 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
We investigated flexible liposomes as a potential oral drug delivery system. However, enhanced membrane fluidity and structural deformability may necessitate liposomal surface modification when facing the harsh environment of the gastrointestinal tract. In the present study, silica-coated flexible liposomes loaded with curcumin (CUR-SLs) having poor water solubility as a model drug were prepared by a thin-film method with homogenization, followed by the formation of a silica shell by the sol-gel process. We systematically investigated the physical properties, drug release behavior, pharmacodynamics, and bioavailability of CUR-SLs. CUR-SLs had a mean diameter of 157 nm and a polydispersity index of 0.14, while the apparent entrapment efficiency was 90.62%. Compared with curcumin-loaded flexible liposomes (CUR-FLs) without silica-coatings, CUR-SLs had significantly higher stability against artificial gastric fluid and showed more sustained drug release in artificial intestinal fluid as determined by in vitro release assays. The bioavailability of CUR-SLs and CUR-FLs was 7.76- and 2.35-fold higher, respectively, than that of curcumin suspensions. Silica coating markedly improved the stability of flexible liposomes, and CUR-SLs exhibited a 3.31-fold increase in bioavailability compared with CUR-FLs, indicating that silica-coated flexible liposomes may be employed as a potential carrier to deliver drugs with poor water solubility via the oral route with improved bioavailability.
Liposomes are colloidal vesicular structures composed of self-assembled lipid bilayers generated when phospholipids are hydrated in an aqueous media. Different classes of drug molecules can be encapsulated by liposomes, with hydrophobic drugs typically inserted into the lipid bilayers and hydrophilic drugs loaded within the aqueous interior.Citation1–Citation3
Although liposomes have been extensively studied as drug carriers for parenteral administration, their potential application in oral drug delivery has also been studied.Citation4–Citation6 Oral liposome carriers may provide increased solubility and protection of encapsulated drugs from the harsh environment of the gastrointestinal tract. The fluidity of the lipid bilayers and the relatively small size of the liposomes greatly facilitate oral absorption.Citation6 However, the instability of conventional liposomes under the physiological conditions typically found in the gastrointestinal tract has hindered clinical applications, and therefore a number of approaches have been developed in attempts to address this problem.Citation7–Citation9
The most frequently used PEGylation method has been reported to produce stable liposomes and enhance the oral bioavailability of poorly soluble and low-bioavailability drugs.Citation10,Citation11 In these cases, the poly(ethylene glycol)s (PEGs) were chemically linked to the hydrophilic amino termini of phospholipids, forming phospholipid-PEG conjugates for physical liposomal structural stabilization and efficient steric protection. Similar methods have been used to construct oral liposomes, such as polysaccharide-anchored liposomes using synthesized glycolipids.Citation12 The above methods can be summarized as liposomal surface premodification because the lipid conjugates are first synthesized, followed by formulation preparation. Researchers have also developed alternative postmodification methods, including direct encapsulation of liposomes by biomacromolecules or through polymerization of functional monomers.Citation13–Citation18 For existing drug-loaded liposomes, postmodification can be performed directly upon the liposomal surface. The silica-coated liposome Liposil, proposed by Devoisselle et al,Citation15 represents one of several promising approaches to liposome postmodification developed in recent years.Citation15–Citation18 These silica-based particles were obtained via liposome templating in a sol-gel reaction. Preliminary tests demonstrated that the silica shell was stable at pH 1.2 and protected the inner structure, while hydrolysis of the silica shell occurred at pH 7.4 and triggered drug release. Because sol-gel-derived silica is biocompatible, chemically inert, and inexpensive, the silica-coating method may provide a potential route for the development of oral liposome formulations.Citation18
Flexible liposomes (FLs), or transfersomes, originally developed for transdermal drug delivery, are liposome-based vesicles mainly composed of phospholipids and surfactants, such as bile salts.Citation19,Citation20 Compared with conventional liposomes, the membrane flexibility and permeability of FLs are greatly increased due to the existence of bilayer softening components. FL vesicles can therefore easily and rapidly adapt their shape to the surrounding environment. It is well known that bile salts and phospholipids enhance oral absorption. Moreover, bile salts may induce the phase transition of liposomal vesicles and create mixed micelles that are excellent vehicles for poorly water-soluble drug molecules, as demonstrated by in vitro assays under simulated gastrointestinal environments.Citation21,Citation22 Consequently, FLs containing bile salts have been explored as a potential drug delivery system for oral administration.Citation6,Citation23–Citation25 For example, the highly lipophilic drug fenofibrate was encapsulated by FLs, greatly increasing the oral bioavailability to a significantly higher level than a conventional liposomal formulation.Citation6 A similar result was obtained using hexamethylmelamine as a model drug.Citation25 However, FLs may also encounter problems with stability, even more severely than conventional liposomes, because of their enhanced membrane fluidity and structural deformability. Oral administration requires a highly stable structure and leakage of reservoir contents before arrival at the absorption sites will significantly decrease the bioavailability and pharmacological effect. Thus, modification of the surface of FLs is necessary to enhance stability and structural integrity against the hostile bioenvironment encountered after oral administration.
In this study, poorly water-soluble curcumin was used as a model drug. Silica-coated FLs containing curcumin were prepared and evaluated both in vitro and in vivo. The oral bioavailability of curcumin from silica-coated FLs, FLs, and curcumin suspensions was measured and compared. Our results clearly demonstrate the efficient combination of silica coatings with FLs for improved oral bioavailability of a poorly water-soluble drug.
Materials and methods
Materials
Curcumin was obtained from Rongsheng Biotechnology Co, Ltd (Xi’an, China). Soybean phosphatidylcholine (SPC) was supplied by AVT Pharmaceuticals, Ltd (Shanghai, China). Sodium deoxycholate (SDC) was purchased from TCI Chemical Reagent Co, Ltd (Tokyo, Japan). Tetraethoxysilane (TEOS) was purchased from J&K Scientific Ltd (Beijing, China). All other chemicals were of analytic grade.
Preparation of CUR-FL and CUR-SL
CUR-FLs were prepared by the dry-film dispersion method. Curcumin, SPC, and SDC (SPC/SDC at 85:15% weight [w]/w) were dissolved in methanol/chloroform (1:1 volume [v]/v) and evaporated under vacuum in a water bath at 25°C. After complete removal of the solvent and a thin film had formed, 10 mL of phosphate buffer (pH 7.4) was added to the lipid film and hydrated for 30 minutes to obtain a crude dispersion of liposomes containing 30 mg curcumin. The liposomal dispersion was then homogenized by a high-pressure homogenizer (ATS, AH100B, Ontario, Canada) at a pressure of 1000 bars for 10 cycles, and purified by passing through a Sepharose CL-4B (Sigma-Aldrich, St Louis, MO, USA) gel-filtration column to remove free curcumin.
CUR-SLs were prepared after obtaining CUR-FLs. The key step was silica shell formation. To achieve this, the inorganic precursor TEOS was added dropwise to the liposome suspension at room temperature to synthesize silica shells, and the mixture (TEOS–SPC at a molar ratio of 8:1)Citation15 was gently stirred for 48 hours (initial pH 8.5, with 0.1 mol · L−1 NaOH used to adjust the pH). To counter the possibility of curcumin leakage during the silica condensation step, which could adsorb onto the external surface of the silica shell, the samples were purified by a Sepharose CL-4B column to remove nonencapsulated curcumin and nondeposited silica.
Characterization of CUR-FL and CUR-SL
The particle size and the polydispersity index (PDI) for each of the liposome preparations were determined by dynamic light scattering (Zetasizer Nano ZS; Malvern Instruments, Malvern, UK) at 25°C. Each experiment was carried out in triplicate. The microstructures of liposomes were analyzed by transmission electron microscope (TEM) (Philips, Amsterdam, The Netherlands) using the negative-stain method. Samples were diluted in deionized water and dropped onto a carbon-coated copper grid coated with carbon film. The generated constructs were then stained with 2% phosphotungstic acid solution and air dried at room temperature. After these steps, TEM observation was carried out in bright field mode by electron diffraction at an operating voltage of 200 kV.
Infrared spectra were obtained from discs containing SPC or silica-coated liposome samples and potassium bromide (KBr) powder. Ten scans over the range of 400–4000 cm−1 were performed at a resolution of 4 cm−1 with 99.999% nitrogen.
Entrapment efficiency
The percentage of curcumin encapsulated by silica-coated FLs was measured in two steps according to the preparation process. First, the entrapment efficiency of CUR-FL was determined by gel filtration. The obtained CUR-FLs before chromatographic purification were divided into two equal parts. One part was purified using the Sephadex CL-4B column and the eluent collected. The other part was diluted into the same volume of the eluent above. The two samples were then treated with 10% Triton X-100 to disrupt the vesicular structure to release curcumin. Entrapment efficiency of CUR-FL (EECUR-FL%) was calculated as
where W and W0 are the curcumin content in purified FLs and total drug in dispersion, respectively. The content of curcumin was measured by high pressure liquid chromatography (HPLC) (L-2000; Hitachi Ltd, Tokyo, Japan) with a mixture of acetonitrile and 2% acetic acid (51:49, v/v) as the mobile phase at a flow rate of 0.7 mL · min−1 and detected at 428 nm. The calibration curve was linear over a concentration range of 3.125ng/mL−1 with a correlation coefficient of >0.999. In the second step, the CUR-SL was purified using the Sephadex CL-4B column, and the eluent was sonicated and incubated with saline at 37°C under vigorous overnight stirring. The sample was then treated with 10% Triton X-100, and the released curcumin was detected by HPLC using the method described above. The apparent entrapment efficiency of CUR-SL (EECUR-SL%) was calculated as
where W′ and W0′ are the curcumin content in purified silica-coated FLs and total drug in dispersion, respectively.
In vitro release assay
In vitro release of curcumin from formulations was evaluated by dynamic dialysis.Citation6 Before the release test, 0.5 mL of curcumin formulation was diluted to 3 mL in the release medium and placed into a dialysis bag (molecular weight cut off 10,000 Da) and incubated in 150 mL of release medium at 37°C in shaking mode. At designated time intervals, 300 μL of release sample was withdrawn and analyzed by HPLC. CUR-FL and CUR-SL were then compared under different simulated bioenvironments.
Determination of curcumin in mice plasma by RP-HPLC
Curcumin in mice plasma was analyzed by HPLC using a mixture of acetonitrile and 2% acetic acid in a volume ratio of 51:49 (v/v) as the mobile phase at a flow rate of 1.0 mL · minute−1. For the measurement of plasma curcumin, 50 μL of internal standard solution (emodin, 4 μg · mL−1 in ethanol) was added to 0.2 mL plasma. After vortex mixing for 1 minute, 500 μL of anhydrous diethyl ether was added and again vortex-mixed for 1 minute. After centrifugation at 10,000 rpm for 10 minutes, the supernatant was withdrawn and evaporated under a light stream of nitrogen at room temperature. The residue was dissolved in 100 μL of the mobile phase and again centrifuged at 10,000 rpm for 10 minutes. Twenty microliters of the supernatant was then injected for HPLC analysis. Quantification was based on the peak area ratio R (ACUR/AEMO). The calibration curve obtained was R = 0.0023C + 0.0335 (r = 0.9998, n = 5). The linear range was from 3.125 to 400 ng·mL−1, with a correlation coefficient > 0.999. The lower limit of quantification was 3.1 ng · mL−1. The accuracy and precision determination were performed with five replicates of three different concentrations – low, medium, and high – of quality control samples. Accuracy of the determination of curcumin in mice plasma at the low quality control (LQC) level was 92.12% ± 3.50%; at the medium quality control (MQC) level, it was 109.32% ± 3.76%; and at the high quality control (HQC), the level was 103.32% ± 0.52%. Within-day and between-day precisions were all below 4.0%. The average extraction recovery of curcumin in mice plasma (n = 5) at the LQC level was 97.49% ± 2.04%; at the MQC level was 96.66% ± 3.03%, and at the HQC level was 97.08% ± 0.38%.
Bioavailability studies
Bioavailability of CUR-SL was compared with those of curcumin-loaded FLs and suspensions. Twenty-four Sprague Dawley rats weighing 200 ± 20 g were randomly divided into three treatment groups and given CUR-SLs, CUR-FLs, and curcumin suspensions. The rats were fasted for 12 hours before the experiment. The dose of each formulation (50 mg · kg−1, expressed as curcumin equivalent doses) was given through gavage administration. Subsequently, 200 μL of blood sample was collected from the post-eyelid vein into heparinized tubes at indicated time intervals. Plasma samples were separated by centrifugation at 10,000 rpm for 10 minutes, then frozen and stored at −20°C until analysis. Curcumin pharmacokinetic parameters were determined using the computer software Kinetica 4.4 (Innaphase, Philadelphia, PA). Cmax and Tmax were observed as raw data. The area under the plasma concentration–time curve from zero to the last measurable plasma concentration (AUC0–t) was calculated by the linear trapezoidal rule.
Statistical analysis
Raw data were analyzed using the SPSS statistical software (version 11.0, IBM Corporation, Armonk, NY). The Student’s t-test was applied, and P < 0.05 was considered statistically significant.
Results and discussion
Preparation and characterization of CUR-SL
In recent decades, silica-based drug delivery has been a leading topic in pharmaceutical science. While traditional organic systems (eg, liposomes, micelles, and polymeric nanoparticles) suffer limitations, such as low physicochemical stability and rapid elimination by the immune system, silica particles offer a potentially biocompatible, stable, and stealth alternative.Citation26 A large number of silica-based pharmaceutical studies have been reported, including controlled and targeted drug delivery, even at the subcellular level.Citation27–Citation29 In contrast to most of the other reports in this field, Dessivio et alCitation15 proposed a hybrid strategy to take advantage of both silica and organic systems. The constructed silica-coated liposomes, called “Liposil,” possessed the high stability of silica materials and versatile drug-loading capabilities of liposomes, providing a novel potential carrier for drug delivery, especially in hostile bioenvironments. The mechanism behind the formation of the stable silica shell upon the liposomal surface was first explained as directed by attraction to the quaternary ammonium surface of the phospholipids on the silica. This was further explained by stabilizing effects achieved through the presence of water molecules located between the head groups of the phospholipids and the silica surface, forming an interfacial H-bonded water layer.Citation30 The silica shell formation can be obtained by either a prehydrolysis or a hydrolysis process of the inorganic precursor TEOS, according to methods described in the literature. While the former process includes a preliminary 48-hour hydrolysis of TEOS before incubation with liposomes for another 72 hours, the latter is based on a synchronous process of precursor hydrolysis and surface precipitation for 60 hours.Citation15,Citation17 In our case, we chose the second method and further reduced the silica shell formation time to 48 hours.
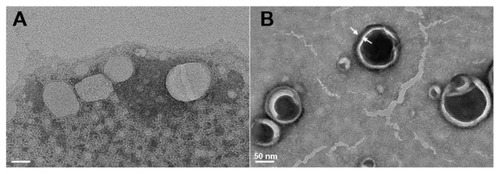
According to the dynamic light scattering (DLS) measurement, CUR-SL has a narrow size distribution around 157 nm with a polydispersity index of 0.14, while freshly prepared samples of CUR-FL had a diameter of 91 nm (). The precipitation of silica on the surface of the FLs resulted in the formation of a core–shell-like structure, according to TEM (). A more spherical shape was observed for the silica-coated liposomes than for conventional FLs, suggesting a possible external structural reinforcement of the ultradeformable lipid bilayer by the silica shell.
Figure 1 Vesicle sizes of (A) CUR-FL and (B) CUR-SL as determined by dynamic light scattering.
Abbreviations: CUR-FL, curcumin-loaded flexible liposomes; CUR-SL, silica-coated flexible liposomes loaded with curcumin.
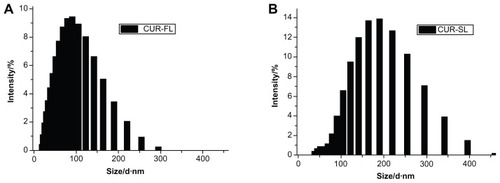
Figure 2 Morphologies of flexible liposome (A) and silica-coated flexible liposomes (B), measured by transmission electron microscopy (bar = 50 nm).
A Fourier transform infrared (FT-IR) spectroscopy study was carried out to confirm the presence of the silica coating in the formulation. shows the FT-IR spectra of blank FLs and silica-coated FLs without curcumin (SLs). For FLs, the peak observed at 2924 cm−1 was attributed to methylene C-H antisymmetric stretching, and the peak at 1087 cm−1 was due to the P-O of SPC.Citation31,Citation32 The FT-IR spectra of SLs showed vibrational absorption bands centered around 1080 cm−1 (asymmetric Si-O-Si stretching), 859 cm−1 (Si-O-Si bending vibration), and 470 cm−1 (Si-O bending), providing direct evidence confirming the formation of the silica shell.Citation16–Citation18
Figure 3 (A) FT-IR spectra of SL and FL (B) FT-IR spectra of CUR, CUR-FL, and CUR-SL.
Abbreviations: FT-IR, Fourier transform infrared; SL, silica-coated flexible liposomes; FL, flexible liposomes; CUR, curcumin; CUR-FL, curcumin-loaded flexible liposomes; CUR-SL, silica-coated flexible liposomes loaded with curcumin.
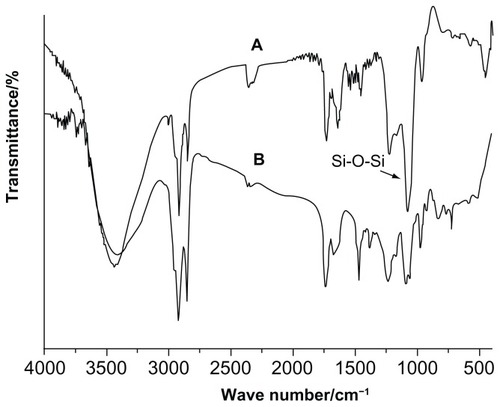
The FT-IR spectra of curcumin, CUR-FL, and CUR-SL are shown in . The bands at 3444 cm−1 (O-H), 1508 cm−1 (C=C), and 1280 cm−1 (C=O) of curcumin correspondingly shifted to 3421 cm−1, 1635 cm−1, and 1237 cm−1 of CUR-FL, suggesting the encapsulation of curcumin in liposomes.Citation33 In addition, CUR-SL showed Si-O-Si stretching through a band around 1080 cm−1, which was similar to that for SLs.
The encapsulation efficiency of CUR-FL was 93.28% ± 2.51% (n = 3). No obvious drug leakage or liposome structure destabilization was observed under the mild silica-coating conditions. Because of sensitivity of silica coatings to ultrasonic sound and the hydrolysis of silica coatings at neutral pH,Citation15 we chose a method combining sonication and incubation at neutral pH to completely remove the silica shell from CUR-SLs. The concentrations of CUR-FL before and after silica encapsulation, referred to as C0′ and C′, varied little, and the ratio of C′ to C0′ was about 0.97. The mean value of the apparent encapsulation efficiency in CUR-SL, based on the value in CUR-FL, was calculated as 90.62% (n = 3).
In vitro release and stability
In vitro release under a simulated bioenvironment has been used as an important indicator of in vivo performance. We compared in vitro release profiles of CUR-SL and CUR-FL (), using 2% sodium dodecyl sulfate in both artificial gastric and intestinal fluids to serve as a surfactant to improve the solubility of curcumin. To distinguish free curcumin molecules from those associated with liposomes, a dynamic dialysis method was used to prevent the escape of liposomal particles into the outer release medium.
Figure 4 In vitro release profiles in (A) 2% SDS artificial gastric juice for CUR-FL (-▬-) and CUR-SL (
Note: *P < 0.05.
Abbreviations: SDS, sodium dodecyl sulfate; CUR-FL, curcumin-loaded flexible liposomes; CUR-SL, silica-coated flexible liposomes loaded with curcumin.
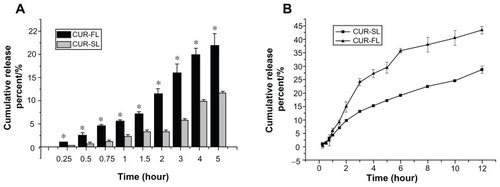
The release assay in artificial gastric fluid was performed within 5 hours because normal stomach emptying has several hours duration. The amount of drug release from FLs was significantly reduced when encapsulated by the silica shell, indicating the protection of the inner liposome structure in acidic conditions by the silica coating. In the artificial intestinal fluid, a more sustained drug release was observed for silica-coated FLs, possibly due to the gradual rupture of the silica shell on the liposomal surface.
In vitro stability was also determined. After maintenance at room temperature for one month, the particle size of CUR-FL increased 5.85-fold, while that of CUR-SL increased almost 40%. A similar trend was observed at 4°C for CUR-FL and CUR-SL (254.73 versus 19.89% change in particle sizes, P < 0.01). These results demonstrate an improvement in the release properties and the superiority of CUR-SLs over CUR-FLs.
Bioavailability studies
To further investigate the role of silica shell, the oral bioavailability of CUR-SL in rats was compared with those of curcumin suspensions and FLs. Mean concentration data were plotted against time to create plasma curcumin concentration versus time profiles (). The double-peak phenomenon observed was possibly due to the enterohepatic circulation of curcumin.Citation34
Figure 5 Mean curcumin plasma concentration profiles of CUR-SL (-▲-), CUR-FL (-▲-), and CUR-SU (-■-) following oral administration at a dose of 50 mg · kg−1 (n = 8).
Abbreviations: CUR-SL, silica-coated flexible liposomes loaded with curcumin; CUR-FL, curcumin-loaded flexible liposomes; CUR-SU, curcumin suspensions.
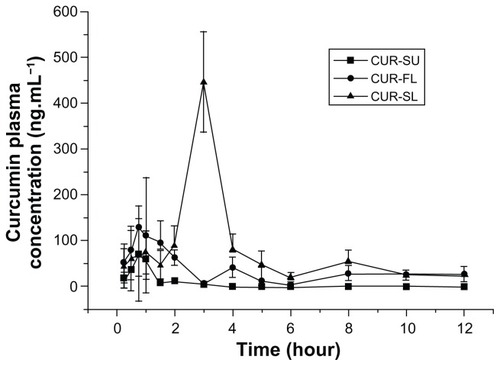
After gavage administration, liposomal formulations showed significantly improved absorption files over a plain curcumin suspension. After normalization by the AUC0−t value of the curcumin suspension, the bioavailability of CUR-FL was approximately 2.35-fold higher. Furthermore, CUR-FL showed greater absorption than a curcumin suspension, as indicated by their respective Cmax values (128.78 vs 71.35 ng · L−1; P < 0.01). FLs containing bile salts have been reported to enhance the oral absorption of poorly water-soluble drugs. Possible mechanisms include but are not limited to an enhancement effect by sodium deoxycholate on liposome vesicle–micelle phase transition and carrier-mediated transmembrane absorption conferred by the ultradeformability of FLs.Citation6,Citation21,Citation22 In this study, a positive effect by FLs on the poorly water-soluble curcumin was observed, but the unstable structure of the FLs necessitated surface modification for further application in drug delivery. After the silica cladding on the liposomal surface, the peak in plasma curcumin with CUR-SL delivery was delayed compared with CUR-FL (3 hours versus 0.75 hour; P < 0.01), with more sustainable plasma concentrations. The highest absorption was exhibited by CUR-SL, with greater Cmax and AUC0−t values than CUR-FL (446.66 vs 128.78 ng · L−1 (P < 0.01) and 673.79 vs 203.64 ng · hour · L−1; P < 0.01). It is remarkable that the silica-coating strategy improved the properties of FLs, both in vitro and in vivo.
Conclusion
Curcumin has aroused great interest worldwide because of its various biological activities. However, its relative insolubility in water and instability cause extremely low oral absorption, which greatly restricts its clinical application. Therefore, multidisciplinary methods have been explored over several decades to improve the in vivo behavior of curcumin. For example, structural modification or cyclodextrin complexation has been used as molecular strategies to enhance the oral bioavailability of curcumin.Citation35,Citation36 Researchers have also investigated nanoscale formulations of curcumin, such as liposomes, microemulsions, and micelles, for potential therapeutic applications.Citation37–Citation42 The nanoscale effect by which such drug delivery systems improve the oral performance of curcumin involves increasing the surface area and interactions of curcumin, thereby providing better water dispersibility to enhance its absorption.Citation38
In this study, a novel silica-coating liposomal strategy was used to construct a nanohybrid formulation for curcumin. Silica-coated FLs were prepared by the dry-film dispersion method, combined with a sol–gel precipitation upon the liposomal surface. The stability of the resultant formulation was significantly improved but retained the existing advantages of liposomal delivery. In vivo assays showed enhanced oral bioavailability of curcumin by FLs surrounded by a silica shell compared with conventional FLs and plain suspensions of curcumin. This indicates that silica-coated FLs have potential applications as a novel oral delivery system, especially for poorly water-soluble drugs.
Acknowledgments
The authors are grateful for financial support from the National Natural Science Foundation of China (No 81102404), the National Creating New Drug Program of China (No 2010-ZX-09401-306-1-4), the Doctoral Fund of Ministry of Education of China (20110182120015), the Natural Science Foundation of Chongqing (cstc2011 jjA1585), a special fund of the Chongqing Key Laboratory (CSTC), Foundation for University Key Teachers by the Chongqing Municipal Education Commission (2011), the Fundamental Research Funds for the Central Universities (XDJK2013A010), and the doctoral fund of the Southwest University (SWU110028).
Disclosure
The authors report no conflicts of interest in this work.
References
- TorchilinVPRecent advances with liposomes as pharmaceutical carriersNat Rev Drug Discov2005414516015688077
- YingXWenHLuWLDual-targeting daunorubicin liposomes improve the therapeutic efficacy of brain glioma in animalsJ Control Release201014118319219799948
- QinYChenHZhangQLiposome formulated with TAT-modified cholesterol for improving brain delivery and therapeutic efficacy on brain glioma in animalsInt J Pharm201142030431221945185
- FrickerGKrompTWendelAPhospholipids and lipid-based formulations in oral drug deliveryPharm Res2010271469148620411409
- RogersJAAndersonKEThe potential of liposomes in oral drug deliveryCrit Rev Ther Drug Carrier Syst1998154214809822867
- ChenYLuYChenJEnhanced bioavailability of the poorly water-soluble drug fenofibrate by using liposomes containing a bile saltInt J Pharm200937615316019394416
- ParmentierJBeckerMMHeintzUStability of liposomes containing bio-enhancers and tetraether lipids in simulated gastro-intestinal fluidsInt J Pharm201140521021721145956
- AriënAGoigouxCBaqueyCStudy of in vitro and in vivo stability of liposomes loaded with calcitonin or indium in the gastrointestinal tractLife Sci199353127912908412488
- ChiangCMWeinerNGastrointestinal uptake of liposomes. I. In vitro and in situ studiesInt J Pharm1987377585
- MinatoSIwanagaKKakemiMApplication of polyethyleneglycol (PEG)-modified liposomes for oral vaccine: effect of lipid dose on systemic and mucosal immunityJ Control Release20038918919712711443
- LiHSongJHParkJSPolyethylene glycol-coated liposomes for oral delivery of recombinant human epidermal growth factorInt J Pharm2003258111912753749
- SihorkarVVyas1SPPotential of polysaccharide anchored liposomes in drug delivery, targeting and immunizationJ Pharm Pharmaceut Sci20014138158
- MuXZhongZPreparation and properties of poly(vinyl alcohol)-stabilized liposomesInt J Pharm2006318556116624507
- TakeuchiHYamamotoHToyodaTPhysical stability of size controlled small unilameller liposomes coated with a modified polyvinyl alcoholInt J Pharm2006318556116624507
- BeguSAubert-PouesselALernerDALiposil, a promising composite material for drug storage and releaseJ Control Release20071181617250924
- QiJJYueXLWangYEncapsulation and relese of doxorubicin form silica-coated liposomeJ Clin Rehab Tissue Eng Res20091341854188
- BéguSDurandRLernerDAPreparation and characterization of siliceous material using liposomes as templateChem Commun20037640641
- DwivediNArunagirinathanMASharmaSSilica-coated liposomes for insulin deliveryJ Nanomaterials2010Article ID 65204810.1155/2010/652048
- CevcGSchätzleinARichardsenHUltradeformable lipid vesicles can penetrate the skin and other semi-permeable barriers intact. Evidence from double label clsm experiments and direct size measurementsBiochim Biophys Acta20021564213012100992
- CevcGLipid vesicles and other colloids as drug carriers on the skinAdv Drug Deliv Rev20045667571115019752
- PorterCJTrevaskisNLCharmanWNLipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugsNat Rev Drug Discov2007623124817330072
- AndrieuxKForteLLesieurSSolubilisation of dipalmitoyl phosphatidyl-choline bilayers by sodium taurocholate: a model to study the stability of liposomes in the gastrointestinal tract and their mechanism of interaction with a model bile saltEur J Pharm Biopharm20097134635518835441
- NiuMMLuYHovgaardLLiposomes containing glycocholate as potential oral insulin delivery systems: preparation, in vitro characterization, and improved protection against enzymatic degradationInt J Nanomedicine201161155116621822379
- GuanPPLuYQiJPEnhanced oral bioavailability of cyclosporine A by liposomes containing a bile saltInt J Nanomedicine2011696597421720508
- SunJDengYWangSLiposomes incorporating sodium deoxycholate for hexamethylmelamine (HMM) oral delivery: development, characterization, and in vivo evaluationDrug Deliv20101716417020196698
- ChristopheBJohnBLinggenKSilica particles: a novel drug-delivery systemAdv Mater20041618
- TimoLChristopheJJensMNanostructured silica materials as drug-delivery systems for doxorubicin: single molecule and cellular studiesNano Lett200992877288319572735
- ZhangYWangJBaiXMesoporous silica nanoparticles for increasing the oral bioavailability and permeation of poorly water soluble drugsMol Pharm2012950551322217205
- PanLHeQLiuJNuclear-targeted drug delivery of TAT peptide-conjugated monodisperse mesoporous silica nanoparticlesJ Am Chem Soc20121345722572522420312
- FollietNRoilandCBéguSInvestigation of the interface in silica-encapsulated liposomes by combining solid state NMR and first principles calculationsJ Am Chem Soc2011133168151682721899369
- CavallaroGMannaGLLiveriVTStructural investigation of Water/ Lecithin/ Cyclohexane Microemulsions by FT-IR SpectroscopyJ Colloid Interf Sci1995176281285
- DreherFWaldePWaltherPInteraction of a lecithin microemulsion gel with human stratum corneum and its effect on transdermal transportJ Control Release199745131140
- DasRKKasojuNBoraUEncapsulation of curcumin in alginate-chitosan-pluronic composite nanoparticles for delivery to cancer cellsNanomedicine2009615316019616123
- BelkacemiADogguiSDaoLChallenges associated with curcumin therapy in Alzheimer diseaseExpert Rev Mol Med201113e3422051121
- ParvathyaKSNegibPSSrinivasaPCurcumin–amino acid conjugates: Synthesis, antioxidant and antimutagenic attributesFood Chem2010120523530
- YadavVRSureshSDeviKEffect of cyclodextrin complexation of curcumin on its solubility and antiangiogenic and anti-inflammatory activity in rat colitis modelAAPS Pharm Sci Tech200910752762
- TangHMurphyCJZhangBAmphiphilic curcumin conjugate-forming nanoparticles as anticancer prodrug and drug carriers: in vitro and in vivo effectsNanomedicine2010585586520735222
- TsaiYMChang-LiaoWLChienCFEffects of polymer molecular weight on relative oral bioavailability of curcuminInt J Nanomedicine201272957296622745556
- TakahashiMUechiSTakaraKEvaluation of an oral carrier system in rats: bioavailability and antioxidant properties of liposome-encapsulated CurcuminJ Agric Food Chem2009579141914619757811
- ZhongfaLChiuMWangJEnhancement of curcumin oral absorption and pharmacokinetics of curcuminoids and curcumin metabolites in miceCancer Chemother Pharmacol20126967968921968952
- CuiJYuBZhaoYEnhancement of oral absorption of curcumin by self-microemulsifying drug delivery systemsInt J Pharm200937114815519124065
- WangKZhangTLiuLNovel micelle formulation of curcumin for enhancing antitumor activity and inhibiting colorectal cancer stem cellsInt J Nanomedicine201274487449722927762