdfAccessed 24 April, 2011
- StoppiniLBuchsPAMullerDA simple method for organotypic cultures of nervous tissueJ Neurosci Methods19913721731821715499
- FrozzaRLHornAPHoppeJBA comparative study of beta-amyloid peptides Aβ1-42 and Aβ25–35 toxicity in organotypic hippocampal slice culturesNeurochem Res200934229530318686032
- HoppeJBFrozzaRLHornAPAmyloid-β neurotoxicity in organotypic culture is attenuated by melatonin: involvement of GSK-3β, tau and neuroinflammationJ Pineal Res201048323023820136701
- PaxinosGWatsonCThe Rat Brain in Stereotaxic Coordinates5th edBurlingtonElsevierAcademic Press2005
- HughesRNThe value of spontaneous alternation behavior (SAB) as a test of retention in pharmacological investigations of memoryNeurosci Biobehav Rev200428549750515465137
- BevinsRABesheerJObject recognition in rats and mice: a one-trial non-matching-to-sample learning task to study ‘recognition memory’Nat Protocol20061313061311
- EnnaceurADelacourJA new one-trial test for neurobiological studies of memory in rats. 1. Behavioral dataBehav Brain Res198831147593228475
- PetersonGLReview of the folin-phenol protein quantification method of Lowry, Rosebrough, Farr and RandallAnal Biochem19791002201220393128
- WalshDMSelkoeDJDeciphering the molecular basis of memory failure in Alzheimer’s diseaseNeuron200444118119315450169
- Wyss-CorayTInflammation in Alzheimer’s disease: driving force bystander or beneficial response?Nat Med20061291005101516960575
- WalshDMKlyubinIFadeevaJVNaturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivoNature2002416688053553911932745
- FerreiraSTKleinWLThe Aβ oligomer hypothesis for synapse failure and memory loss in Alzheimer’s diseaseNeurobiol Learn Mem201196452954321914486
- SelkoeDJResolving controversies on the path to Alzheimer’s therapeuticsNat Med20111791060106521900936
- MuckeLSelkoeDJNeurotoxicity of amyloid β-protein: synaptic and network dysfunctionCold Spring Harb Perspect Med201227a00633822762015
- ShankarGMLiSMehtaTHAmyloid-b protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memoryNat Med200814883784218568035
- FrotscherMZafirovSHeimrichBDevelopment of identified neuronal types and of specific synaptic connection in slice cultures of rat hippocampusProg Neurobiol19954561431647598766
- HolopainenIEOrganotypic hippocampal slice cultures: a model system to study basic cellular and molecular mechanisms of neuronal cell death, neuroprotection, and synaptic plasticityNeurochem Res200530121521152816362771
- Woodruff-PakDSAnimal models of Alzheimer’s disease: therapeutic implicationsJ Alzheimers Dis200815450752119096153
- JongDFJansenRHoefnagelsWNo effect of one-year treatment with indomethacin on Alzheimer’s disease progression: a randomized controlled trialPlos One200831e147518213383
- PotschkaHTargeting the brain – surmounting or bypassing the blood– brain barrierHandb Exp Pharmacol201019741143120217538
- KeanWFBuchananWWThe use of NSAIDs in rheumatic disorders: a global perspectiveInflammopharmacology200513434337016354389
- BernardiAZilbersteinACCVJagerEEffects of indomethacin-loaded nanocapsules in experimental model of inflammation in ratsBr J Pharmacol200915841104111119422380
- DavdaJLabhasetwarVCharacterization of nanoparticle uptake by endothelial cellsInt J Pharm20022331–2515911897410
- KayedRHeadEThompsonJLCommon structure of soluble amyloid oligomers implies common mechanism of pathogenesisScience2003300561848648912702875
- MedeirosRPredigerRDPassosGFConnecting TNF-α signaling pathways to iNOS expression in a mouse model of Alzheimer’s disease: relevance for the behavioral and synaptic deficits induced by amyloid β proteinJ Neurosci200727205394540417507561
- PassosGFFigueiredoCPPredigerRDSChemokine receptor 5 signaling pathway in the neuroinflammatory response and cognitive deficits induced by β-amyloid peptideAm J Pathol200917541586159719729478
- LockmanPROyewumiMOKoziaraJMRoderKEMumperRJAllenDDBrain uptake of thiamine-coated nanoparticlesJ Control Release200393327128214644577
- SahniJKDogguiSAliJBabootaSDaoLRamassamyCNeurotherapeutic applications of nanoparticles in Alzheimer’s diseaseJ Control Release2011152220823121134407
- RamírezGToroRDöbeliHvon BernhardiRProtection of rat primary hippocampal cultures from AB cytotoxicity by pro-inflammatory molecules is mediated by astrocytesNeurobiol Dis2005191–224325415837580
- SchultzbergMLindbergCAronssonAFHjorthESpulberSDOpricaMInflammation in the nervous system – physiological and pathophysiological aspectsPhysiol Behav2007921–212112817597167
- Bozyczko-CoyneDO’KaneTMWuZLCEP-1347/KT-7515, an inhibitor of SAPK/JNK pathway activation, promotes survival and blocks multiple events associated with Abeta-induced cortical neuron apoptosisJ Neurochem200177384986311331414
- WaetzigVHerdegenTNeurodegenerative and physiological actions of c-Jun N-terminal kinases in the mammalian brainNeurosci Lett20043611–3646715135894