Abstract
A novel nanocarrier system was designed and developed with key components uniquely structured at the nanoscale for early cancer diagnosis and treatment. In order to perform magnetic resonance imaging, hydrophilic superparamagnetic maghemite nanoparticles (NPs) were synthesized and coated with a lipophilic organic ligand. Next, they were entrapped into polymeric NPs made of biodegradable poly(lactic-co-glycolic acid) linked to polyethylene glycol. In addition, resulting NPs have been conjugated on their surface with a 2,2′-(7-(4-((2-aminoethyl)amino)-1-carboxy-4-oxobutyl)-1,4,7-triazonane-1,4-diyl)diacetic acid ligand for subsequent 68Ga incorporation. A cell-based cytotoxicity assay has been employed to verify the in vitro cell viability of human pancreatic cancer cells exposed to this nanosystem. Finally, in vivo positron emission tomography-computerized tomography biodistribution studies in healthy animals were performed.
Introduction
Despite impressive progress in basic and clinical medicine, cancer still ranks as one of the leading causes of mortality in Europe and in the United States.Citation1,Citation2 Consequently, the development of imaging agents and diagnostic methods for detecting cancer at an early presymptomatic stage as well as new therapeutic agents able to overcome the problems commonly associated with traditional treatments is urgently required.
Cancer diagnosis utilizes anatomical or molecular imaging methods such as magnetic resonance imaging (MRI), positron emission tomography (PET), single photon emission computerized tomography (CT), ultrasound imaging, optical imaging. Each of these modalities has its own limitations in terms of spatial resolution, sensitivity, and depth of signal penetration that might be overcome by the combination of two or more such modalities.Citation3 Therefore, dual or multimodal imaging are currently being explored to enhance the quality and specificity of imaging methods in order to achieve proper visualization of organs or to better localize the accumulation of a diagnostic or therapeutic agent. Concomitantly, the development of multimodal imaging probes has attracted much attention during the last years.
Recently, nanotechnology has emerged as an excellent tool for the generation of nanoparticles (NPs) for diagnostic (imaging) or therapeutic purposes. Among all NPs, inorganic ones have attracted great interest, given their intrinsic physical properties and compatible size. The best example can be found in magnetic NPs (MNPs) which have been used as contrast agents for MRI for more than 20 years.Citation4 However, the pursuit of multimodal imaging agents requires the development of organic ligands that are able to make the MNPs lipophilic (coated with an organic molecule), and that entrap them into a polymeric matrix acting as a targetable water soluble nanocarrier. In ideal planning, this nanocarrier would be further functionalized with an appropriate labeling agent (eg, a positron emitter), to allow multi-modal in vivo imaging. In order to achieve efficient distributions in the targeted tumor lesions for early cancer diagnosis and treatment, the colloidal nanocarriers are required to be highly monodisperse and smaller than 200 nm.Citation5
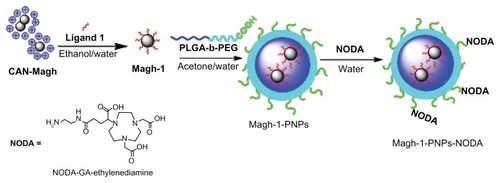
Here we describe the preparation and characterization of a novel, versatile, and biocompatible hybrid nanocarrier for dual PET/MRI imaging. This nanocarrier contains a superparamagnetic maghemite core consisting of crystalline, as well as hydrophilic and strongly positively charged ceric ammonium nitrate (CAN)-stabilized maghemite NPs for MRI contrast,Citation6 which were encapsulated in the well-known poly(D,L-lactide-co-glycolide)-block-poly(ethylene glycol) copolymer (PLGA-b-PEG-COOH). Polyethyleneglycol (PEG)-based polymeric nanoparticles (PNPs), owing to their stealth character,Citation7 are widely used delivery systems for in vivo applications. On the other hand, the PLGA-b-PEG-COOH copolymer self-assembles to form targetable PNPs consisting of a hydrophobic poly(D,L-lactide-co-glycolide) (PLGA) core and a hydrophilic PEG corona-like shell. Interestingly, the presence of active sites (COOH) on the surface allows surface modification with 2,2′-(7-(4-((2-aminoethyl)amino)-1-carboxy-4-oxobutyl)-1,4,7-triazonane-1,4-diyl)diacetic acid (NODA) to chelate 68Ga for PET imaging (see for the schematic representation of the fabrication of this nanocarrier). Since the resulting composite presented a high T2 relaxivity, such a system should be suitable as an MRI contrast agent. Furthermore, the 68Ga-radiolabeled NPs proved to be stable and allowed in vivo biodistribution studies in healthy animals using PET-CT. Further attachment of functional groups that can act as targeting moieties or therapeutic agents could be pursued in the near future using the functional groups present on the NP surface, leading to multimodal PET-MRI diagnostic agents. The combination of both PET and MRI imaging capabilities should provide images with high anatomical resolution of MRI at the sensitivity of PET (10−9 to 10−12 M).Citation8
Figure 1 Schematic representation of the synthesis of Magh-1-PNPs-NODA.
Abbreviations: CAN, ceric ammonium nitrate; PNPs, polyethyleneglycol-based nanoparticles; NODA, 2,2′-(7-(4-((2-aminoethyl)amino)-1-carboxy-4-oxobutyl)-1,4,7-triazonane-1,4-diyl)diacetic acid; PLGA-b-PEG, poly(D,L-lactide-co-glycolide)-block-poly(ethylene glycol).
Despite the suitability of the nanoplatforms reported here for the preparation of a wide range of diagnostic multimodal agents, one first application is expected in the field of pancreatic cancer diagnosis in the frame of the SaveMe project (EU-FP7 project number CP-IP 263307-2). Thus, a preliminary investigation (in vitro toxicity tests) of the developed nanosystem has been performed.
An application to pancreatic ductal adenocarcinoma (PDAC) has been preliminarily investigated. PDAC is one of the most aggressive malignancies with an overall 5-year survival rate of only 3%–5% and a median survival time after diagnosis of 6 months.Citation9 This highly lethal disease is usually diagnosed in an advanced stage when there are few or no effective therapies available.Citation10
Materials and methods
All chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA) and used as received. Poly(D,L-lactide-co-glycolide) (50/50) with carboxylic acid end group (PLGA-COOH, pharmaceutical grade, inherent viscosity 0.12 dL/g, molecular weight (MW) ~7 kDa) was purchased from Lakeshore Biomaterials (Lakeshore Biomaterials, Inc, Birmingham, AL, USA). PEG with amino and carboxylic acid end groups (NH2-PEG-COOH, >95%, MW ~3 kDa) was purchased from Rapp Polymere GmbH (Tübingen, Germany). All aqueous solutions were prepared with deionized water obtained using an ultrafiltration system (Milli-Q, Millipore Corporation, Billerica, MA, USA) with a measured resistivity above 18 MΩ/cm (referred in the text as double distilled [dd] water). Tetrahydrofuran was distilled from sodium/benzophenone just prior to use and stored under Ar. CH2Cl2 and CHCl3 were passed through basic alumina prior to use. Transmission electron microscopy (TEM) was conducted on a JEOL JEM 2010 at 200 keV (JEOL, Tokyo, Japan). Samples for TEM analyses were prepared by spreading a small drop of the NP dispersion on amorphous carbon-coated copper grids (Formvar carbon 400 mesh grids) followed by air-drying. Inductively coupled plasma (ICP) was performed on diluted acidic aqueous solutions using a Jobin Yvon Ultima 2 device (HORIBA, Ltd, Fukuoka, Japan). 1H-NMR (hydrogen-1 nuclear magnetic resonance) and 13C-NMR (carbon nuclear magnetic resonance) spectra were recorded using CDCl3 or dimethyl sulfoxide solutions at 300, 400, and 600 MHz for 1H, and 75.46, 100.6, and 150.92 MHz for 13C. Chemical shifts (δ) are reported in ppm relative to CHCl3 (δ = 7.26 for 1H and δ = 77.0 for 13C). Fourier transform infrared spectra were recorded on a PerkinElmer Spectrum 2000 (PerkinElmer, Waltham, MA, USA). Dynamic light scattering (DLS) measurements were performed on a Malvern Zetasizer nano-S working with a 532 nm laser beam (Malvern Instruments Ltd, Malvern, UK). Moreover, ζ potential measurements were conducted in DTS1060C-clear disposable zeta cells at 25°C. SpectraAA 100 Varian (Palo Alto, CA, USA) was used for atomic absorption spectroscopy analyses. Thermogravimetric analyses were carried out on a TA Instruments 1GA Q500 (TA Instruments, New Castle, DE, USA) apparatus (nitrogen flow, isothermal temperature accuracy of 0.1°C for a weighting precision of 0.1 μg).
Synthesis of ethyl 12-((3,4-dihydroxyphenethyl)amino)-12-oxododecanoate 1 was conducted as reported in the steps 2–4 of the supplementary material (see also Figures S2–S4).
Synthesis of Magh-1
CAN-maghemite NPs were synthesized according to a known procedure that was recently developed in our laboratory with slight modifications, using preformed magnetite NPs as starting nanoscale material (see supporting information for details concerning the all synthesis pathways).Citation6
Accordingly, the original CAN-Magh solution was diluted with water and added to an ethanolic solution of ethyl 12-([3,4-dihydroxyphenethyl]amino)-12-oxododecanoate (EDAO) 1. The mixture was sonicated in an ultrasound bath for 1 hour, and it was left to react overnight at room temperature under mechanical stirring. Afterward, the solution was decanted magnetically.
Synthesis of Magh-1-PNPs
A total of 560 mg of PLGA-b-PEG-COOH (10 kDa) was added to 56 mL of an acetone solution of Magh-1. The organic phase was mixed to 560 mL of ultrapure water under vigorous stirring, maintaining the water/organic ratio of 10/1 with constant removal of the solution. The mixture was kept for 30 minutes under vigorous stirring. The residual organic solvent was evaporated under reduced pressure. The solution was concentrated to a volume of 100 mL using a tangential flow filter (Pellicon XL filter device, Biomax membrane with 500.000 NMWL; Millipore Corporation) following by filtration using a syringe filters SterivexTM-GP of polyethersulfone (0.22 μm, Millipore Corporation).
Synthesis of Magh-1-PNPs-NODA
The linkage of NODA on the surface of the particle was carried out through a classic peptide formation. In particular, Magh-1-PNP water solutions were stirred for 2 hours in the presence of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide. Then, NODA-GA-ethylenediamine were added, and the reaction mixture was kept under stirring for an additional 2 hours. The particles were purified with a centrifugal filter device.
Nuclear magnetic resonance (NMR) relaxivity
Relaxivity measurements were performed on a Bruker Minispec TD-NMR system (Bruker Optik GmbH, Ettlingen, Germany) at 37°C operating at 1.5 Tesla and 60 MHz proton resonance frequency. The longitudinal relaxation times (T1) and transverse relaxation times (T2) were determined at the following concentrations for each nanocomposite: 0.01, 0.03, 0.1, 0.3, 1, and 3 mM Fe in 50% D2O/H2O. A linear regression of the concentration versus T1 relaxation time or T2 relaxation time yielded R1 and R2 values [mmol−1 s−1], as summarized in .
Table 1 Relaxivity values obtained for Magh, Magh-1-PNPs, and Magh-1-PNPs-NODA
Radiolabeling of NPs with 68Ga
The 68Ga (T1/2 = 68 minutes, β+ = 89%, Electronic Capture (EC) = 11%) was obtained from an iThemba 68Ge/68Ga generator system (IDB Holland bv, Baarle-Nassau, Netherlands) with a nominal 68Ge activity of 740 MBq installed at CIC biomaGUNE (Guipuzcoa, Spain). The generator was eluted with 0.6 M of HCl solution (6 mL). Aqueous HCl solution (37%, 2.5 mL) was added to the generator eluate, giving a final HCl concentration of 4 M. The resulting solution (total volume = 8.5 mL) was passed through a Chromafix® 30-PS-HCO3-anion-exchange column (MACHEREY-NAGEL GmbH and Co, Düren, Germany; 45 mg, conditioned sequentially with 3 mL of purified water and 3 mL of 4 M aqueous HCl solution) at a flow rate of 3 mL/minute at room temperature and dried under air. The 68Ga was then eluted with 2 mL of purified water at a flow rate of 0.7 mL/min and the eluate (~220 MBq) was collected in a vial precharged with 45 mg of 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES). After the addition of 1 M NaOH solution (5–10 μL, final pH = 3.5) to the 68Ga eluate, 200 μL of NPs (Magh-1-PNPs-NODA) solution were added and the vial, and was heated at 60°C for 30 minutes. The reaction crude was cooled at room temperature and 68Ga-labeled NPs were filtered under centrifugation (10,000 rpm, 10 minutes) using Amicon® Ultra-4 Centrifugal Filter Units (10 kDa, Millipore Corporation). The residue was washed twice with HEPES solution (pH = 3.5) and finally suspended in physiologic saline solution. All fractions were measured in a dose calibrator (CRC® – 25PET Dose Calibrator, Capintec, Inc, Ramsey, NJ). The labeling yield of the NPs was calculated as the ratio between the amount of radioactivity in the NPs solution and the total amount of radioactivity in all fractions.
Quality control of the NPs was performed by thin layer chromatography (TLC). A fraction of the NPs solution (100 μL) was mixed with a solution containing ethylenediaminetetraacetic acid (EDTA, 1 mg/mL). After shaking for 10 seconds, 5 μL were deposited on a silica gel 60 F254 plate (Macherey-Nagel) and eluted with a mixture of buffered 0.1 M aqueous NaOAc solution (pH = 4.5)/0.1 M aqueous EDTA solution 90/10. After complete elution, the plates were dried at 60°C and analyzed using a radio-TLC reader (miniGITA Star, Raytest Isotopenmessgeräte GmbH, Straubenhardt, Germany) and the fractions corresponding to labeled NPs (Rf < 0.1) and free gallium (Rf ~0.35) were quantified. The same analytical procedure and conditions were used to assess the stability of labeled NPs in physiologic saline solution and in rat serum; in this case, the NPs solution was analyzed at different times after preparation (15, 30, 60, and 90 minutes).
Toxicity assays
Cell lines
Human PDAC-derived cell lines Panc-1, ASPC-1, MiaPaCa, Su86.86, T3M4, BxPC3, and Colo-357 were kindly provided by Dr Klaus Felix (University of Heidelberg, Heidelberg, Germany). ASPC-1, Su86.86, T3M4, BxPC3, and Colo-357 cell lines were maintained in culture in Roswell Park Memorial Institute medium supplemented with 10% fetal bovine serum, L-glutamine, penicillin, and streptomycin. Panc-1 and MiaPaCa cell lines were maintained in DMEM supplemented with 10% fetal bovine serum, L-glutamine, penicillin, and streptomycin.
Viability assay
PDAC cells were seeded in flat-bottom 96-well plates at 5000 cells/well in a volume of 200 μL of supplemented medium. After 72 hours of culture in the presence/absence of increasing dosage of Magh-1-PNPs-NODA NPs, 25 μL of 5 mg/L 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma-Aldrich, St Louis, MO) was added directly to the cells followed by an additional 4 hours of incubation, then 100 μL of DMSO was added. The optical density of individual wells was measured at a wavelength of 550 nm with Infinite® F500 (Tecan Group, Ltd, Mannedorf, Switzerland). All assays were performed in triplicate.
Imaging studies
Image acquisition
Male rats weighting 325–375 g (Sprague–Dawley, Harlan Laboratories, Udine, Italy) were used to perform PET studies. The animals were maintained and handled in accordance with the Guidelines for Accommodation and Care of Animals (European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes) and in accordance with internal guidelines, and experimental procedures were previously approved by local authorities. PET studies were performed using an eXploreVista-CT small animal PET-CT system (GE Healthcare, Little Chalfront, England). During PET studies, rats were kept normothermic using a heating blanket (Homoeothermic Blanket Control Unit; Bruker Optik GmbH). Three animals were submitted to whole body scans to assess the biodistribution pattern of labeled NPs. In all cases, anesthesia was induced with 3% isoflurane and maintained by 1.5% to 2% of isoflurane in 100% O2. The tail vein was catheterized with a 24-gauge catheter for intravenous administration of 68Ga-labeled NPs (7.4 ± 2.4 MBq, corresponding to 3.8 ± 1.3 μg of Fe2O3, resuspended in 300 μL of physiologic saline solution), which was injected concomitantly with the start of a PET dynamic acquisition.
Dynamic images (32 frames: 4 × 5 seconds, 4 × 10 seconds, 6 × 30 seconds, 6 × 60 seconds, 6 × 150 seconds, and 6 × 300 seconds) were acquired in four bed positions in the 400–700 keV energetic window, with a total acquisition time of 220 minutes. After each PET scan, CT acquisitions were also performed providing anatomical information as well as the attenuation map for the later image reconstruction. Dynamic acquisitions were reconstructed (decay and CT-based attenuation corrected) with filtered back projection using a ramp filter with a cut-off frequency of 1 Hz.
Image analysis
PET images were analyzed using PMOD image analysis software (PMOD Technologies Ltd, Zürich, Switzerland). Volumes of interest were manually drawn in the lungs, liver, kidneys, brain, heart, spleen, bladder, and bone using the CT images as anatomical reference. Volumes of interest were then transferred to the PET images, and time activity curves (decay corrected) were obtained for each organ as cps/cm3. Injected dose and organ mass normalizations were finally applied to data to get time activity curves as standardized uptake values for each organ.
Results and discussion
Chemistry and nanotechnology
In a previous work, the production of hydrophilic water-compatible maghemite (γ-Fe2O3) was described.Citation6 Accordingly, the experimental conditions did not involve any surface-passivating bifunctional ligand, nor routinely used physically adsorbed natural/nonnatural polymer. More specifically, this fabrication process comprises the high-power ultrasonic irradiation of preformed 10–15 nanosized magnetite (Fe3O4) NPs together with the strong oxidant ceric ammonium nitrate [CAN, CeIV(NH4)2(NO3)6] in a 1/1 v/v acetone/H2O mixture.Citation6 Under these conditions, the simultaneous oxidation of the magnetite NPs to maghemite NPs and CAN-mediated modification(s) of the NP surface charge/functionality resulted in crystalline, hydrophilic, and strongly positively charged CAN-stabilized maghemite NPs that formed extremely stable colloidal water dispersions due to electrostatic repulsive interactions. These conditions also promote the formation of a polyCOOH-containing organic shell on the NP surface, which allows further grafting of the particles.Citation11
These NPs have an average hydrodynamic radius of 44–55 nm (DLS measurements), a ζ potential of ~40–50 mV, and are 8–12 nm in average size as measured from TEM (TEM micrograph analysis of more than 100 particulate objects). In addition, 57Fe Mössbauer spectroscopy performed at 300 K afforded a spectrum that discloses asymmetrical sextets of broadened lines that were attributed to significant superparamagnetic fluctuations. The NPs superparamagnetism feature that is essential for MRI application, was also confirmed by superconductive quantum interference device.Citation6 The so-formed CAN-maghemite NPs are water-dispersible, and in these conditions (ie, without any surface modification), their entrapment into polymeric NPs is rather hard. In order to solve this phase compatibility problem, the maghemite NP surface was coated by a hydrophobic organic ligand (ethyl 12-((3,4-dihydroxyphenethyl)amino)-12-oxododecanoate (herein referred as ligand 1) specially designed to ensure lipophilicity to the iron oxide NPs. Ligand 1 possesses a catechol unit at one end, a central aliphatic chain, and a terminal ester group. On the one hand, the catechol unit is well known to present a strong affinity to iron due to both the σ and π donor power of oxygen atoms.Citation12 On the other hand, the aliphatic chain is necessary to confer stability to the systems thanks to interchain hydrogen bonding, while the terminal ester group should provide good solubility of the NPs in common organic solvents. To prove this idea, a solubility test of CAN-Magh versus Magh-1 in a water/dichloromethane mixture was preformed (see Supplementary Material, Figure S6A). As could be expected upon coating of CAN-Magh NPs with ligand 1, the NPs readily transferred from the water phase to the dichloromethane organic layer.
IR analysis (see Supplementary Material, Figure S5) confirmed the successful coating of the maghemite NPs with ligand 1: in the region between 3300 and 3500 cm−1, there is a strong absorption due to the hydroxyl groups present on the surface of the maghemites that make impossible the identification of polar organic moieties. However, the stretching of aliphatic −CH of the ligand can be observed in region between 2800 and 3000 cm−1. Furthermore, at 1700 and 1640 cm−1, the stretching of the two C=O (ester and amide groups) can be easily recognized. In addition, thermogravimetric analysis confirmed the presence of an organic coating of 35.6% in mass (see Figure S3B).
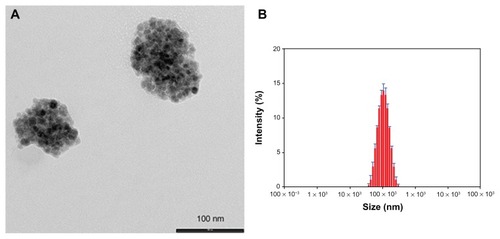
Following our interest in the entrapment of lipophilic MNPs,Citation13–Citation15 the hydrophobic Magh-1 NPs were incorporated into polymeric micelles using the nanoprecipitation technique.Citation15,Citation16 The amphiphilic copolymer PLGA-b-PEG-COOH served as a biocompatible coating that may enable blood circulation. Briefly, an acetone solution of Magh-1 and copolymer was added dropwise to dd water under vigorous stirring leading to the formation of Magh-1-PNPs. The as-synthesized polymeric NPs were characterized by means of DLS (). The analysis showed a hydrodynamic radius of 95.32 ± 0.88 nm with a polydispersity index (PDI) value of 0.153 ± 0.007. The NPs proved to be stable in an aqueous environment with a ζ-potential value of -39.2 mV at pH = 5.45. TEM observations confirmed the formation of polymeric shells around clusters of magh-1 ().
Figure 2 TEM image and relative DLS measurements of Magh-1-PNPs. TEM image (A) and relative DLS measurements (B) of Magh-1-PNPs.
Abbreviations: TEM, transmission electron microscopy; PNPs, polyethyleneglycol-based nanoparticles; DLS, dynamic light scattering.
The concentration of iron was investigated by means of atomic absorption spectrophotometry, and it was found to be 112 ppm.
In order to add the second contrast agent to obtain the dual modal imaging platform, the polymeric NP surface was functionalized with the 68Ga chelating agent, NODA. The carboxylic acid originating from the free end of the PEG building block was utilized for this functionalization reaction: first, it was activated with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) in water. In the next step, NODA was introduced in the reaction mixture and an amidation reaction between the activated acid and NODA amine function occurred. The resulting Magh-1-PNPs-NODA possesses a hydrodynamic radius of 92.34 ± 0.72 nm with a polydispersity index value of 0.167 ± 0.011 (DLS analysis), and a ζ-potential value of −44 mV at pH = 6.30. The concentration of iron in this case was found to be of 134 ppm with a total yield in iron of 64%.
Magh-1-PNPs-NODA in vitro toxicity in PDAC-derived cell lines
One potential field of application of 68Ga-labeled NPs is the clinical and preclinical diagnosis and evaluation of PDAC, which is one of the most deadly tumors, for which detection of neoplastic lesions at an early stage might permit life-saving interventions.Citation10
Thus, we evaluated the toxicity of Magh-1-PNPs-NODA NPs on different human PDAC-derived cell lines (ASPC-1, Panc-1, MiaPaCa, Su86.86, T3M4, BxPC3, Colo-357) using cell growth inhibition assays (MTT test, and Figure S1). Magh-1-PNPs-NODA NPs caused a modest dose-dependent decrease of cell proliferation.
Figure 3 Cellular viability rate versus maghemite concentration after 72 hour incubation with Magh-1-PNPs-NODA NPs.
Notes: Relative values were normalized to the values from the corresponding untreated cells and are shown as percent of survival. Results are expressed as a mean percentage of three independent experiments performed in triplicate ± SD.
Abbreviations: NPs, nanoparticles; PNPs, polyethyleneglycol-based nanoparticles; NODA, 2,2′-(7-(4-((2-aminoethyl)amino)-1-carboxy-4-oxobutyl)-1,4,7-triazonane-1,4-diyl)diacetic acid; SD, standard deviation.
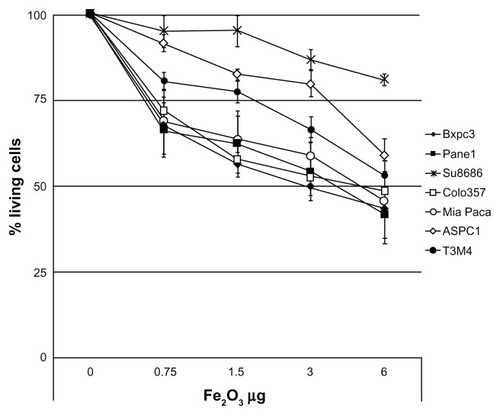
Despite the fact that a considerable variability among cell lines was observed, about a 50% reduction of the proliferation rate was observed within 72 hours of incubation in the majority of the culture cells when 3–6 μg of Fe2O3 was present. Notably, at higher concentrations, the Su8686 cell growth was only marginally affected by Magh-1-PNPs-NODA NPs.
NMR relaxivity
Although, in general, cells can survive short-term exposure to low concentrations of NPs, several groups have found cytotoxic effects to emerge in a dose- and time-dependent manner for most type of NPs. This is generally due to NP internalization inside cells, which elicits inflammatory responses, cell-cycle irregularities, and gene expression alterations.Citation17 Although a certain extent of Magh-1-PNP-induced cytotoxicity in the present study occurred, it is important to keep in mind that in vitro results can differ from what is found in vivo, and that toxicity in vitro may not necessarily be clinically relevant. This depends on whether repetitive NP administration is required, as well as on the NP bioavailability at the target site.
All nanocomposites were very weak T1 relaxing agents, and as expected for maghemite NPs, they were strong T2 relaxation agents (). The obtained values indicate that the polymer shell enhances R2 relaxivity, making the NODA functionalized compound ideally suitable as a T2/T2* MRI contrast agent.
Radiolabeling of NPs with 68Ga
The attachment of positron emitters to macromolecules is usually approached by the chelation of a (positron emitter) metallic atom using a bifunctional polyazacarboxylate- or tetraaza-macrocycle.Citation18,Citation19 This strategy can be transferred to the incorporation of radioisotopes into NPs, because contrary to the case of the radio-halogens (eg, 18F), the labeling reaction usually takes place under mild conditions without damaging the NPs.
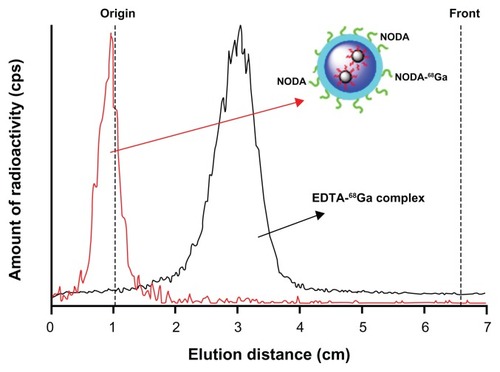
A number of metallic PET isotopes have been used to date for NP labeling, including 64Cu,Citation20 and 68Ga.Citation21 Copper-64 has probably been the most widely used due to its relatively long half life (12.7 hours), which permits PET evaluation of slow biochemical pathways. However, the production of 64Cu requires the irradiation of 64Ni with protons, and a laborious purification process using anion exchange chromatography.Citation22 Due to this fact 68Ga, which can be easily obtained from a commercially available 68Ge/68Ga generator, is gaining interest in the development of multimodal imaging agents, and its incorporation into NPs using bifunctional chelating agents has been already reported in the literature.Citation23 In the current work, a similar procedure was followed but a preconcentration step was applied to the 68Ga3+ solution obtained from the generator.Citation23 Under optimized experimental conditions (T = 60°C, t = 30 minutes, pH = 3.5), the labeling yield of Magh-1-PNPs-NODA, as measured by gamma-counting after filtration, was 84% ± 6%. Quality control of the NPs performed by TLC (, red line) showed no peak corresponding to free 68Ga after filtration and reconstitution in physiologic saline. The stability of labeled NPs in physiologic saline solution and in rat serum was also determined by TLC. The fraction of free gallium at t = 90 minutes was <5% in both media (not shown in ). These results confirm the stability of the 68Ga-NODA complex, which is very appropriate for subsequent in vivo studies.
Figure 4 TLC profiles obtained for 68Ga-labelled NPs (red line, Rf = 0) and free EDTA-68Ga complex (black line, Rf = 0.35).
Note: The complex EDTA-68Ga is generated in situ.
Abbreviations: TLC, thin layer chromatography; NPs, nanoparticles; EDTA, ethylenediaminetetra-acetic acid; NODA, 2,2′-(7-(4-((2-aminoethyl)amino)-1-carboxy-4-oxobutyl)-1,4,7-triazonane-1,4-diyl)diacetic acid.
Imaging studies
In , an image corresponding to the biodistribution pattern at long times after administration (frames 10–32) is presented. High accumulation in the liver and spleen can be visualized. The significant accumulation observed in the lungs might suggest the presence of a small percentage of aggregates, probably formed after administration of the NPs. The significant levels of radioactivity detected in the heart, especially at long times after administration of the labeled NPs, point towards a slow clearance from the bloodstream (). The low level of radioactivity in the bladder () indicates a partial (and slow) elimination via urine.
Figure 5 PET image and CT time-activity curves of 68Ga. PET image of 68Ga-labeled NPs signal corresponding to averaged time frames at t > 20 minutes after IV injection (A). CT images were adjusted on the Y-axis for an appropriate fitting with the tracer distribution. Time-activity curves for heart (B), brain (C), lungs (D), kidneys (E), spleen (F), and liver (G) after IV administration of 68Ga-labeled NPs.
Note: Average values +/− standard deviation (n = 3) are shown.
Abbreviations: PET, positron emission tomography; CT, computerized tomography; NPs, nanoparticles; IV, intravenous; ID, injective dose.
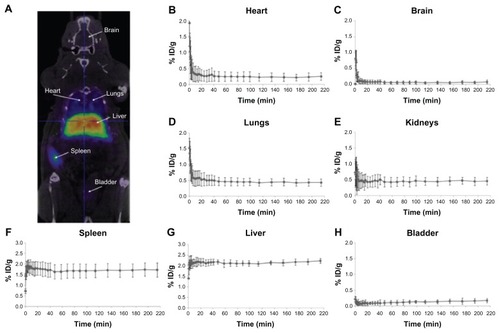
Time–activity curves for the heart, brain, lungs, kidney, spleen, liver, and bladder are shown in . Following tail vein injection, dynamic imaging revealed a steep peak in the heart, brain, and lungs (, respectively) as expected from a transient increase in blood pool activity. Brain activity decreased to negligible values (<0.1% injected dose g−1) 10 minutes after injection; higher values were reached in the heart (~0.25% ID g−1) and lungs (~0.45% ID g−1). Uptake into the spleen () and liver () was rapid. Within 5 minutes of the appearance of radioactivity in the blood, liver, and spleen, levels reached a plateau at 1.7% and 2.1% injected dose g−1, respectively. This level was maintained until the end of the study (4 hours after administration of NPs).
Accumulation of radioactivity in the bladder () slowly increased along the scan time up to 0.2% ID g−1 at the end of the study, while the accumulation of radioactivity in bones (results not shown) was very low at all times (<0.1% of ID/g all along the study). The results obtained in the current work demonstrate the suitability of 68Ga-labelling for subsequent determination of biodistribution patterns. Although the NPs described here did not have any functionalization to target specific organs or tissues, the same labeling and quantification strategy could be applied to targeted NPs to assess the potentially selective accumulation in specific organs or tumors. Therefore, the NPs presented here are an excellent platform for further functionalization in order to become suitable PET/MRI dual imaging contrast agents.
Indeed, our in vitro growth inhibition studies indicate an IC50 value for PDAC cell lines of approximately 3.0–6.0 μg of F2O3 (). This is a considerably high value considering the dosage of NPs injected in vivo for imaging experiments (2.67 μg of F2O3 injected into animals, ) suggesting a low grade of toxicity of Magh-1-PNPs-NODA NPs. It is well known that the physicochemical characteristics of NPs are often changed when they come into contact with the blood. In particular, NP interactions with proteins in the blood both quantitatively and qualitatively affect NP half-lives and biodistribution. Surface protein deposition may induce NP aggregation, and large aggregates may be trapped in the capillaries. Also, surface opsonization is an issue (ie, the deposition of proteins that facilitate NP recognition and clearance from the blood by circulating phagocytes or tissue macrophages in direct contact with the blood). All these events contribute to the reduction of NP bioavailability at the target site. A very recent reference discussed these issues.Citation24
Thus further investigation can be carried out on Magh-1-PNPs-NODA NPs for the application of such NPs for the early diagnosis and evaluation of pancreatic neoplasms.
Conclusion
We have developed a novel multifunctional biocompatible hybrid nanocarrier for dual PET/MRI imaging. This nanocarrier is composed of a polymer shell containing maghemite NPs at its lipophilic core and bearing 68Ga chelating group on the outer surface. The T2 relaxivity values show promising perspectives for the NPs to be used as MRI contrast agents; moreover, the incorporation of the positron emitter 68Ga, and the stability of the resulting NP-NODA-68Ga complex, confirm the suitability of the labeled NPs as contrast agents for in vivo PET imaging. The values at which Magh-1-PNPs-NODA proved to be suitable for MRI/PET analysis were found to be not toxic in vitro on seven cell lines of PDAC. Thus, the nanoplatform presented here is a promising tool for the future development of innovative PET/MRI diagnostic agents. Additional applications of this nanocarrier in nanomedicine are currently under investigation.
Results
Kaiser testCitation25 of NPs after surface derivatization with 1,4 diaminobutane afforded average values of 0.163 mmol of accessible COOH/g NPs. ICP measurements: Fe concentration was 3.24 mg/mL while Ce concentration was 1.053 μg/mL (Weight ratio Ce/Fe = 0.000325).
2. Acetyl chloride (20 μL, 0.28 mmol) was added to a stirred solution of 12-hydroxydodecanoic acid (1.00 g, 4.63 mmol) in 40 mL of ethanol under nitrogen atmosphere. The mixture was heated to reflux for 24 hours, then, the solvent was removed under vacuum and the resulting oil (1.02 g, 4.47 mmol, yield = 90%) was used for the next step without further purification.
Hydrogen-1 nuclear magnetic resonance (300 MHz, CDCl3): δ = 4.09 (q, 2H, CH2), 3.59 (t, 2H, CH2), 2.23 (t, 2H, CH2), 1.58 (bs, 6H, CH2), 1.25 (bs, 15H, CH2 + CH3).
Carbon nuclear magnetic resonance: (75 MHz, CDCl3): δ = 174.023 (C=O), 61.835 (CH2), 60.170 (CH2), 34.035 (CH2), 32.578 (CH2), 29.560 (CH2), 29.477 (CH2), 29.385 (2CH2), 29.210 (CH2), 29.047 (CH2), 25.583 (CH2), 24.913 (CH2), 14.200 (CH3).
3. A suspension of periodic acid (2.13 g, 9.33 mmol) in 40 mL of acetonitrile was left to react for 20 minutes at room temperature. Then, a solution of ethyl 12-hydroxydodecanoate (1.02 g, 4.16 mmol) and pyridinium chlorochromate (20 mg, 0.091 mmol) in 10 mL of acetonitrile was slowly added, keeping the temperature at 0°C with an ice bath. Once completed, the addition the ice bath was removed and the mixture was left to react for 24 hours at room temperature.
Afterward, 40 mL of ethyl acetate were added, and the mixture was first washed with water (3 × 40 mL), then with an aqueous solution of sodium hydrogen sulfate (40% m/V, 3 × 40 mL), and finally again with water (2 × 40 mL). The organic phase was dried with anhydrous sodium sulfate, and it was filtered and evaporated in vacuo to obtain the product as a white solid (1.00 g, 3.87 mmol, yield = 93%). melting point: 51°C–53°C.
Hydrogen-1 nuclear magnetic resonance (300 MHz, CDCl3): δ = 4.12 (q, 2H, CH2), 2.37 (t, 2H, CH2), 2.29 (t, 2H, CH2), 1.61 (bs, 4H, CH2), 1.26 (bs, 15H, CH2 + CH3).
Carbon nuclear magnetic resonance: (150 MHz, CDCl3): δ = 180.046 (C=O), 174.003 (C=O), 60.169 (CH2), 34.342 (CH2), 34.041 (CH2), 29.441 (CH2), 29.295 (CH2), 29.156 (2CH2), 29.060 (CH2), 28.987 (CH2), 24.918 (CH2), 24.625 (CH2), 14.198 (CH3).
4. To a stirred solution of 12-ethoxy-12-oxododecanoic acid (3.87 mmol, 1.00 g) in 30 mL of anhydrous tetrahydrofuran (THF) under nitrogen, 1,1-carbonyldiimidazole (6.17 mmol, 0.99 g) was slowly added. The mixture was stirred for 1.5 hours, diluted with an additional 70 mL of anhydrous THF, and added dropwise (4 hours) to a suspension of dopamine hydrochloride (3.87 mmol, 0.73 g) and pyridine (5.8 mmol, 0.47 mL) in the same solvent (30 mL of anhydrous THF), maintaining a nitrogen atmosphere. The mixture was left to react for 1 hour, and then the solvent was removed under vacuum.
The solid was redispersed in ethyl acetate and washed three times with water. The organic phase was dried over MgSO4, filtered and evaporated in vacuo. The crude product was then purified with column chromatography (EtOAc/Pet ether, 3:1) to provide the final ligand (0.72 g, 1.83 mmol, yield = 47%). mp: 60°C–62°C.
Hydrogen-1 nuclear magnetic resonance (300 MHz, CDCl3): δ = 6.81 (d, 1H, Ar), 6.78 (m, 1H, Ar), 6.59 (d, 1H, Ar), 5.65 (bs, 1H, NH), 4.15 (q, 2H, CH2), 3.45 (q, 2H, CH2), 2.70 (t, 2H, CH2), 2.30 (t, 2H, CH2), 2.18 (t, 2H, CH2), 1.60 (bs, 4H, CH2), 1.22 (bs, 15H, CH2 + CH3).
Carbon nuclear magnetic resonance (150 MHz, CDCl3): δ = 174.538 (C=O), 174.373 (C=O), 144.518 (C), 143.225 (C), 130.490 (C), 120.373 (CH), 115.479 (CH), 155.183 (CH), 60.373 (CH2), 41.000 (CH2), 36.777 (CH2), 34.898 (CH2), 34.404 (CH2), 29.309 (CH2), 29.294 (CH2), 29.180 (CH2), 29.115 (2CH2), 29.067 (CH2), 25.730 (CH2), 24.950 (CH2), 14.251 (CH3).
Gas chromatography–mass spectrometry ES+: 416 (M + Na).
Coating of CAN-Magh with ligand
Original CAN-Magh solution (10 mL) was diluted with 30 mL of water and added to a solution of 1 (300 mg, 0.762 mmol) in 40 mL of ethanol. The mixture was sonicated in an ultrasound bath for 1 hour, and it was left to react overnight at room temperature under mechanical stirring. Afterward, the solution was decanted magnetically (strong external magnet) and the supernatant was removed while the brownish solid was washed with ethanol (2 × 30 mL). Finally, the solid (Magh-1) was redispersed in 80 mL of acetone, sonicated for 30 minutes (bath sonicator, Branson Ultrasonics Corporation, Danbury, CT) and immediately used in the following steps.
5. Poly(D,L-lactide-co-glycolide)-block-poly(ethylene glycol) was synthesized through the activation of carboxyl- capped PLGA-COOH (MW ~7 kDa) with N-hydroxysuccinimide using dicyclohexylcarbodiimide coupling chemistry, and then conjugated to the amine bifunctional NH2-PEG-COOH (MW ~3 kDa), in chloroform and in the presence of N,N-Diisopropylethylamine.Citation15
6. Synthesis of Magh-1-PNPs-NODA
The linkage of 2,2′-(7-(4-((2-aminoethyl)amino)-1-carboxy-4-oxobutyl)-1,4,7-triazonane-1,4-diyl)diacetic acid (NODA) on the surface of the particle was carried out through a classic peptide formation. In particular, 35 mL of a 0.28 mM Magh-1-polyethyleneglycol-based nanoparticle water solution (0.010 mmol of D,L-lactide-co-glycolide)-block-poly(ethylene glycol) were stirred for 2 hours in the presence of 0.010 mmol of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (1.7 mL of a 5.7 mM water solution neutralized with 0.1 M HCl solution up to pH = 7.60). Then, 0.010 mmol of NODA-GA-ethylenediamine was added (6.1 mL of a 1.6 mM water solution neutralized with 0.1 M NaOH solution up to pH = 7.15) and the reaction mixture was kept under stirring for additional 2 hours. The particles were purified with a centrifugal filter device (Amicon Ultra, Ultracel membrane with 100.000 normal molecular weight limit; Millipore Corporation, Billerica, MA) and redispersed in 25 mL of water.
7. Synthesis of Magh-1-PNPs-NODA
The linkage of 2,2′-(7-(4-((2-aminoethyl)amino)-1-carboxy-4-oxobutyl)-1,4,7-triazonane-1,4-diyl)diacetic acid (NODA) on the surface of the particle was carried out through a classic peptide formation. In particular, 35 mL of a 0.28 mM Magh-1-polyethyleneglycol-based polymeric nanoparticle water solution (0.010 mmol of poly(D,L-lactide-co-glycolide)-block-poly(ethylene glycol)) was stirred for 2 hours in the presence of 0.010 mmol of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (1.7 mL of a 5.7 mM water solution neutralized with 0.1 M HCl solution up to pH = 7.60). Then, 0.010 mmol of NODA-GA- ethylenediamine was added (6.1 mL of a 1.6 mM water solution neutralized with 0.1 M NaOH solution up to pH = 7.15), and the reaction mixture was kept under stirring for additional 2 hours. The particles were purified with a centrifugal filter device (Amicon Ultra, Ultracel membrane with 100.000 normal molecular weight limit; Millipore Corporation, Billerica, MA) and redispersed in 25 mL of water.
Figure S1 Transmission electron microscopy image of ceric ammonium nitrate (CAN)-Magh nanoparticles (NPs).

Figure S2 Synthesis of ethyl 12-hydroxydodecanoate.
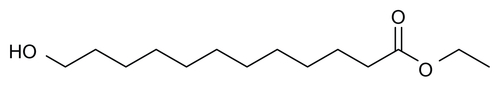
Figure S3 Synthesis of 12-ethoxy-12-oxododecanoic acid.
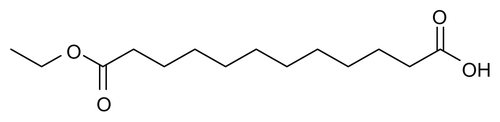
Figure S4 Synthesis of ethyl 12-((3,4-dihydroxyphenethyl)amino)-12-oxododecanoate (EDAO, 1).
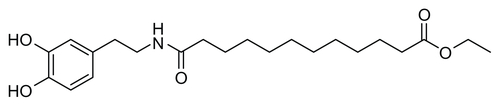
Figure S5 Infrared spectra of ceric ammonium nitrate (CAN)-Magh (black line) and Magh-1 (blue line).
Abbreviation: T, transmittance.
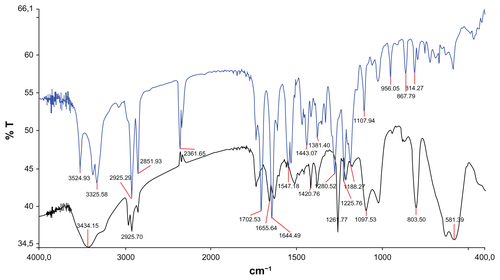
Figure S6 Solubility test of ceric ammonium nitrate (CAN)-Magh versus Magh-1 (A) and thermogravimetric analysis of Magh-1 (B).
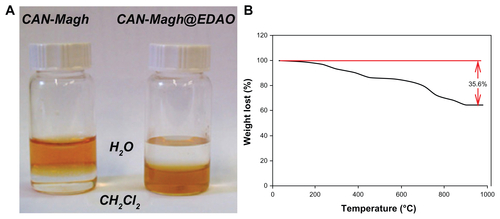
Figure S7 Synthesis of Magh-1-PNPs-NODA.
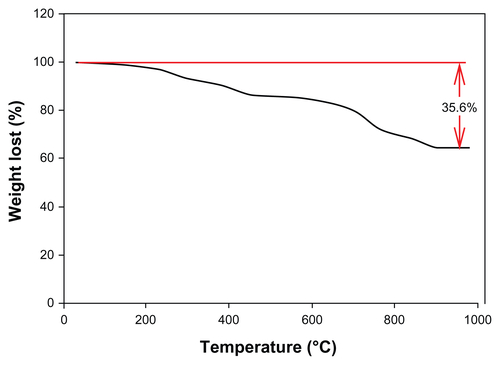
Table S1 Statistical analysis of the mean values reported in
Acknowledgments
This work has been supported with the funding of the EU-FP7 European project SaveMe (contract number CP-IP 263307-2).
The authors would like to thank Maria Puigivila, Boguslaw Szczupak, and Enrique Alonso for technical support. Lorena Passoni receives funding from Fondazione Umberto Veronesi (Milano, Italy).
Disclosure
The authors report no conflicts of interest in this work.
Supplementary materials
1. Derivatization of maghemite NPs with 1,4-diaminobutane for Kaiser test
N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), 50 mg (0.26 mmol), was added to 1 mL of maghemite NPs suspended in doubly distilled water (ddH2O), and the reaction mixture was shaken for 40 minutes at room temperature. Then, the excess of EDC was washed out by centrifugation at 8000 rpm (8 minutes, 5°C, three times). Then, 1 mL of ddH2O and 0.25 mL (3.22 mmol) of 1,4-diaminobutane were added and the solution was shaken for 1.5 hours. The excess of amine was removed by centrifugation at 12,500 rpm (5°C, and three times).Citation25
References
- Karim-KosHEde VriesESoerjomataramILemmensVSieslingSCoeberghJWRecent trends of cancer in Europe: a combined approach of incidence, survival and mortality for 17 cancer sites since the 1990sEur J Cancer200844101345138918280139
- JemalASiegelRXuJWardECancer statistics, 2010CA Cancer J Clin201060527730020610543
- CheonJLeeJHSynergistically integrated nanoparticles as multimodal probes for nanobiotechnologyAcc Chem Res200841121630164018698851
- CorotCRobertPIdéeJMPortMRecent advances in iron oxide nanocrystal technology for medical imagingAdv Drug Deliv Rev200658141471150417116343
- MoghimiSMHunterACMurrayJCLong-circulating and target-specific nanoparticles: theory to practicePharmacol Rev200153228331811356986
- HavivAHGrenècheJMLelloucheJPAggregation control of hydrophilic maghemite (gamma-Fe2O3) nanoparticles by surface doping using cerium atomsJ Am Chem Soc201013236125191252120735060
- GrefRMinamitakeYPeracchiaMTTrubetskoyVTorchilinVLangerRBiodegradable long-circulating polymeric nanospheresScience19942635153160016038128245
- CherrySRFundamentals of positron emission tomography and applications in preclinical drug developmentJ Clin Pharmacol200141548249111361044
- Bond-SmithGBangaNHammondTMImberCJPancreatic adenocarcinomaBMJ2012344e247622592847
- ZafarSFEl-RayesBFChemotherapeutic strategies in advanced or metastatic pancreatic adenocarcinomaAm J Clin Oncol Epub May 24, 2012
- EsmanNHavivALelloucheJPMagnetically responsive polypyrrole nanotubes using Ce(III)-stabilized maghemite nanoparticlesNanotechnology2011222828560421642756
- YuenAKHuttonGAMastersAFMaschmeyerTThe interplay of catechol ligands with nanoparticulate iron oxidesDalton Trans20124192545255922241454
- Comes FranchiniMBaldiGBonacchiDBovine serum albumin-based magnetic nanocarrier for MRI diagnosis and hyperthermic therapy: a potential theranostic approach against cancerSmall20106336637020020469
- BaldiGBonacchiDFranchiniMCSynthesis and coating of cobalt ferrite nanoparticles: a first step toward the obtainment of new magnetic nanocarriersLangmuir20072374026402817335257
- Comes FranchiniMBoniniBFCamaggiCMDesign and synthesis of novel 3,4-disubstituted pyrazoles for nanomedicine applications against malignant gliomasEur J Med Chem20104552024203320129719
- ChengJTeplyBASherifiIFormulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug deliveryBiomaterials200728586987617055572
- LewinskiNColvinVDrezekRCytotoxicity of nanoparticlesSmall200841264918165959
- KukisDLDirilHGreinerDPA comparative study of copper-67 radiolabeling and kinetic stabilities of antibody-macrocycle chelate conjugatesCancer199473Suppl 37797868306260
- BoswellCASunXNiuWComparative in vivo stability of copper-64-labeled cross-bridged and conventional tetraazamacrocyclic complexesJ Med Chem20044761465147414998334
- HuangXZhangFLeeSLong-term multimodal imaging of tumor draining sentinel lymph nodes using mesoporous silica-based nanoprobesBiomaterials201233174370437822425023
- KoHYChoiKJLeeCHKimSA multimodal nanoparticle-based cancer imaging probe simultaneously targeting nucleolin, integrin αvβ3 and tenascin-C proteinsBiomaterials20113241130113821071077
- MatarreseMBedeschiPScardaoniRAutomated production of copper radioisotopes and preparation of high specific activity [(64)Cu]Cu-ATSM for PET studiesAppl Radiat Isot201068151319736021
- VelikyanIBeyerGJLångströmBMicrowave-supported preparation of (68)Ga bioconjugates with high specific radioactivityBioconjug Chem200415355456015149183
- MoghimiSMHunterACAndresenTLFactors controlling nanoparticle pharmacokinetics: an integrated analysis and perspectiveAnnu Rev Pharmacol Toxicol20125248150322035254
- KaiserEColescottRLBossingerCDCookPIColor test for detection of free terminal amino groups in the solid-phase synthesis of peptidesAnal Biochem19703425955985443684