 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Bile salt/phospholipid mixed micelles (MMs) are potent carriers used for oral absorption of drugs that are poorly soluble in water; however, there are many limitations associated with liquid formulations. In the current study, the feasibility of preparing bile salt/phospholipid MM precursor (preMM) pellets with high oral bioavailability, using fluid-bed coating technology, was examined. In this study, fenofibrate (FB) and sodium deoxycholate (SDC) were used as the model drug and the bile salt, respectively. To prepare the MMs and to serve as the micellular carrier, a weight ratio of 4:6 was selected for the sodium deoxycholate/phospholipids based on the ternary phase diagram. Polyethylene glycol (PEG) 6000 was selected as the dispersion matrix for precipitation of the MMs onto pellets, since it can enhance the solubilizing ability of the MMs. Coating of the MMs onto the pellets using the fluid-bed coating technology was efficient and the pellets were spherical and intact. MMs could be easily reconstituted from preMM pellets in water. Although they existed in a crystalline state in the preMM pellets, FB could be encapsulated into the reconstituted MMs, and the MMs were redispersed better than solid dispersion pellets (FB:PEG = 1:3) and Lipanthyl®. The redispersibility of the preMM pellets increased with the increase of the FB/PEG/micellar carrier. PreMM pellets with a FB:PEG:micellar carrier ratio of 1:1.5:1.5 showed 284% and 145% bioavailability relative to Lipanthyl® and solid dispersion pellets (FB:PEG = 1:3), respectively. Fluid-bed coating technology has considerable potential for use in preparing sodium deoxycholate/phospholipid preMM pellets, with enhanced oral bioavailability for poorly water-soluble drugs.
Introduction
The most preferable route for drug administration is oral, due to high patient compliance and safety. It was estimated that sales of oral formulations of drugs in the United States alone would reach $56.7 billion in 2012.Citation1 Thus, the development of novel drugs and technologies that are effective when administered orally is a mainstay of pharmaceutical research. However, approximately 40% of the drugs in development and approximately 60% of the drugs coming directly from synthesis are poorly soluble.Citation2 Oral delivery of these drugs is always plagued by low bioavailability, erratic absorption, high intra- and inter- subject variation, and lack of dose proportionality.Citation3 Therefore, delivering drugs via the oral route presents great challenges.
Although some strategies, such as salt formation,Citation4 micronization,Citation5 solid dispersion,Citation6–Citation8 and cyclodextrin complexation,Citation9–Citation11 have shown potential to improve the oral bioavailability of Biopharmaceutical Classification System II drugs,Citation12 they are often limited by the physicochemical properties or the size of drug molecules and are only successful in some instances.Citation13,Citation14 The ability of lipids to enhance the oral absorption of poorly water-soluble drugs has been well known for many years.Citation15 The digestion of lipids promotes the secretion of bile salts and phospholipids, which, together with the products of lipid digestion, form lyotropic colloidal carriers, including multilamellar and unilamellar vesicles, mixed micelles (MMs), and micelles.Citation16 Natural existence of these nanostructures provides a rationale for adopting these carriers for use in order to improve oral absorption of poorly water-soluble drugs.
Recently published data suggest that micelles may be particularly useful for the oral absorption of poorly soluble drugs due to their potential for intercellular permeability, absorption through membrane internalization and lymphatic transportation due to the extremely fine particle size.Citation16,Citation17 Bile salts are the hepatic metabolites of cholesterol and can form stable MMs with phospholipids, glycerides, and fatty acids.Citation18 Such micellar systems are usually larger and more fluid than simple micelles and hence have better solubilizing ability for lipophilic compounds.Citation19 Due to their good physiological compatibility and excellent solubilizing capacity, bile salt micelles and derived mixed systems have been intensively investigated as drug-carrier systems.Citation20–Citation22 Previous investigations into bile salt/phospholipid MMs has mainly focused on the solubilization of insoluble drugs,Citation18,Citation23–Citation25 structural identification,Citation26 physicochemical properties,Citation27 and transmembrane characteristics;Citation17 however, bile salt/phospholipid MMs may also be good candidates for oral delivery of poorly water-soluble drugs.
At present, micellar drug delivery systems are usually prepared as colloidal solutions; however, this is inconvenient for administration and there are stability concerns about the solution state. Inspired by the concept of solid self-nanoemulsifying drug-delivery systems,Citation3,Citation28 the bile salt/phospholipid MMs can be prepared as solid precursor MMs (preMMs), which are hypothesized to reconstitute and form MMs under normal gastrointestinal physiology activity in the presence of body fluid. In previous studies, we developed a fluid-bed coating technique to prepare solid dosage forms of solid dispersion,Citation6–Citation8 cyclodextrin inclusion complexes,Citation9–Citation11 and self-nanoemulsifying drug-delivery systems.Citation3,Citation28 The process involves spraying the solution of the drug/carrier and the subsequent deposition of the coprecipitate onto nonpareil pellets in the drying airflow in a fluid-bed coater. Similarly, the fluid-bed coating technique can also precipitate the drug-loaded MMs onto nonpareil pellets to form preMM pellets and this will overcome the disadvantages of liquid preparations. Moreover, the prepared preMM pellets can be put directly into capsules, thus avoiding problems associated with post-processing such as mixing, grinding, granulation, and tableting.
Fenofibrate (FB) is a Biopharmaceutical Classification System II drug that is used clinically to reduce blood lipids, though its in vivo performance is limited by poor water solubility. Although micronization and nanonization have been implemented to significantly improve the oral bioavailability of FB,Citation29 it has also been reported that the absorption of FB can be significantly increased by food.Citation30,Citation31 The effect of food intake on FB absorption is likely due to the promotion of endogenous bile salt secretion as a result of food digestion;Citation32 thus, bile salt/phospholipid MMs may be good carriers for oral administration of FB.
In the current study, sodium deoxycholate (SDC) was used as the bile salt in this process. Based on the ternary phase diagram and the phase solubility behavior of SDC/phospholipids, FB-loaded SDC/phospholipid preMM pellets were prepared using a fluid-bed coating technique. The resultant preMM pellets were characterized using scanning electron microscopy (SEM), reconstitution, redispersion, and powder X-ray diffractometry (PXRD). The oral bioavailability of preMM pellets was also studied.
Materials and methods
Materials
FB was purchased from Enhua Pharmaceutical Corporation (Xuzhou, People’s Republic of China). Phospholipids were supplied by Shanghai Taiwei Pharmaceutical Company Limited (Shanghai, People’s Republic of China). SDC was purchased from Sinopharm Chemical Reagent Co, Ltd (Shanghai, People’s Republic of China). Polyethylene glycol (PEG) 6000 was purchased from Polymer Pharmaceutical and Excipient Co (Shanghai, People’s Republic of China). Nonpareil pellets (sugar spheres PF101, 710–850 μm in diameter) were provided by Gaocheng Biotech and Health Co, Ltd (Hangzhou, People’s Republic of China). Lipanthyl® capsules, a product of Laboratoires Fournier SA (Dijon, France), were purchased from a local distributor (Zhongshan Hospital Fudan University, Shanghai, People’s Republic of China). Fenofibric acid was provided by Weile Chemical Scientific Co (Shanghai, People’s Republic of China). Deionized water was prepared using a Milli-Q purification system (EMD Millipore, Billerica, MA, USA). High-performance liquid chromatography (HPLC)-grade methanol was obtained from Tedia Co (Fairfield, OH, USA). All other chemicals used in the study were of analytical grade.
Ternary phase diagram of the SDC/phospholipids/water solution
SDC and phospholipids were mixed at weight ratios of 1:9, 2:8, 3:7, 4:6, 5:5, 6:4, 7:3, 8:2, and 9:1. The mixture was first dissolved in a methanol/methylene chloride mixed solution (1:1, volume:volume [v:v]) and then rotary evaporated to dry (IKA RV10; IKA® Werke GmbH & Co, KG, Staufen, Germany) in order to obtain a homogeneous mixture. The residue was titrated with deionized water at 37°C. The mixtures were vortex-mixed and then observed visually to identify the formation of MMs that presented as clear or translucent liquids. The weight of added water was recorded at the phase transition point based on which ternary phase diagram was drawn.
Effects of hydrophilic polymers on solubilization of MMs to FB
In order to precipitate the MMs onto the pellets, hydrophilic polymers are applied as a dispersion matrix, which may affect the solubilization of MMs to FB. Thus, phase solubility studies were performed in polyvinyl pyrrolidone (PVP), hydroxypropyl methyl cellulose (HPMC), and PEG aqueous solutions, respectively, at concentrations of 0%, 0.5%, 1%, 3%, and 5%. The process was the same as that used in the phase solubility studies of cyclodextrins.Citation33 In brief, SDC and phospholipids were mixed in a weight ratio of 4:6 and dissolved in a methanol/methylene chloride mixed solution (1:1, v:v) and then dried with a rotary evaporator. The resulting film was hydrated with a polymer solution for 1 hour and then diluted with the same solution to obtain MM suspensions with concentrations of 1, 10, 20, 30, 40, 50, 60, 70, and 80 mg/mL. An excess amount of FB was added to the 5 mL MM suspensions, which were then shaken in a water bath shaker at 37°C for 60 hours until equilibrium was reached. The suspensions were then filtered using 0.20 μm polycarbonate membranes. The filtrate was diluted with methanol and assayed for FB. Phase solubility diagrams were plotted with FB solubility versus MM concentration.
Effects of hydrophilic polymers on critical micelle concentration (CMC) of MMs
The effects of PVP, HPMC, and PEG aqueous solutions (1%, weight:volume) on the CMC of the MMs (SDC:phospholipids = 4:6) were studied using the standard pyrene method.Citation18,Citation34 In brief, 50 μL pyrene methanol solution (400 μmol/L) was put into a 10 mL volumetric flask and methanol was removed by vacuum drying. A specific volume of MM suspension (10 mg/mL, prepared as described above) was added to the residue and diluted to 10 mL with the corresponding solution to obtain a micellar concentration range of 0.01–0.22 mg/mL. The mixtures were ultrasonically treated for 0.5 hours followed by 8 hours of shaking at 37°C. Free pyrene was removed by filtration through 0.22 μm polycarbonate membranes. The fluorescence of the filtrate was scanned 30 nm/minute, at an excitation wavelength of 335 nm, using the LS55 fluorescence spectrophotometer (PerkinElmer, Waltham, MA, USA). The slit for excitation and emission was set as 15.0 and 2.5 nm, respectively. The fluorescence intensity of I1 and I3 were measured at wavelengths of 372 and 384 nm, respectively. The pyrene 1:3 ratio (I1:s3) was plotted with the concentration of MMs. The plot was fitted with the Boltzmann equation, as follows, and the CMC was denoted with x0 value in the equation:Citation33
Preparation of FB-loaded SDC/phospholipid preMM pellets
FB-loaded SDC/phospholipid preMM pellets were prepared with a Mini-Glatt fluid-bed coater (Wurster insert; Glatt GmbH, Binzen, Germany) based on previously established procedures, with modifications.Citation3,Citation6–Citation11,Citation28 SDC and phospholipids with weight ratio of 4:6 served as micellar carriers and were dissolved in an isopropanol-water (3:7, v:v) mixed solution. FB and PEG were then put into the solution and dissolved using sonication. The solid content of the solution was set as 10% (weight:volume). The resulting solution was sprayed through a nozzle (0.5 mm in diameter) onto the fluidized nonpareil pellets to obtain a coating weight gain of 100%. The detailed operating conditions were as follows: product temperature, 30°C−34°C; air flow rate, 97–103 m3/hour; spray rate, 0.5 mL/minute; atomizing air pressure, 1.4–1.5 bar. The pellets were further dried for 15 minutes after coating completion.
The preMM pellets were first prepared with FB:total carrier weight ratios of 1:1, 1:2, 1:3, and 1:4 at a fixed PEG:micellar carrier ratio of 1:1 (weight:weight). The preMM pellets were then prepared with PEG:micellar carrier weight ratios of 1:2 and 0:3 respectively at a fixed FB:total carrier ratio of 1:3 (weight:weight). A solid dispersion pellet, with an FB:PEG weight ratio of 1:3, was also prepared using the same technology and served as the control.
The preMM and solid dispersion were also prepared by depositing them onto inert PVC pellets under the same coating conditions in order to circumvent the interference of nonpareil cores with PXRD. The outer layer of the pellets was peeled off carefully and ground into fine powder for analysis.
SEM
Both the surface and cross-section morphologies of the preMM pellets were observed using an XL30 scanning electron microscope (Royal Philips Electronics, Amsterdam, The Netherlands). The samples were fixed on a brass stub using double-sided tape and then gold-coated in a vacuum by a sputter coater. The photographs were taken at an excitation voltage of 20 kV.
PXRD
A Shimadzu X-ray diffractometer (XRD-6000; Kyoto, Japan) was employed to study the PXRD patterns of the FB, PEG, physical mixture (FB:PEG:micellar carrier = 1:1.5:1.5), solid dispersion and the preMMs over a 2.5°–50° 2θ range and at a scan rate of 0.3°/minute. The tube anode was Cu with a Ka of 0.154 nm. The instrument operated at 40 kV of tube voltage and 60 mA of tube current. The PXRD patterns were collected in a step scan mode (step size 0.02°).
Reconstitution
The preMM pellets were put into 25 mL of ultrapure water (37°C) and stirred for 30 minutes. The suspension was filtered with 0.22 μm polycarbonate membranes and the filtrate was diluted with ten volumes of ultrapure water for particle-size determination using a NICOMP™ 380 ZLS Particle Sizer (NICOMP Particle Sizing Systems, Santa Barbara, CA, USA).
Redispersibility test and in vitro determination of FB
Since the preMM pellets could reconstitute and form MMs in water, FB was actually encapsulated in the reconstituted MMs during dissolution, thus the term “redispersibility” was preferable to “dissolution.”Citation3 The redispersibility was performed using a ZRS-8G dissolution tester (Tianda Tianfa Technology Co, Ltd Tianjin, People’s Republic of China) based on the Chinese Pharmacopoeia Method I (the rotating basket method). Lipanthyl®, the physical mixture (FB:PEG:micellar carrier = 1:1.5:1.5), solid dispersion pellets, and preMM pellets, equivalent to 30 mg FB, were sealed in #0 capsules (Suzhou Capsugel Ltd., Suzhou, People’s Republic of China) for the redispersibility study. SDS aqueous solution (1%, weight:volume) was selected as the redispersing medium and 900 mL was verified to reach sink condition.Citation34 The capsules were put into the baskets and immersed into the redispersing medium, thermostatically maintained at 37°C ± 0.5°C and stirred at 100 rpm. At predetermined intervals, 5 mL samples were withdrawn and filtered with 0.22 μm polycarbonate membranes. The filtrates were assayed for FB content by HPLC. In the meantime, 5 mL of fresh redispersing medium at an equal temperature was added to keep the volume constant.
An HPLC-ultraviolet (UV) method was applied to detect FB content in phase solubility and redispersion studies. The Agilent 1200 HPLC system (Agilent Technologies, Santa Clara, CA, USA) was composed of a quaternary pump, a degasser, an autosampler, a column heater, and a tunable ultraviolet detector. FB was separated on a Diamonsil-C18 column (5 μm, 4.6 mm × 250 mm; Dikma Technologies Inc, Beijing, People’s Republic of China) guarded with an ALLTECH pre-column (C18, 5 μm, 1.0 mm × 20 mm; Grace Davison Discovery Sciences, Deerfield IL, USA) at 40°C and detected at 286 nm. The mobile phase was composed of methanol and water at a volume ratio of 90:10 and pumped at 1.0 mL/minute.
Bioavailability study
The bioavailability study was carried out on six male Beagle dogs weighing 10 ± 0.5 kg using a crossover experimental design. The Laboratory Animal Service Center of Fudan University provided the animals. Animals were fasted for 12 hours prior to the experiment and had free access to water. All animal procedures followed the principles outlined in the Declaration of Helsinki for all animal experimental investigations. There was a 7-day washout period between the two treatments.
The solid dispersion pellets and Lipanthyl® were selected as controls to study the relative bioavailability of the preMM pellets (FB:PEG:micellar carrier = 1:1.5:1.5). Both types of pellets had been previously put into #0 capsules. The preparations, at doses of 30 mg/kg, were placed at the pharyngeal site and swallowed spontaneously by the animals. Two-milliliter blood samples were collected from the antebrachium veins of the dogs and placed into heparinized tubes at 0 (pre-dose), 0.25, 0.5, 0.75, 1, 1.25, 1.5, 2, 4, 6, 8, 10, and 12 hours post-dose. Blood samples were centrifuged at 4000 × g for 10 minutes and plasma was withdrawn and stored at −18°C until analysis.
Determination of fenofibric acid in dog plasma
Due to the fast metabolism of FB to fenofibric acid in vivo, pharmacokinetic evaluation of FB is based on quantification of fenofibric acid in plasma.Citation34 The same Agilent 1200 HPLC system as was used for in vitro determination was used to quantify fenofibric acid in dog plasma. The mobile phase was composed of methanol, deionized water, and 10% phosphoric acid with volume ratio of 70:30:1. The flow rate of the mobile phase was fixed at 1.0 mL/minute and the column temperature was set at 30°C. Fenofibric acid was separated on a Diamonsil-C18 column guarded with an ALLTECH precolumn (C18, 5 μm, 1.0 mm × 20 mm) and detected at 287 nm.
Liquid-liquid extraction procedures were used to extract fenofibric acid from dog plasma. Internal standard solution (10 μg/mL indomethacin in methanol) of 100 μL and 20 μL hydrochloric acid aqueous solution (1 mol/L) was successively added to 0.8 mL plasma and vortex-mixed for 1 minute. Then 5 mL of diethyl ether anhydrous was added to the suspension and the solution was vortex-mixed for an additional 10 minutes. After centrifuging at 4000 × g for 15 minutes, the organic layer was transferred to another tube and evaporated under a light stream of nitrogen at 40°C. The residue was dissolved by 100 μL methanol under vortex mixing for 2 minutes and centrifuged at 15000 × g for 10 minutes. A volume of 20 μL of the supernatant was taken for HPLC analysis. Quantification was based on the peak area ratio of fenofibric acid to the internal standard (R, AFA:AIS). In the concentration (C) range of 0.0625–2.50 μg/mL, linearity was observed with correlation coefficients of over 0.99 (a typical calibration curve: R = 1.507C − 0.0130 − r = 0.9984, n = 7). Accuracy for the determination of fenofibric acid in dog plasma (n = 9) was 94.86% ± 3.33%. Intra- and inter-day precisions were all below 5%, and the extraction recovery of fenofibric acid in dog plasma (n = 9) was 74.67% ± 2.46%.
Data and statistical analysis
Pharmacokinetic analysis was performed using DAS® 2.0 (Boying Corporation, Beijing, People’s Republic of China). Maximum concentration (Cmax) and time of maximum concentration (tmax) were observed as raw data. The linear trapezoidal rule was used to calculate area under the curve to the last measurable concentration (A∪C0–t). Area under the curve extrapolated to infinity (A∪C0–∞) was calculated as:
Results and discussion
Ternary phase diagram of the SDC/phospholipids/water solution
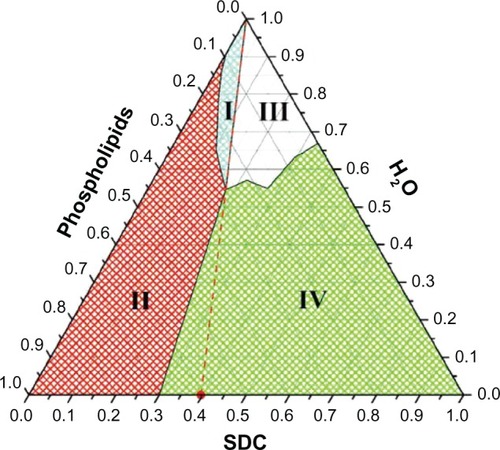
The phase diagram of the SDC/phospholipids/water solution can be divided into four regions by appearance (). When the weight ratio of the SDC:phospholipids was lower than 3:7, a turbid milky-white solution accompanied with a solid SDC:phospholipid mixture was formed (region II). With the addition of water, the excess SDC/phospholipids could be dispersed and a uniform milky white solution was formed (region I). However, when the weight ratio of the SDC:phospholipid exceeded 3:7, a clear and transparent solution could be formed, although there were still two regions (III and IV) depending on amount of water added. Based on the phase diagram, a clear and transparent phase can be formed when the weight ratio of SDC:phospholipids exceeds 3:7. Phospholipids alone form insoluble liquid crystalline bilayer structures rather than micelles when added to water,Citation35 while bile salts can solubilize phospholipids by surrounding the perimeter of the phospholipid bilayer to form MMs.Citation24,Citation36 However, the ability of bile salts to solubilize phospholipids depends on their amounts.Citation37 At low ratio of SDC:phospholipids (less than 3:7), phospholipid aggregates formed with a milky appearance. Based on the phase diagram, when the SDC:phospholipid weight ratio exceeded 4:6 (molar ratio 1:1, indicated as the region to the right of the red dashed line), the amount of SDC was sufficient to solubilize the phospholipids and could be diluted infinitely without any obvious change in appearance. This result coincides with reported values.Citation25 Since the bile salt toxicity to gastrointestinal epithelia and membranes can be counteracted by equimolar or greater concentrations of phospholipids,Citation39 SDC:phospholipids with weight ratios of 4:6 were fixed for the follow-up studies.
Figure 1 The ternary phase diagram of the SDC/phospholipids/water solutions.
Abbreviation: SDC, sodium deoxycholate.
Solubilization of MMs on FB and CMC of the MMs
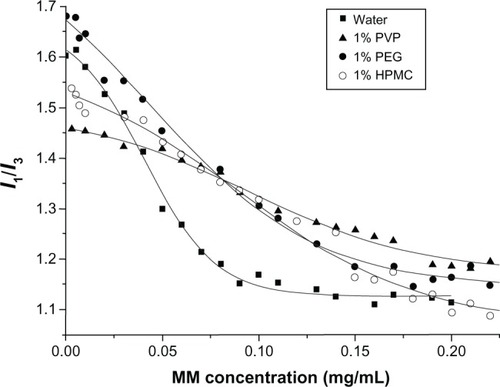
The SDC/phospholipid MMs showed significant solubilizing ability for FB, as indicated by the linear increase in the FB solubility with a corresponding increase in the concentration of MMs (). Hydrophilic polymers had a great effect on the solubilizing ability of MMs. The addition of HPMC/PVP decreased the solubilizing effects of MMs on FB, however, PEG improved the solubilizing ability (). This could be due to a CMC alteration of the MMs resulting from the addition of the polymer. Increased CMC will decrease the quantity of micelles in the solution if the concentration of micelles does not change, which decreases the solubilization ability of micelles. The CMC value of SDC/phospholipid MMs was determined in water and then compared with that in PVP, HPMC, and PEG aqueous solutions (1%, w:v). Plots of pyrene 1:3 ratios versus concentration of SDC/phospholipids in different media are presented in , the center of which denotes the CMC value. Compared to the plot from water, plots from polymer solutions seemed to be compressed and the inflection points were not so obvious. The fitted results from the Boltzmann equation are shown in . The CMC values for SDC/phospholipid MMs were more or less increased in the polymer solutions. This was ascribed to the decreased polarity of the media by the polymers. The CMC value was doubled in the PVP and HPMC solutions compared to in water. The increased CMC will inevitably decrease the quantity of micelles in the solution and the solubilizing ability of the micelles, thus HPMC and PVP decreased the solubilizing effects of the MMs on FB. Although PEG increased the CMC of the MMs, the alteration was not obvious and the CMC value was still at the same level as in water. However, PEG can associate with the micelle corona and provide an expanded region of reduced polarity for drug solubilization.Citation40 Consequently, PEG improved the solubilizing ability of the MMs for FB, thus PEG was adopted as the dispersion matrix to precipitate the MMs onto the pellets.
Table 1 The fitted results of the Boltzmann equation and the CMC of the mixed micelles in different solutions
Figure 2 Effects of HPMC (A), PVP (B), and PEG (C) on the solubilizing ability of MMs to FB (37°C, n = 3).
Abbreviations: FB, fenofibrate; HPMC, hydroxypropyl methyl cellulose; MMs, mixed micelles; PVP, polyvinyl pyrrolidone; PEG, polyethylene glycol.
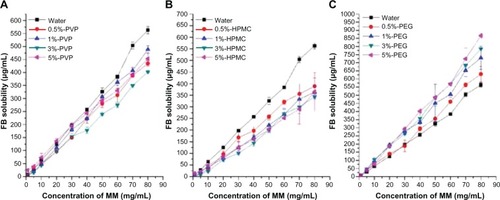
Figure 3 Plots of pyrene in a 1:3 ratio versus concentration of MMs in 1% HPMC, PVP, and PEG aqueous solutions.
Abbreviations:I, fluorescence intensity; HPMC, hydroxypropyl methyl cellulose; MM, mixed micelle; PVP, polyvinyl pyrrolidone; PEG, polyethylene glycol.
Preparation of FB loaded SDC/phospholipid preMM pellets
Coating of MMs onto the pellets was effective, under the coating conditions in this study, with recoveries of over 90%. Under visual observation, the pellets were all spherical in shape and intact. The SEM photographs of the preMM pellets with FB:PEG:micellar carrier weight ratios of 1:1.5:1.5, 1:1:2, and 1:0:3 are shown in . The surface of the pellets, without PEG as the dispersion matrix, was imperfect under SEM observation. Phospholipids are in a semi solid state and soft at room temperature. Without PEG as the dispersion matrix, the phospholipids were exposed on the surface of the pellets, so that the pellets were soft and sticky by touch. The adhesions between the pellets damaged the surfaces. Since the preMMs were dispersed into the PEG matrix in the other two pellets, the surfaces of these pellets were rigid and not sticky so that the pellets with MMs dispersed into PEG matrix were intact. In addition, the cross-sectional view confirmed a layer of tightly coated preMMs around the nonpareil core (–).
Figure 4 SEM photographs of the surface (A–C, ×50) and cross-section (D–F, ×200 of A–C, respectively) of the preMM pellets prepared with FB:PEG:micellar carrier = 1:1.5:1.5 (A and D), 1:1:2 (B and E) and 1:0:3 (C and F), respectively.
Abbreviations: FB, fenofibrate; MM, mixed micelle; PEG, polyethylene glycol; SEM, scanning electron microscopy.

Reconstitution
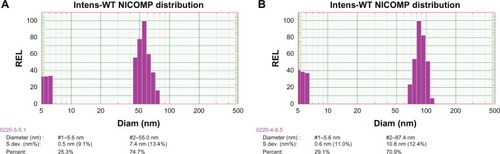
The preMM pellets (FB:PEG:micellar carrier = 1:1.5:1.5) were readily dispersed in ultrapure water. The redispersed solution was as clear and transparent as the appearance of the original MM solution. The Nicomp distribution of the particle size for the original and reconstituted MMs is shown in . There were two peaks in the particle size distribution of both the original and the reconstituted MMs. The smaller size (~5 nm) might be derived from SDC micelles. Approximately 74.7% of the original MMs were around 55 nm in size (). The particle size of the reconstituted MMs was slightly increased, with around 70.9% at approximately 87.4 nm in size (). These results indicate that the preMMs can be reconstituted to form MMs without significant variation in the particle size of the MM counterparts.
Figure 5 Particle size distribution for original (A) and reconstituted (B) MMs.
Abbreviations: Diam, diameter; MMs, mixed micelles; S.Dev, standard deviation.
Effects of total carrier amount and the weight ratio of PEG:micellar carrier on redispersibility
The weight ratio of the FB:total carrier had a great effect on the redispersibility of the preMM pellets. The redispersion rate and degree was increased with increased carrier amount as shown in the redispersion profiles (). However, when the amount of total carrier exceeded that of FB by two-fold, the redispersion rate and degree seemed to be maximized and did not increase further. At FB:total carrier weight ratios of 1:3 and 1:4, the degree of redispersion reached approximately 95% in 30 minutes. Although micronization can enhance the dissolution of drugs that are poorly soluble in water, it cannot increase their solubility. Thus, Lipanthyl® (micronized FB) could only dissolve less than 80% in 120 minutes. However, FB was redispersed through the encapsulation into the reconstituted mixed micelles. The MMs enhanced the dissolution rate and degree of FB only if the micellar amount was enough to encapsulate FB (ie, FB:total carrier weight ratio was 1:3 and 1:4). This was also the reason that the redispersibility of the preMM pellets increased with the increase in the carrier amount. In addition, since a 1.5-fold amount of micellar carrier to FB was used in the physical mixture, the physical mixture showed superior redispersibility than did the preMM pellets with the FB:PEG:micellar carrier weight ratio of 1:0.5:0.5. However, the physical mixture was only 40% redispersed within 120 minutes; far less than that of the preMM pellets with the same FB/carrier weight ratio. These results indicate that the simple blending of SDC and phospholipids does not provide quick and complete reconstitution. The homogeneous mixing of SDC and phospholipids in the preMMs by fluid-bed coating technology may be an essential prerequisite to obtain favorable redispersibility.
Figure 6 Redispersion profiles of preMM pellets with different FB:PEG:micellar carrier weight ratios (values displayed in brackets) and dissolution of FB from physical mixture, solid dispersion pellets (FB:PEG = 1:3) and Lipanthyl® (Laboratoires Fournier SA, Dijon, France).
Abbreviations: PM, physical mixture; FB, fenofibrate; MM, mixed micelle; PEG, polyethylene glycol.
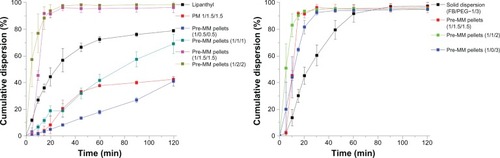
The effect of PEG content on redispersibility was also studied at a fixed FB:total carrier ratio of 1:3 (weight:weight). A previous study showed that an FB:PEG weight ratio of 1:3 is adequate to form a solid dispersion and that microcrystalline FB existed in it.Citation35 Thus, the solid dispersion pellets, which dissolved about 90% in 60 minutes, showed superior dissolution when compared to Lipanthyl®, likely due to the higher dispersion state (). In spite of this fact, the preMM pellets still had the highest redispersion rate, which could be due to the encapsulation of FB into the reconstituted MMs during redispersion instead of the solid dispersion of FB in PEG. However, dispersion of the preMMs into the PEG matrix is favorable for redispersibility, since the preMM pellets containing PEG as the distribution matrix redispersed faster than those without PEG. Due to coverage by a large quantity of phospholipids, the surfaces of the preMM pellets without PEG become viscous in water, which is unfavorable for redispersion. The preMM pellets containing PEG were highly dispersed into the PEG matrix and had a faster reconstitution rate based on the solid dispersion mechanism.
PXRD
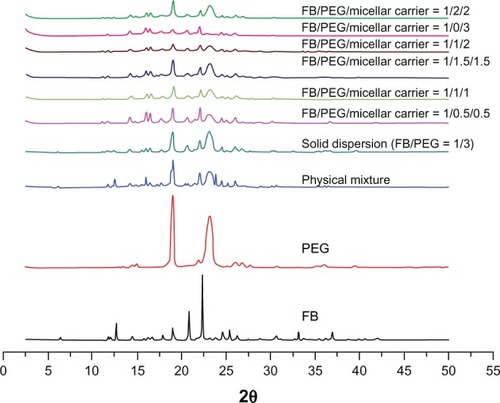
The X-ray powder diffraction patterns of FB, PEG, physical mixture (FB:PEG:micellar carrier = 1:1.5:1.5), solid dispersion and preMMs are shown in . FB and PEG demonstrated typical diffraction peaks ranging from 2.5° to 50° (2θ), respectively. Diffraction peaks for the physical mixture of both FB and PEG were discernible with the diffractogram, looking more like the superimposition of each of the components. Since FB forms eutectic mixtures with PEG at a weight ratio of 1:3,Citation35 both the characteristic peaks of FB and PEG could be identified in the diffraction curves of the solid dispersion. However, the diffraction peaks of FB remained in all preMM pellets, indicating that the FB was crystalline in the preMM pellets. The solution of MMs evaporated by the fluid-bed coating process during the preparation, thus the micellar structure could not be maintained. The preMMs actually existed as a lipid film and were dispersed in a PEG matrix. When the preMM pellets come in contact with water, MMs can be reconstituted and FB is encapsulated. This is the same as the thin-film evaporation technique used to prepare micelles except that the lipid film is formed on the surface of the pellets instead of the round-bottom flask.Citation18,Citation24,Citation41
Figure 7 X-ray powder diffraction patterns of FB, PEG, physical mixture, solid dispersion (FB:PEG = 1:3) and preMMs with different FB:PEG:micellar carrier weight ratios.
Abbreviations: FB, fenofibrate; MMs, mixed micelles; PEG, polyethylene glycol.
Bioavailability
The plasma fenofibric acid concentration versus time after oral administration of the preMM pellets, solid dispersion pellets, or Lipanthyl® is shown in . The preMM pellets showed a similar absorption phase as the solid dispersion pellets though they presented a slower elimination phase. In general, the plasma concentrations for these two preparations were higher than that of Lipanthyl® at the same time points. Double peaks of maximum concentrations were observed in the profile of the preMM pellets, which seemed to be a common phenomenon in the bile salt/phospholipid MMs and was ascribed to the enterohepatic circulation of bile salt or phospholipids. The pharmacokinetic parameters for the three preparations are shown in . Lipanthyl® had the shortest tmax of 1.063 ± 0.239 hours, which corresponded with faster dissolution at earlier times as shown in . The overall absorption of FB was significantly improved regarding Cmax and A∪C. Lipanthyl® showed the smallest Cmax, A∪C0–t and A∪C0–∞ values of 0.397 ± 0.162 μg/mL, 1.451 ± 0.394 μg/mL ⋅ hour, and 2.033 ± 1.200 μg/mL ⋅ hour, respectively. FB is a Biopharmaceutical Classification System II drug, and the limiting step for its absorption is the dissolution. Compared to Lipanthyl®, the solid dispersion pellets had better dissolution and therefore had higher Cmax, A∪C0–t and A∪C0–∞values of 0.662 ± 0.222 μ/mL, 2.837 ± 1.459 μg/mL ⋅ hour, and 5.755 ± 4.104 μg/mL ⋅ hour, respectively. The preMM pellets had the fastest redispersibility among the preparations, and thus had the highest Cmax, A∪C0–t, and A∪C0–∞ values, reaching 0.874 ± 0.395 μg/mL, 4.123 ± 2.263 μg/mL ⋅ hour, and 7.200 ± 5.347 μg/mL ⋅ hour, respectively. Based on the A∪C0–t value, the relative bioavailability of the pro-MM pellets was 284% and 145% relative to Lipanthyl® and the solid dispersion pellets, respectively.
Table 2 The main pharmacokinetic parameters of preMM pellets (FB:PEG:micellar carrier = 1:1.5:1.5), solid dispersion pellets (FB:PEG = 1/3) and Lipanthyl® (Laboratoires Fournier SA, Dijon, France), after a single dose
Figure 8 Mean plasma FB acid concentration-time curves in beagle dogs after oral administration of preMM pellets (FB:PEG:micellar carrier = 1:1.5:1.5), solid dispersions pellets (FB:PEG = 1:3) and Lipanthyl® (Laboratoires Fournier SA, Dijon, France) (n = 6).
Abbreviations: FB, fenofibrate; MM, mixed micelle; PEG, polyethylene glycol.
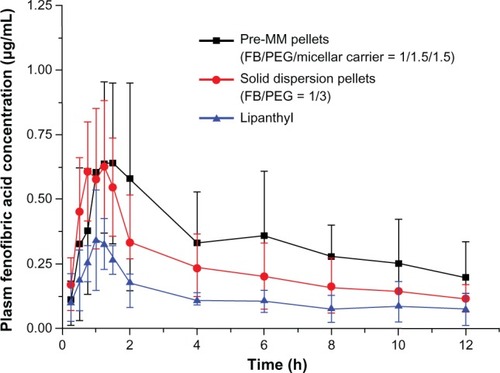
Micronization and solid dispersion technology are the main contributors to the enhanced dissolution rate. However, mixed micelles can increase not only the redispersion rate, but also the degree of redispersion, which provides them with the highest bioavailability. The unstirred water layer composes the diffusional barrier for the transport of lipophilic drugs from the bulk fluid phase of the small intestine lumen to the brush border membrane.Citation42 Micellar encapsulation greatly enhances the mass transport of lipophilic materials,Citation16 which also contributes to the enhanced absorption of FB. However, it has been suggested that micelles are not absorbed intact across the brush border membrane and that lipophilic drugs may dissociate from the mixed micellar phase before being absorbed across the apical membrane.Citation16 In this case, the MMs act as a drug reservoir and the encapsulated drugs are released constantly in the brush border. Therefore, the preMM pellets presented a slower elimination phase than the solid dispersion pellets.
Conclusion
SDC and phospholipids can form MMs when their weight ratio exceeds 3:7. PEG increased the solubilizing ability of the MMs to FB. Solid preMM pellets were successfully prepared by fluid-bed coating technology and could easily be reconstituted in water and form MMs. The solidifying process did not significantly increase the particle size of the reconstituted MMs. Although it existed in the crystalline state in the preMM pellets, FB was quickly redispersed due to having been encapsulated into the reconstituted MMs. When compared to SB/PEG solid dispersion and Lipanthyl®, the oral bioavailability of FB was significantly improved by the preMM pellets.
Acknowledgments
This work was financially supported by the National Key Basic Research Program of People’s Republic of China (2009CB930300). Dr Wei Wu would like to thank the Shanghai Municipal Commission of Education (10GS05) and Ministry of Education (NCET-11-0114) for personnel fostering funding.
Disclosure
The authors report no conflicts of interest in this work.
References
- DesaiPPDateAAPatravaleVBOvercoming poor oral bioavailability using nanoparticle formulations – opportunities and limitationsDrug Discov Today201192e87e95
- KeckCMMüllerRHDrug nanocrystals of poorly soluble drugs produced by high pressure homogenisationEur J Pharm Biopharm200662131616129588
- LeiYLuYQiJSolid self-nanoemulsifying cyclosporin A pellets prepared by fluid-bed coating: preparation, characterization and in vitro redispersibilityInt J Nanomedicine2011679580521589647
- SerajuddinATSalt formation to improve drug solubilityAdv Drug Deliv Rev200759760361617619064
- MunozAGuichardJPReginaultPMicronised fenofibrateAtherosclerosis1994110SupplS45S487857384
- ZhangXSunNWuBLuYGuanTWuWPhysical characterization of lansoprazole/PVP solid dispersion prepared by fluid-bed coating techniquePowder Technol20081823480485
- SunNZhangXLuYWuWIn vitro evaluation and pharmacokinetics in dogs of solid dispersion pellets containing Silybum marianum extract prepared by fluid-bed coatingPlanta Med200874212613218240100
- SunNWeiXWuBChenJLuYWuWEnhanced dissolution of silymarin/polyvinylpyrrolidone solid dispersion pellets prepared by a one-step fluid-bed coating techniquePowder Technol200818217280
- ChenZLuYQiJWuWEnhanced dissolution, stability and physicochemical characterization of ATRA/2-hydroxypropyl-β-cyclodextrin inclusion complex pellets prepared by fluid-bed coating techniquePharm Dev Technol201318113013622304703
- LuYZhangXLaiJYinZWuWPhysical characterization of meloxicam-β-cyclodextrin inclusion complexes pellets prepared by a fluid-bed coating methodParticuology20097118
- ZhangXWuDLaiJLuYYinZWuWPiroxicam/2-hydroxypropyl-beta-cyclodextrin inclusion complex prepared by a new fluid-bed coating techniqueJ Pharm Sci200998266567518543294
- DahanAMillerJMAmidonGLPrediction of solubility and permeability class membership: provisional BCS classification of the world’s top oral drugsAAPS J200911474074619876745
- Merisko-LiversidgeELiversidgeGGCooperERNanosizing: a formulation approach for poorly-water-soluble compoundsEur J Pharm Sci200318211312012594003
- PatravaleVBDateAAKulkarniRMNanosuspensions: a promising drug delivery strategyJ Pharm Pharmacol200456782784015233860
- DahanAHoffmanAThe effect of different lipid based formulations on the oral absorption of lipophilic drugs: the ability of in vitro lipolysis and consecutive ex vivo intestinal permeability data to predict in vivo bioavailability in ratsEur J Pharm Biopharm20076719610517329087
- PorterCJTrevaskisNLCharmanWNLipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugsNat Rev Drug Discov20076323124817330072
- YanoKMasaokaYKataokaMSakumaSYamashitaSMechanisms of membrane transport of poorly soluble drugs: role of micelles in oral absorption processesJ Pharm Sci20109931336134519743502
- ZhuYYuJTongSPreparation and in vitro evaluation of povidone-sodium cholate-phospholipid mixed micelles for the solubilization of poorly soluble drugsArch Pharm Res201033691191720607496
- HendradiEObataYIsowaKNagaiTTakayamaKEffect of mixed micelle formulations including terpenes on the transdermal delivery of diclofenacBiol Pharm Bull200326121739174314646182
- DongowskiGFritzschBGiesslerJHartlAKuhlmannONeubertRHThe influence of bile salts and mixed micelles on the pharmacokinetics of quinine in rabbitsEur J Pharm Biopharm200560114715115848066
- HammadMAMüllerBWSolubility and stability of lorazepam in bile salt/soya phosphatidylcholine-mixed micellesDrug Dev Ind Pharm199925440941710194595
- GuoJWuTPingQChenYShenJJiangGSolubilization and pharmacokinetic behaviors of sodium cholate/lecithin-mixed micelles containing cyclosporine ADrug Deliv2005121353915801719
- NagataMYotsuyanagiTIkedaKSolubilization of vitamin K1 by bile salts and phosphatidylcholine-bile salts mixed micellesJ Pharm Pharmacol198840285882897458
- DuanRLSunXLiuJGongTZhangZRMixed micelles loaded with silybin-polyene phosphatidylcholine complex improve drug solubilityActa Pharmacol Sin201132110811521170082
- HammadMAMüllerBWIncreasing drug solubility by means of bile salt-phosphatidylcholine-based mixed micellesEur J Pharm Biopharm19984633613679885310
- RanganathanRVautier-GiongoCBakshiMSBalesBLHajduJPhospholipid containing mixed micelles. Characterization of diheptanoyl phosphatidylcholine (DHPC) and sodium dodecyl sulfate and DHPC and dodecyl trimethylammonium bromideChem Phys Lipids200513519310415854628
- SinghJUnluZRanganathanRGriffithsPAggregate properties of sodium deoxycholate and dimyristoylphosphatidylcholine mixed micellesJ Phys Chem B2008112133997400818335917
- LeiYQiJNieSSolid self-nanoemulsifying cyclosporine A pellets prepared by fluid-bed coating: stability and bioavailability studyJ Biomed Nanotechnol20128351552122764422
- KeatingGMOrmrodDMicronised fenofibrate: an updated review of its clinical efficacy in the management of dyslipidaemiaDrugs200262131909194412215067
- BalfourJAMcTavishDHeelRCFenofibrate. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in dyslipidaemiaDrugs19904022602902226216
- YunHYJoo LeeEYoun ChungSThe effects of food on the bioavailability of fenofibrate administered orally in healthy volunteers via sustained-release capsuleClin Pharmacokinet200645442543216584288
- Guivarc’hPHVachonMGFordyceDA new fenofibrate formulation: results of six single-dose, clinical studies of bioavailability under fed and fasting conditionsClin Ther20042691456146915531008
- LuYTangNQiJWuWPhase solubility behavior of hydrophilic polymer/cyclodextrin/lansoprazole ternary system studied at high polymer concentration and by response surface methodologyPharm Dev Technol201217223624121067338
- AguiarJCarpenaPMolina-BolívarJACarnero RuizCOn the determination of the critical micelle concentration by the pyrene 1:3 ratio methodJ Colloid Interface Sci20032581116122
- TangNLaiJChenYLuYWuWFenofibrate solid dispersion pellets prepared by fluid-bed coating: physical characterization, improved dissolution and oral bioavailability in beagle dogsJ Chin Pharm Sci2009182156161
- SmallDMPhase equilibria and structure of dry and hydrated egg lecithinJ Lipid Res1967865515576057484
- MazerNABenedekGBCareyMCQuasielastic light-scattering studies of aqueous biliary lipid systems. Mixed micelle formation in bile salt-lecithin solutionsBiochemistry19801946016157356951
- RuppCSteckelHMüllerBWSolubilization of poorly water-soluble drugs by mixed micelles based on hydrogenated phosphatidylcholineInt J Pharm20103951–227228020580793
- DialEJRooijakkersSHDarlingRLRomeroJJLichtenbergerLMRole of phosphatidylcholine saturation in preventing bile salt toxicity to gastrointestinal epithelia and membranesJ Gastroenterol Hepatol200823343043617868333
- OliveiraCPRibeiroMERicardoNMThe effect of water-soluble polymers, PEG and PVP, on the solubilisation of griseofulvin in aqueous micellar solutions of Pluronic F127Int J Pharm2011421225225722001534
- JinSFuSHanJImprovement of oral bioavailability of glycyrrhizin by sodium deoxycholate/phospholipid-mixed nanomicellesJ Drug Target201220761562222726209
- ThomsonABSchoellerCKeelanMSmithLClandininMTLipid absorption: passing through the unstirred layers, brush-border membrane, and beyondCan J Physiol Pharmacol19937185315558306192