 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Background and methods
A new cyclosporin A-loaded, PEGylated chitosan-modified lipid-based nanoparticle was developed to improve upon the formulation of cyclosporin A. PEGylated chitosan, synthesized in three steps using mild reaction conditions, was used to modify the nanoparticles. Cyclosporin A-loaded, PEGylated chitosan-modified nanoparticles were prepared using an emulsification/solvent evaporation method. The drug content and encapsulation efficiency of the cyclosporin A-loaded, PEGylated chitosan-modified nanoparticles were measured by high-performance liquid chromatography. The average size of the nanoparticles was determined by transmission electron microscopy and dynamic light scattering. The pharmacokinetic behavior of the nanoparticles was investigated in rabbits after intravenous injection. Cyclosporin A concentrations in a whole blood sample were analyzed by high-performance liquid chromatography using tamoxifen as the internal standard. The pharmacokinetic parameters were calculated using the 3p87 software program.
Results
Fourier transform infrared spectroscopy and nuclear magnetic resonance confirmed the structure of PEGylated chitosan. The drug content and encapsulation efficiency of the cyclosporin A-loaded, PEGylated chitosan-modified nanoparticles were 37.04% and 69.22%, respectively. The average size of the nanoparticles was 89.4 nm. The nanoparticles released 30% cyclosporin A-loaded in 48 hours in vitro, with no initial burst release. The mode of release in vitro was prone to bulk erosion. The in vivo results showed the biological half-life of the elimination phase (t1/2β) of the nanoparticles was 21 times longer than that of the cyclosporin A solution, and the area under the curve for the nanoparticles was 25.8 times greater than that of the cyclosporin A solution.
Conclusion
Modification of PEGylated chitosan prolonged the retention time of the nanoparticles in the circulatory system and improved the bioavailability of cyclosporin A.
Introduction
Cyclosporin A is one of the most effective immunosuppressive drugs. It inhibits T lymphocyte function, which plays an important role in induction of the immune response. Immune responses usually occur following organ transplants, such as liver, kidney, and bone marrow.Citation1–Citation3 However, cyclosporin A is a poorly water-soluble cyclic peptide comprising 11 amino acids. Use of cyclosporin A has been limited by several disadvantages, including low bioavailability, a narrow therapeutic window, nephrotoxicity, hepatotoxicity, and neurotoxicity.Citation4–Citation7 Moreover, cyclosporin A injection is limited in patients who are unable to take oral preparations because it carries a risk of anaphylactic shock and nephrotoxicity due to Cremophor EL®, a solubilizing agent used in the commercial intravenous formulation.Citation8–Citation10 Because of the disadvantages of the commercial products discussed above, there is great interest in development of alternative dosage forms,Citation11 including a solid dispersion,Citation12 polymer nanoparticles,Citation13 and lipid-based formulations.Citation14,Citation15 Many papersCitation16,Citation17 have verified that the cyclosporin A released from nanoparticle formulations maintain or improve the efficiency of the immune response.
Lecithin vesicles can be used in lipid-based formulations. Lecithin derived from biological phospholipids is biodegradable, shows a relative lack of immunogenicity, and has low intrinsic toxicity.Citation18 However, lecithin vesicles are detected by the host defense system rapidly because they are absorbed by proteins/ cells and lack steric stability in the systemic circulation.Citation19 Therefore, certain biodegradable polymers can be used to modify the surface of lipid-based nanoparticles. These modified lipid-based nanoparticles may enhance the delivery of drugs, increase drug uptake, and reduce drug toxicity. Poly(ethylene glycol) (PEG) is nontoxic and is eliminated by a combination of renal and hepatic pathways, which makes it ideal for use in pharmaceutical applications. It may be dissolved in both organic solvents and water. The US Food and Drug Administration has approved PEG for intravenous, oral, and dermal applications in humans.Citation20 PEG has the lowest level of protein and cellular absorption among the known polymers.Citation21 These properties have been exploited in numerous ways, including grafting/ mixing PEG to the surfaces of micro/nanoparticles to prevent deposition of proteinaceous material.Citation22,Citation23 ChitosanCitation24–Citation27 is also a compatible, biodegradable, and nontoxic polymer. Its cationic properties may be useful for neutralizing the negative surfaces of lipid-based nanoparticles. PEGylated chitosan is a graft polymer that has been synthesized by our group, as previously reported.Citation28 It was synthesized using a novel procedure and linkage by our group. We hypothesized that using PEGylated chitosan would enable nontoxic cyclosporin A-loaded, lipid-based nanoparticles to circulate for a long time in the blood system and increase the stability of the nanoparticles.
In this study, we prepared cyclosporin A-loaded, PEGylated chitosan-modified, lipid-based nanoparticles. The morphology of the nanoparticle and its in vitro release was measured. The pharmacokinetic behavior of the nanoparticles in rabbits was also evaluated.
Materials and methods
Materials
Cyclosporin A was kindly donated by the Huadong Pharmaceutical Factory (Hangzhou, People’s Republic of China). Chitosan (molecular weight 120,000 with 75%–85% deacetylation) and mPEG5000 were purchased from Sigma-Aldrich Chemical Company (St Louis, MO, USA). Soybean lecithin was purchased from the Lipoid Company (Ludwigshafen, Germany). All other chemicals were of analytical or chromatographic grade and used without further purification.
Synthesis of mPEG-grafted chitosan
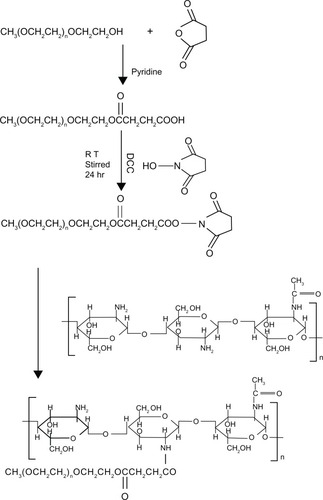
Methoxypolyethyleneglycol (mPEG)-grafted chitosan was obtained in three steps using the method described in our previous paper.Citation28 In brief, the hydroxyl groups at the end of mPEG5000 were converted to carboxyl groups and succinyl esters. mPEG was first converted to mPEG-Su-COOH using succinic anhydride. The condensation compound, mPEG-Su-COONSu, was then obtained via the reaction between mPEG-Su-COOH and N-hydroxy-succinamide catalyzed by N,N′-dicyclohexyl carbodiimide. Finally, PEGylated chitosan was obtained by a condensation reaction between the –NH2 group on chitosan and the –COONSu group on mPEG-Su-COONSu. The whole synthetic scheme for mPEG-CO-(NH-chitosan) is shown in .
Figure 1 Synthetic scheme for mPEG-CO-(NH-chitosan).
Abbreviation: RT, room temperature.
Fourier transform infrared analyses of mPEG, mPEG-COOH, mPEG-Su-COONSu, and mPEG-CO-(NH-chitosan) were performed using an infrared spectrophotometer (IR-460, Shimadzu, Japan). The 1H nuclear magnetic resonance (NMR) spectrum for mPEG-CO-(NH-chitosan) was recorded using an Avance DMX500 spectrometer (Bruker, Ettlingen, Germany) at 500 mHz in dimethyl sulfoxide.
Many papersCitation29,Citation30 have reported that PEGylated chitosan has low cytotoxicity. The method used for calculating the ratio of PEG grafted to the chitosan backbone was as follows:
where N0 is the molar number of the NH2 groups on chitosan at the beginning of synthesis, and N1 is the molar number of PEG chains grafted to chitosan after the complete reaction. The weight of the grafted PEG chains was obtained from the difference between the weight of PEGylated chitosan and the initial weight of chitosan.
Preparation of cyclosporin A-loaded, PEGylated chitosan-modified nanoparticles
The nanoparticles were prepared using a modified emulsification/solvent evaporation method. Cyclosporin A and soybean lecithin were dissolved in the solvent mixture (methylene chloride to acetone ratio, 3:1) as solution A. PEGylated chitosan and Poloxamer were dissolved in an aqueous solution as solution B. Solution A was injected slowly into Solution B under fast magnetic stirring, followed by ultrasonic dispersion with a probe-type ultrasonicator (400 W, JY92-II, Ningbo Xinzhi Scientific Instrument Institute, Zhejiang, People’s Republic of China). The ultrasonic treatments were performed 60 times, with one second of sonication followed by a 3-second interval in an ice bath. The methylene chloride and acetone in the resulting emulsion were removed under reduced pressure using a rotovap (RE-52C, Ya-Rong Biochemical Instrument Factory, Shanghai, People’s Republic of China) at room temperature. The whole dispersion system was then centrifuged (Hereaus Instruments, Thermo, Germany) at 12,000 × g for 30 minutes at 4°C to obtain the precipitates. An aqueous solution was obtained by dissolving the precipitate in water. A cellulose ester filter (0.22 μm, Millipore, Billerica, MA, USA) was used to separate microspheres or agglomerates from this solution. The final product was freeze-dried in the presence of 10% (w/v) sorbitol.
Morphology of cyclosporin A-loaded, PEGylated chitosan-modified nanoparticles
Transmission electron microscopy (TEM) was performed using a JEM-1200EX instrument (JEOL, Tokyo, Japan) operating at 100 kV. The TEM samples were obtained by diluting a 1 mg/mL suspension of cyclosporin A-loaded, PEGylated chitosan-modified nanoparticles with phosphate-buffered saline at pH 7.4 to 2.5% (w/v) with ultrapure water. The sample solution was stained with 1% osmium tetroxide. The solution was then dropped onto carbon-coated copper grids and dried at room temperature before measurement.
The mean size and size distribution of the cyclosporin A-loaded, PEGylated chitosan-modified nanoparticles were measured by dynamic light scattering using a Zetasizer (3000HS, Malvern Instruments Ltd, Worcestershire, UK) in ultrafine water. The zeta potential of the nanoparticles was also measured using the Zetasizer. Each sample was measured three times, and the values are reported as the mean diameter ± standard deviation.
Determination of drug content and encapsulation efficiency
The cyclosporin A concentration in the preparation and in vitro release samples was analyzed by high-performance liquid chromatography performed on an Agilent system consisting of an 1100 isopump (Hypersil 5 μm ODS2 4.6 × 250 mm). The mobile phase was a mixture of acetonitrile and water (85:15 v/v) with 0.16% phosphoric acid. The detection wavelength was 221 nm. The column temperature was 70°C, the flow rate was 1.0 mL per minute, and the injected volume of the sample was 20 μL. The retention time of cyclosporin A in this mobile phase was 8.161 minutes. The drug content and encapsulation efficiency were determined by high-performance liquid chromatography measurement.
Acetonitrile was used to extract cyclosporin A thoroughly from the nanoparticles. The mixture of cyclosporin A nanoparticles in acetonitrile was vortexed and centrifuged at 5000 rpm for 10 minutes to remove the precipitated excipients. The supernatant was collected for measurement. The diluted cyclosporin A concentration was calculated according to a cyclosporin A standard curve. The encapsulation efficiency and drug content of the cyclosporin A-loaded nanoparticles were calculated as reported elsewhere.Citation31,Citation32 The equations were as follows:
In vitro release studies
Release of cyclosporin A from the nanoparticles was determined by the dialysis method. A suspension of cyclosporin A-loaded, PEGylated chitosan-modified nanoparticles in 100 mL of 0.1 M phosphate buffer (pH 7.4) was transferred into a dialysis bag (molecular weight cutoff 3500). This bag was placed in a brown flask filled with 0.1 M phosphate buffer (pH 7.4). The total concentration of cyclosporin A in this flask was maintained under sink conditions, meaning that the total cyclosporin A concentration in this flask was maintained at 1/3–1/7 of the concentration of cyclosporin A in saturated solution. The whole release system was stirred at room temperature. At a predetermined time point, 1 mL of solution was sampled from the flask and the same volume of fresh medium was added. The amount of cyclosporin A released into the medium was analyzed by high-performance liquid chromatography.
Bioavailability study
Five rabbits weighing approximately 2 kg were selected for this study. The rabbits were fasted for 12 hours before administration of the trial preparation and for the whole experiment (only water was permitted during this period). A single intravenous dose equivalent to 5 mg cyclosporin A per kg weight of the rabbit was injected into the auricular vein in the form of cyclosporin A-loaded, PEGylated chitosan-modified nanoparticles, cyclosporin A-loaded, chitosan-modified nanoparticles, or cyclosporin A solution (formulation: 50 mg cyclosporin A was dissolved in 0.32 mL of ethanol and 650 mg of Cremophor was added to assist the solubility of cyclosporin A). Prior to injection, the solution was diluted to 5 mL by sterile saline (0.9% NaCl). At predetermined time intervals (0, 2, 5, 10, 20, and 40 minutes, and at 1, 2, 4, 8, 12, and 24 hours), blood samples were drawn from the auricular artery of the rabbits into heparin-containing tubes.
To determine the concentration of cyclosporin A in the blood samples correctly, tamoxifen was used as an internal standard for high-performance liquid chromatography measurement.Citation33 A 50 μL tamoxifen/methanol (2.5 μg/mL) internal standard solution was added to a 400 μL blood sample. After a few minutes, 1.75 mL of deionized water was added. The red solution turned brown after 200 μL of NaOH (1 M) and 4 mL of ether-methanol (95:5) were added. Cyclosporin A and tamoxifen were extracted into the organic layer. The organic layer was separated by vortexing for 30 seconds. Organic solvents, including ether and methanol, were evaporated in a N2 atmosphere in a 50°C water bath. The resulting deposit, ie, the cyclosporin A and tamoxifen extract, was stored at −20°C.
Before measurement using high-performance liquid chromatography, the cyclosporin A and tamoxifen extract were resuspended in 200 μL of acetonitrile:0.5% (v/v) phosphoric acid solution (65:35, v/v). To this resuspension, 1 mL of n-hexane was added, and the solution was vortexed. After centrifuging, the bottom layer of the solution was used for high-performance liquid chromatographic analysis.
High-performance liquid chromatographic analysis of the in vivo samples was performed on an Agilent system (Hypersil 5 μm ODS2 4.6 × 250 mm column) with a 1100 isopump. The mobile phase was a mixture of methanol:acetonitrile: monopotassium phosphate 0.01 M (57:25:18, v/v/v). The detection wavelength was 205 nm. The flow rate was 1.0 mL per minute, and the injected volume of the sample was 20 μL. The retention time of cyclosporin A was 9.948 minutes, while the retention time of tamoxifen was 13.89 minutes.
Results and discussion
Preparation of cyclosporin A-loaded, PEGylated chitosan-modified nanoparticles
As reported in our previous paper,Citation28 the structure of PEGylated chitosan was confirmed by Fourier transform infrared spectroscopy and NMR experiments (). This synthetic procedure and the linkage between PEG and chitosan were reported by us for the first time. The conditions used in the synthesis were mild, although the time taken to finish the whole process was lengthy. The cyclosporin A-loaded nanoparticles were prepared using a modified emulsion/solvent evaporation technique.
In this formulation, soybean lecithin was selected as a carrier. The crystal nucleus of cyclosporin A was first separated out following evaporation of the organic solvent. Lecithin was deposited on the cyclosporin A nucleus as a result. As this occurred, interactions developed between the polar groups of lecithin and the amino/hydroxyl groups of PEGylated chitosan. These two segments wrapped around each other and changed the electronic properties of the nanoparticle surface. Moreover, the solubility of cyclosporin A in lecithin was increased because of the similar polar properties of these two substances.
PEGylated chitosan has the hydrophilic PEG segment and the hydrophobic backbone of chitosan, so it acts not only as a cationic polymer to coat the lecithin carrier but also as an emulsion stabilizer in this formulation. The Poloxamer present in this formulation increased the PEGylated chitosan dissolution in the Poloxamer/water mixture and enhanced the stability of the emulsion. Therefore, the function of PEGylated chitosan in our formulation was used to modify the lipid-based nanoparticles. PEGylated chitosan-modified nanoparticles are different from nanoparticles using PEGylated chitosan as a carrierCitation23 and those using chitosan to coat lipid nanoparticles.Citation25
Morphology of cyclosporin A-loaded, PEGylated chitosan-modified nanoparticles
and show the morphology of the cyclosporin A-loaded, PEGylated chitosan-modified nanoparticles. Dynamic light scattering revealed that the average diameter of the majority of the PEGylated chitosan-modified nanoparticles was 89.3 nm. The polydispersity index of the nanoparticles was 0.456. The diameters of nanoparticles measured by TEM were smaller than the data obtained from dynamic light scattering analysis because of the vacuum-drying conditions required for TEM. Moreover, although the zeta potential of the cyclosporin A-loaded, PEGylated chitosan-modified nanoparticles was low (–8.5 mV), a majority of the nanoparticles had a uniform, monodispersed, nonaggregated morphology. This unusual morphology may be in part due to the nonreactive PEG chains. There are many hydroxyl groups on the PEG chain, which may develop hydrogen bonds with water molecules. Therefore, the conformational entropy is lowered and the interfacial free energy of PEG chains in water is reduced. Thus, a large exclusion volume developed around the nanoparticles. This phenomenon was also called steric hindrance or steric stability. All the nanoparticles dispersed uniformly without adhesion.
Figure 2 Morphology of cyclosporin A-loaded, PEGylated chitosan-modified nanoparticles. (A) Transmission electron microscopy graph (bar, 50 nm) and (B) dynamic light scattering graph.
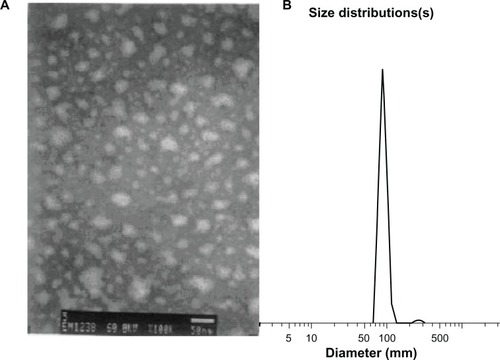
Figure 3 Zeta potential of cyclosporin A-loaded chitosan-modified nanoparticle (left); zeta potential of cyclosporin A-loaded, PEGylated chitosan-modified nanoparticle (right).
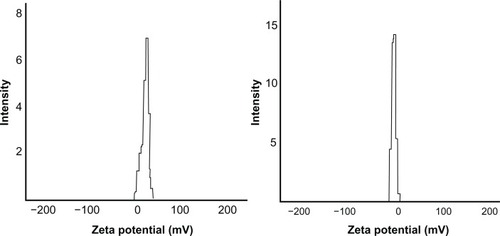
The zeta potential of the cyclosporin A-loaded, PEGylated chitosan-modified nanoparticles and the chitosan-modified nanoparticles was −8.5 mV and +15.2 mV, respectively. In these two formulations, the amount of PEGylated chitosan and chitosan fed was initially the same. Thus, the molar equivalents of the free amine groups of chitosan were more than that of PEGylated chitosan. The zeta potential values were +15.2 mV (chitosan-modified nanoparticles), −8.5 mV (PEGylated chitosan-modified nanoparticles), and −17.5 mV (pure lipid nanoparticles). The near zero value of the zeta potential for the PEGylated chitosan-modified nanoparticles also confirmed that PEGylated chitosan was indeed deposited on the lecithin carrier. The nanoparticles with a near neutral charge were safer than those with a positive charge because cationic nanoparticles and biomaterials would lead to electrostatic interaction with in vivo biomacromolecules, such as protein and DNA, and induce cytotoxicity in nonspecific cells.
Determination of drug content and encapsulation efficiency
The drug content was 37.04% in the cyclosporin A-loaded, PEGylated chitosan-modified nanoparticle formulation, while the encapsulation efficiency was 69.22%. This encapsulation efficiency value is higher than that of nanoparticles prepared by a chitosan-tripolyphosphate cross-link reaction.Citation34,Citation35 The encapsulation efficiency value depended on the high affinity between cyclosporin A and lecithin. Moreover, the low solubility of cyclosporin A in aqueous Poloxamer solution also helped to increase encapsulation efficiency.
In vitro release study
The amount of cyclosporin A released into the medium was analyzed by high-performance liquid chromatography. shows the percentage of cyclosporin A released from the nanoparticles.
Figure 4 In vitro release profiles of cyclosporin A-loaded, PEGylated chitosan-modified nanoparticle (♦) and free cyclosporin A in saturated aqueous solution (▪).
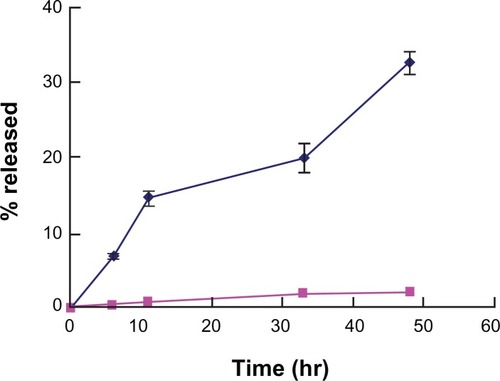
The profile showed that the percentage of cyclosporin A released from the nanoparticles was 32.6% after 48 hours, compared with less than 4% after 52 hours for free cyclosporin A in the saturated solution. When the poorly soluble drug, cyclosporin A, was dispersed in a nanoparticle formulation, the amount of cyclosporin A released from the nanoparticles increased. To understand the mode of drug release from the nanoparticles, the release data were analyzed according to the prediction equation proposed by Korsmeyer et alCitation36 and Lee and Peppas.Citation37
where Mt/M0 is the percentage of drug released, t is time, K is the constant associated with the characteristics of the geometry skeleton of the release system, and n is the release index representing the release mechanism. If n = 0.5, the release mode was predicted to be bulk erosion release, while 0.66 < n < 1 represented a diffusion release system.
The result of the simulation was:
Therefore, n = 1/2. The value of n indicated that the release mode of cyclosporin A from the PEGylated chitosan nanoparticles was prone to bulk erosion release.
Bioavailability study
Because metabolites of cyclosporin A are found in vivo, high-performance liquid chromatography has been used by many researchers to measure cyclosporin A in samplesCitation33,Citation38 because of its sensitivity and specificity. Moreover, to measure the cyclosporin A concentration in blood accurately, tamoxifen is used as the internal standard for high-performance liquid chromatography.
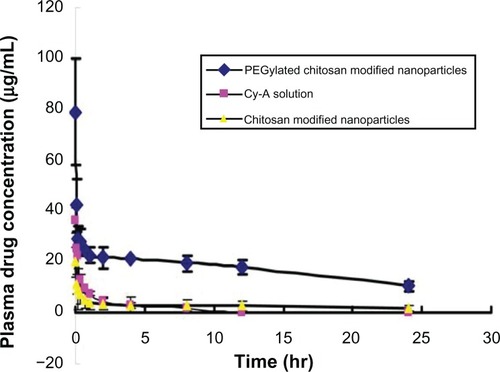
shows the plasma drug concentrations at different time points in rabbits after intravenous injection with cyclosporin A-loaded, PEGylated chitosan-modified nanoparticles, cyclosporin A-loaded chitosan-modified nanoparticles, and cyclosporin A solution.
Figure 5 Plasma drug concentrations at different time points after intravenous injection in rabbits treated with cyclosporin A-loaded, PEGylated chitosan-modified nanoparticles (♦),cyclosporin A-loaded, chitosan-modified nanoparticles (▴), and cyclosporin A solution (▪).
Many mathematical modelsCitation39 including single and two compartment models can be used for pharmacokinetic analysis. The single compartment model depicts the body as a single, kinetically homogeneous unit. The two compartment model assumes that the whole body is divided into two compartments, ie, a central compartment (including the blood and all readily accessible fluids and tissues, such as the liver and kidney) and a peripheral compartment (including all poorly perfused tissues). Compared with the simple single compartment model, the two compartment model is more reasonable and more practical. According to Akaike’s information criterion and the degree of fitting (r2) in pharmacokinetics, the two-compartment open model was chosen to analyze the data in our experiment. 3p87, a practical pharmacokinetic software program devised by the Chinese Pharmacological Society, was used to analyze these data. The pharmacokinetic parameters are shown in .
Table 1 Comparison of pharmacokinetic parameters of cyclosporin A solution, chitosan-modified nanoparticles, and PEGylated chitosan-modified nanoparticles as a two-compartment model after intravenous injection (n= 3–5)
Our in vivo results show that the elimination half-life (t1/2β) of the nanoparticles was 21 times longer than that of the cyclosporin A solution, and the area under the curve for the nanoparticles was 25.8 times larger than that for the cyclosporin A solution. These observations suggest that the PEG chains provided the nanoparticles with stabilizing properties. The conformational clouds of PEG hindered interaction between the proteins in plasma and the nanoparticles, reduced the number of nanoparticles identified and taken up by mononuclear macrophage cells, prolonged the retention time of the nanoparticles in the circulation, and improved their bioavailability.
Conclusion
In this study, we prepared cyclosporin A-loaded, PEGylated chitosan-modified nanoparticles using an emulsification/ solvent evaporation technique with high encapsulation efficiency and monodispersion properties. From the in vitro release test, we found that the release profiles of the nanoparticles approximated bulk erosion release kinetics in the first 52 hours, with a negligible initial burst. These results indicate that PEGylated chitosan-modified, lipid-based nanoparticles can be useful in providing new opportunities to improve cyclosporin A formulations. These formulations may possibly endow nanoparticles with a long circulation time and the bioavailability of cyclosporin A may be increased.
Acknowledgements
We would like to thank the National Natural Science Foundation of China (30973683, 21275038) and Zhejiang Provincial Natural Science Foundation (Y2090477) for their financial support. This work was also sponsored by the Qianjiang Talents Project (2001R10059) from the Department of Science and Technology in Zhejiang Province and Medicine Project (20100633B09) by the Hangzhou Science and Technology Bureau.
Disclosure
The authors report no conflicts of interest in this work.
Supplementary data
Structural characterization of PEGylated chitosan
The structures of mPEG, mPEG-COOH, mPEG-Su-COONSu, and mPEG-CO-(NH-chitosan) were confirmed by Fourier transform infrared data as follows: mPEG (3400 cm−1, broad and blunt peak, hydroxyl group; 2900 cm−1, saturated C-H; 1100 cm−1, CH2OCH2.); mPEG-COOH (3400 cm−1, broad and blunt peak, hydroxyl; 2900 cm−1 saturated C-H; 1110 cm−1, CH2OCH2; 1738 cm−1, C═O.); mPEG-Su-COONSu (3400 cm−1, broad and blunt, hydroxyl group; 2900 cm−1 saturated C-H; 1114 cm−1, CH2OCH2; 1740 cm−1, C═O, 1780 cm−1, 1812 cm−1, succinimide group); mPEG-CO-(NH-chitosan) (3400 cm−1, broad and blunt, hydroxyl; 2900 cm−1 saturated C-H; 1114 cm−1, CH2OCH2; 1740 cm−1, C═O, becomes weaker, 1662 cm−1, 1586 cm−1, the characteristic peak of the amide group of chitosan).
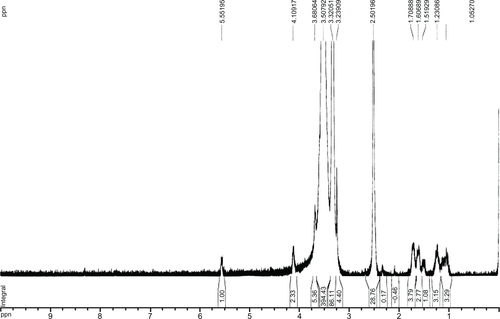
The 1H-NMR spectrum of mPEG-CO-(NH-chitosan) in dimethyl sulfoxide is shown in the . In the spectrum, there are four characteristic peaks, which indicate the target substance. Δ 3.24 ppm (methyl group on head of mPEG); δ 3.24–3.68 ppm (–CH2CH2O, the repeated unit of mPEG); δ 5.55 ppm (hydrogen on the α-C adjacent to oxygen on the saccharide ring of chitosan); δ 8.3 ppm (hydrogen of amide group on the saccharide ring of chitosan). The data presented above indicate that mPEG was grafted successfully to the amino group of chitosan. The graft ratio of PEG to PEGylated chitosan was 16.71%.
Figure S1 1H nuclear magnetic resonance spectra of mPEG-CO-(NH-chitosan) in dimethyl sulfoxide.
References
- PillaiAALevitskyJOverview of immunosuppression in liver transplantationWorld J Gastroenterol2009154225423319750565
- ChadbanSNew-onset diabetes after transplantation – should it be a factor in choosing an immunosuppressant regimen for kidney transplant recipients?Nephrol Dial Transplant2008231816181818326882
- PrzepiorkaDDevineSMFayJWUbertiJPWingardJRPractical considerations in the use of tacrolimus for allogeneic marrow transplantationBone Marrow Transplant1999241053105610578154
- BobadillaNAGambaGNew insights into the pathophysiology of cyclosporine nephrotoxicity: a role of aldosteroneAm J Physiol Renal Physiol2007293F2F917429034
- WijdicksEFNeurotoxicity of immunosuppressive drugsLiver Transpl2001793794211699028
- GinsburgPMThuluvathPJDiarrhea in liver transplant recipients: etiology and managementLiver Transpl20051188189016035068
- TedescoDHaragsimLCyclosporine: a reviewJ Transplant2012201223038622263104
- RobyKAShawLMEffects of cyclosporine and its metabolites in the isolated perfused rat kidneyJ Am Soc Nephrol199341681778400080
- BeauchesnePRChungNSWasanKMCyclosporine A: a review of current oral and intravenous delivery systemsDrug Dev Ind Pharm20073321122017454054
- ItaliaJLBhardwajVRavi KumarMNDisease, destination, dose and delivery aspects of ciclosporin: the state of the artDrug Discov Today20061184685416935754
- AndrysekTImpact of physical properties of formulations on bioavailability of active substance: current and novel drugs with cyclosporineMol Immunol2003391061106512835077
- OnoueSSatoHKawabataYMizumotoTHashimotoNYamadaSIn vitro and in vivo characterization on amorphous solid dispersion of cyclosporine A for inhalation therapyJ Control Release2009138162319376169
- Al-MeshalMAKhidrSHBayomiMAAl-AngaryAAOral administration of liposomes containing cyclosporine: a pharmacokinetic studyInt J Pharm1998168163168
- ItaliaJLBhattDKBhardwajVTikooKRavi KumarMNVPLGA nanoparticles for oral delivery of cyclosporine: nephrotoxicity and pharmacokinetic studies in comparison to Sandimmune Neoral®J Control Release200711919720617399839
- MüllerRHRungeSARavelliVThünemannAFMehnertWSoutoEBCyclosporine-loaded solid lipid nanoparticles (SLN®): drug–lipid physicochemical interactions and characterization of drug incorporationEur J Pharm Biopharm20086853554417804210
- AzziJTangLMooreRPolylactide-cyclosporin A nanoparticles for targeted immunosuppressionFASEB J2010243927393820547662
- Frusic-ZlotkinMSorokaYTivonyRPenetration and biological effects of topically applied cyclosporin A nanoparticles in a human skin organ culture inflammatory model nanoparticles in a human skin organ culture inflammatory modelExp Dermatol20122193894323171455
- VadieiKPerez-SolerRLopez-BeresteinGLukeDRPharmacokinetic and pharmacodynamic evaluation of liposomal cyclosporineInt J Pharm198957125131
- WoodleMCSterically stabilized liposome therapeuticsAdv Drug Deliv Rev199516249265
- VeroneseFMPasutGPEGylation, successful approach to drug deliveryDrug Discov Today2005101451145816243265
- HooftmanGHermanSSchachtEReview: poly(ethylene glycol)s with reactive endgroups. II. Practical consideration for the preparation of protein-PEG conjugatesJ Bioact Compat Polym199611135159
- KataokaKKwonGSYokoyamaMOkanoTSakuraiYBlock copolymer micelles as vehicles for drug deliveryJ Control Release199324119132
- BhattaraiNRamayHRGunnJMatsenFAZhangMPEG-grafted chitosan as an injectable thermosensitive hydrogel for sustained protein releaseJ Control Release200510360962415820408
- KeanTThanouMBiodegradation, biodistribution and toxicity of chitosanAdv Drug Deliv Rev20106231119800377
- PregoCTorresMGarciaDAlonsoMJTransmucosal macromolecular drug deliveryJ Control Release200510115116215588901
- GuzmanECavalloJAChulia-JordanRpH-induced changes in the fabrication of multilayers of poly(acrylic acid) and chitosan: fabrication, properties, and tests as a drug storage and delivery systemLangmuir2011276836684521561105
- GuzmanEChulia-JordanROrtegaFRubioRGInfluence of the percentage of acetylation on the assembly of LbL multilayers of poly(acrylic acid) and chitosanPhys Chem Chem Phys201113182001820721938287
- WeiXHLiangWQThe preparation and characterization of mPEGylated chitosan as a gene vectorChinese J Modern Appl Pharm200320383385
- ChanPKurisawaMChungJEYangY-YSynthesis and characterization of chitosan-g-poly (ethylene glycol)-folate as a non-viral carrier for tumor-targeted gene deliveryBiomaterials20072854054916999995
- JieL-YCaiL-LWangL-JActively-targeted LTVSPWY peptide-modified magnetic nanoparticles for tumor imagingInt J Nanomedicine201273981398922866005
- HuF-QMengPDaiY-QPEGylated chitosan-based polymer micelle as an intracellular delivery carrier for anti-tumor targeting therapyEur J Pharm Biopharm20087074975718620050
- WeiX-HNiuY-PXuY-YDuY-ZHuF-QYuanHSalicylic acid-grafted chitosan oligosaccharide nanoparticle for paclitaxel deliveryJ Bioact Compat Polym201025319335
- ChimalakondaAPShahRBMehvarRHigh-performance liquid chromatographic analysis of cyclosporin A in rat blood and liver using a commercially available internal standardJ Chromatogr B Analyt Technol Biomed Life Sci2002772107114
- WoranuchSYoksanREugeno l-loaded chitosan nanoparticles: I. Thermal stability improvement of eugenol through encapsulationCarbohydr Polym2012
- TripathySDasSChakrabortySPSahuSKPramanikPRoySSynthesis, characterization of chitosan–tripolyphosphate conjugated chloroquine nanoparticle and its in vivo anti-malarial efficacy against rodent parasite: A dose and duration dependent approachInt J Pharm201243429230522664460
- KorsmeyerRWGurnyRDoelkerEBuriPPeppasNAMechanisms of solute release from porous hydrophilic polymersInt J Pharm1983152535
- LeePIPeppasNAPrediction of polymer dissolution in swellable controlled-release systemsJ Control Release19876207215
- ShenJDengYPJinXFPingQESuZGLiLJThiolated nanostructured lipid carriers as a potential ocular drug delivery system for cyclosporine A: improving in vivo ocular distributionInt J Clin Pharm2010402248253
- GibaldiMPerrierDPharmacokinetics. Revised and Expanded2nd edNew York, NYMarcel Dekker1982