 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Background
The aim of this study was to develop an optimized solid self-microemulsifying drug delivery system (SMEDDS) formulation for sirolimus to enhance its solubility, stability, and bioavailability.
Methods
Excipients used for enhancing the solubility and stability of sirolimus were screened. A phase-separation test, visual observation for emulsifying efficiency, and droplet size analysis were performed. Ternary phase diagrams were constructed to optimize the liquid SMEDDS formulation. The selected liquid SMEDDS formulations were prepared into solid form. The dissolution profiles and pharmacokinetic profiles in rats were analyzed.
Results
In the results of the oil and cosolvent screening studies, Capryol™ Propylene glycol monocapry late (PGMC) and glycofurol exhibited the highest solubility of all oils and cosolvents, respectively. In the surfactant screening test, D-α-tocopheryl polyethylene glycol 1000 succinate (vitamin E TPGS) was determined to be the most effective stabilizer of sirolimus in pH 1.2 simulated gastric fluids. The optimal formulation determined by the construction of ternary phase diagrams was the T32 (Capryol™ PGMC:glycofurol:vitamin E TPGS = 30:30:40 weight ratio) formulation with a mean droplet size of 108.2 ± 11.4 nm. The solid SMEDDS formulations were prepared with Sucroester 15 and mannitol. The droplet size of the reconstituted solid SMEDDS showed no significant difference compared with the liquid SMEDDS. In the dissolution study, the release amounts of sirolimus from the SMEDDS formulation were significantly higher than the raw sirolimus powder. In addition, the solid SMEDDS formulation was in a more stable state than liquid SMEDDS in pH 1.2 simulated gastric fluids. The results of the pharmacokinetic study indicate that the SMEDDS formulation shows significantly greater bioavailability than the raw sirolimus powder or commercial product (Rapamune® oral solution).
Conclusion
The results of this study suggest the potential use of a solid SMEDDS formulation for the delivery of poorly water-soluble drugs, such as sirolimus, through oral administration.
Introduction
Sirolimus, also known as rapamycin, is a triene macrolide derived from Streptomyces hygroscopicus.Citation1 It has potent immunosuppressive activity and inhibits T-cell activation.Citation2 Its poor water solubility (2.6 μg/mL at 25°C)Citation3 and low stability are a hurdle to the development of an oral dosage form. Sirolimus is very unstable in electrolyte solution.Citation4 Macrolides such as sirolimus have been shown to be sensitive to both acids and bases, resulting in ring fragmentation and degradation.Citation5–Citation7
The first commercial sirolimus product was Rapamune® oral solution (1 mg/mL of sirolimus; Wyeth, now Pfizer Inc, New York, NY, USA). This product is an oily solution containing a mixture of propylene glycol, phosphatidylcholine, and polysorbate 80.Citation3 It has several disadvantages, such as strict storage conditions, poor taste, and inconvenient usage. In addition, the oral bioavailability is only 14%.Citation8 The tablet formulation of sirolimus was launched in 2002 (Rapamune®) using NanoCrystal® technology acquired by Elan Corporation (Dublin, Ireland).Citation9 However, this formulation requires special production facilities, and the production of nanoparticles consumes large amounts of energy. Its bioavailability is even lower than the general oral dosage form (<17%).Citation10 Therefore, many studies have attempted to improve the bioavailability of sirolimus through the development of forms such as nanocrystals,Citation9 liposomes,Citation11 inclusion complexes,Citation12,Citation13 and solid dispersions.Citation14,Citation15
There has recently been a great deal of attention focused on lipid-based formulations.Citation16 Among the lipid-based formulations, the self-microemulsifying drug delivery system (SMEDDS) is a clear and monophasic mixture of oil and surfactant, sometimes including a cosolvent or cosurfactant. Under gentle agitation similar to the movement of the gastrointestinal tract, the SMEDDS formulation becomes an emulsion. Since SMEDDS are generally thermodynamically stable, they spontaneously produce a stable oil-in-water emulsion.Citation17 The drug in emulsion is presented in a solubilized form, and thus the dissolution and absorption are improved. Therefore, SMEDDS is a potential strategy for enhancing the oral bioavailability of poorly water-soluble drugs.Citation18 However, SMEDDS are liquid formulations, which have several disadvantages, including low stability and portability during the manufacturing process and limited dosage forms, such as soft gelatin capsule. To overcome these problems, solid SMEDDSs have been investigated as alternatives.Citation19
The formulation of sirolimus in SMEDDS has already been performed in other studies.Citation20,Citation21 However, those studies did not consider the effects of surfactants on the stability of sirolimus. In the case of sirolimus, which has low stability, enhancing the stability will contribute to better drug absorption and higher immunosuppressive activity.Citation20 The aim of this study was to develop an optimized formulation of sirolimus solid SMEDDS with enhanced solubility and stability in order to improve oral bioavailability. Excipients that can enhance the solubility and stability of sirolimus were screened. Based on the ternary phase diagram, the optimal formulation was determined and was solidified by granulation.
Materials and methods
Sirolimus (purity 99.4%) was purchased from the Beijing Everbright Science and Trading Co (Beijing, People’s Republic of China). Everolimus (purity 95.9%), tetraglycol (glycofurol), myristic acid isopropyl ester (isopropyl myristate), and zinc sulfate were purchased from Sigma-Aldrich (St Louis, MO, USA). Propylene glycol monocaprylate (Capryol™ PGMC), diethylene glycol monoethyl ether (Transcutol®), lauroyl macrogol-32 glycerides (Gelucire® 44/14), and sucrose monopalmitate (Sucroester 15) were kindly provided by Gattefossé (Lyon, France). D-α-tocopheryl polyethylene glycol 1000 succinate (vitamin E TPGS) was purchased from Eastman Chemical Company (Kingsport, TN, USA). Ethylene oxide propylene oxide block copolymer (poloxamer 407) was purchased from BASF Co, Ltd (Ludwigshafen, Germany). Glycerol triacetate (triacetin) was purchased from Yakuri Pure Chemical (Osaka, Japan). Oleic acid castor oil, and mannitol were purchased from Duksan Pharmaceutical (Seoul, Korea). Triglycerides of caprylic/capric acid (CAPTEX® 300) were purchased from ABITEC Corporation (Columbus, OH, USA). Rapamune® oral solution (lot no A64340), the commercial product of sirolimus, was purchased from Wyeth (now Pfizer Inc). All the organic solvents were high-performance liquid chromatography (HPLC) grade, and the remaining chemicals were reagent grade.
Solubility of sirolimus in various oils and cosolvents
To select a suitable oil and cosolvent for SMEDDS formulation, the solubility of sirolimus in various oils and cosolvents was measured. An excess amount of sirolimus was added to glass tubes containing 500 mg of various oils (triacetin, isopropyl myristate, Capryol™ PGMC, oleic acid castor oil, and CAPTEX® 300) and cosolvents (Transcutol®, glycofurol). The mixtures were sonicated for 10 minutes and then placed in a shaking water bath (60 rpm) at 25°C for 3 days and protected from light. The resulting samples were centrifuged at 15,000 rpm for 20 minutes at 25°C. The supernatant was diluted with methanol, and the concentration of sirolimus was determined by HPLC. HPLC analysis of sirolimus was performed with the same method used in our previous studyCitation15 The HPLC system was composed of a Waters™ (Milford MA, USA) HPLC system with a 5 μm, 4.6 × 250 mm ZORBAX Eclipse XDB-C18 column at 60°C. The eluates were detected at 220 nm. The flow rate was 1.0 mL/minute and the mobile phase used was 84% methanol and 16% water. Fifty microliters of sample were injected for each analysis.
Stability of sirolimus in various surfactants
The stability of sirolimus in various surfactants was analyzed by adding a solution of sirolimus in dimethyl sulfoxide (DMSO) to 50 mL of surfactant dissolved in pH 1.2 simulated gastric fluids (1 mg/mL). The samples were kept at 37°C and protected from light. Suitable aliquots of the samples were collected at the given times (3, 17, 32, 47, 63 minutes) and the concentration of the remaining sirolimus was analyzed by HPLC.
Preparation of the solid SMEDDS formulation
The solid SMEDDS formulation was prepared by dissolving 1 mg sirolimus in a 100 mg mixture of oil, cosolvent, and surfactant. First, sirolimus was dissolved in appropriate amounts of a mixture of oil and cosolvent. Appropriate amounts of surfactant were then added. Each surfactant was preheated at 55°C and used in the melted state. The mixture was then vortex-mixed to ensure homogeneity. The weight ratio of each liquid component (oils, cosolvents, and surfactants) varied by 10% among the different formulations. The total number of formulations was 63 for each surfactant. The formulations were stored for 24 hours at room temperature, and the phase separation was examined prior to the self-emulsification test and particle size analysis. Based on the results, the non-separated samples were chosen for the self-emulsification test.
Visual assessment of the efficiency of self-emulsification
The efficiency of self-emulsification was visually assessed by the following descriptions.Citation22,Citation23 Ten microliters of the SMEDDS formulation was introduced into 20 mL water. When gently stirred using a magnetic bar, the tendency to form an emulsion was judged as “good” when the droplets spread easily in water and were clear or slightly bluish. The tendency was judged as “bad” when there was poor or no emulsion formation with immediate coalescence of oil droplets.
Analysis of the emulsion size
The size of the emulsion of the formulation that was judged as “good” during visual observation was measured. Ten microliters of the SMEDDS formulation was diluted with 20 mL water and gently mixed with a magnetic stirrer. The size distributions of the emulsions were determined by dynamic light scattering (DLS) using ELS-8000 (Otsuka Electronics Co, Ltd, Osaka, Japan). The measured samples were stored at room temperature. After 24 hours, the particle size distributions were re-measured.
Construction of the ternary phase diagram
The ternary phase diagram of the oil, cosolvent, and surfactant was constructed. The volume ratio of water to SMEDDS formulation was fixed at 1000:1. Each component of the SMEDDS formulation (oil, cosolvent, and surfactant) represents an apex of the triangle. Since all formulations contained the same level of drug (1 mg), the weight ratio of drug was omitted in the ternary phase diagram. The sum of ratios of the liquid components (oil, cosolvent, and surfactant) was regarded as being 100%. The corresponding component weight ratio is 100% at each apex and decreases to 0% at the opposite side. Each component increased by 10% compared to the previous formulation, with a total variation range of 0% to 100%. The results of the phase separation test, self-emulsification test, and droplet size analysis were plotted on the ternary phase diagram. All of the ternary phase diagrams were combined into one overall diagram.
Preparation of the solid SMEDDS formulation
The solid SMEDDS formulation was prepared by granulation. Mannitol and sucrose monopalmitate (Sucroester 15), which exhibited a good stabilization of sirolimus in pH 1.2 simulated gastric fluid, were used to prepare the solid SMEDDS formulation. Mannitol (3 g) and sucrose monopalmitate (2 g) were weighed and mixed homogenously Accurately weighted liquid SMEDDS equivalent to 10 mg sirolimus was then added and kneaded. After kneading, the blend was granulated by passing through a 500 μm sieve. The solid SMEDDS formulations were stored at room temperature.
Reconstitution study
Solid SMEDDS (600 mg) were dispersed in 200 mL water and gently vortex-mixed. After mixing, an aliquot of the mixture was filtered through a 1 μm polytetrafluoroethylene (PTFE) membrane filter (Whatman, Kent, UK). The size of droplet in the filtrate was measured by DLS. The results were compared with previous liquid SMEDDS droplet size analysis findings. As a negative control, 600 mg of the mannitol and Sucroester 15 mixture without liquid SMEDDS was analyzed as mentioned above.
In vitro dissolution study
Dissolution profiles of the liquid SMEDDS formulation, the solid SMEDDS formulation, and the raw sirolimus powder were obtained using a VK 7000 dissolution testing station (Varian Inc, Palo Alto, CA, USA) according to the United States Pharmacopeia XXVIII paddle method and operating at a rotation speed of 50 rpm. Each test was performed in 900 mL distilled water and pH 1.2 simulated gastric fluids. The temperature was maintained at 37°C ± 0.1°C.
Samples containing equivalent amounts of sirolimus (1 mg) were placed in dissolution medium, and 4 mL aliquot samples were withdrawn at certain time intervals (0.16, 0.33, 0.5, 0.75, 1, 1.5, 2 hours) and filtered using a 0.45 μm glass microfiber filter. The filtered samples were diluted with methanol, and the concentration of drug was assayed by HPLC.
In vivo study
The in vivo study was performed similarly to our previous studyCitation15 All the experiments were performed according to the guidelines for the care and use of laboratory animals at Chungnam National University, Daejeon, Republic of Korea. Male Sprague-Dawley rats weighing between 200 and 220 g were obtained from Samtaco Bio Korea Inc (Osan, Korea). General and environmental conditions were strictly monitored. The rats were fasted for 24 hours prior to the experiments. After 4 hours of dosing, foods were reoffered. The femoral artery was cannulated with 23-gauge catheters under anesthesia with diethyl ether. After recovery from anesthesia, the rats were administrated with raw sirolimus powder, liquid SMEDDS formulation, solid SMEDDS formulation, and Rapamune® oral solution by oral sonde at a dose of 5 mg/kg. Raw sirolimus powder was suspended in 2 mL 0.2% (w/v) aqueous methylcellulose immediately before dosing. The SMEDDS formulations were accurately weighed and diluted with 2 mL water. Next, 1 mL Rapamune® oral solution was accurately drawn in an enclosed syringe and diluted with 1 mL water. Then, 500 μL blood samples were collected from the femoral artery at certain times (0.33, 0.66, 1, 1.5, 2, 3, 5, 8, 12 hours) and transferred into Eppendorf tubes containing 20 μL of ethylenediaminetetraacetic acid.
The Liquid chromatography-mass spectrometry (LC-MS) analysis of the concentration of sirolimus in the blood samples and the pharmacokinetic data analysis were performed with the same method used in our previous studyCitation15 The whole-blood samples (400 μL) were mixed with 400 μL methanol, 400 μL 6.25% (w/v) zinc sulfate, and 40 μL of an internal standard solution (1 μg/mL everolimus in 50% methanol). After centrifugation at 13,000 rpm for 10 minutes, the supernatants were mixed with NaOH (100 μL, 0.1 M) and 1-chlorobutane and vortexed. After centrifugation at 13,000 rpm for 10 minutes, the supernatants were evaporated to dryness under nitrogen. The residual was dissolved in 100 μL 70% methanol, and 10 μL of the dissolved sample was analyzed using an LCMS-2010A mass spectrometer (Shimadzu Corporation, Kyoto, Japan). The samples were injected using an SIL-10A autoinjector through a SUPELCOSIL™ (Sigma-Aldrich Corporation, St Louis, MO, USA) LC-18 column (150 × 4.6 mm, 3 μm) at 60°C to elute sirolimus ([M–Na+] m/z of 936.00) and everolimus ([M–Na+] m/z of 980.00) using an acetonitrile-water (70:30 v/v) mobile phase at a flow rate of 1.0 mL/minute.
The area under the curve (AUC)0→12 h was calculated by noncompartmental analysis. The values of peak blood concentration (Cmax) and time to peak concentration (Tmax) were obtained directly from the blood data. One-way analysis of variance followed by the least-squares difference test was performed to demonstrate statistically significant differences.
Results and discussion
Solubility of sirolimus in various oils and cosolvents
To develop a SMEDDS of poorly water-soluble sirolimus, the selection of suitable oil, cosolvent, and surfactant is important. For the selection of an appropriate self-emulsifying vehicle, it is important to consider the drug solubility in the vehicle, the efficiency of self-emulsification, and the droplet size distribution of the resulting emulsion.Citation23
In the oils and cosolvent screening, the solubility values of sirolimus in six oils (triacetin, isopropylmyristate, Capryol™ PGMC, oleic acid castor oil, and CAPTEX® 300) and two cosolvents (Transcutol® and glycofurol) were measured. The solubility values of sirolimus in various oils and cosolvents are presented in .
Table 1 Solubility values of sirolimus in various oils and cosolvents
Among the oils, Capryol™ PGMC exhibited the highest solubility of sirolimus at 57.81 mg/g. In the cosolvent screening study, glycofurol demonstrated the highest solubility of sirolimus at 164.42 mg/g. From these results, Capryol™ PGMC and glycofurol were selected as the oil and cosolvent, respectively, for the optimal SMEDDS formulation for further studies.
Effect of surfactants on drug stability
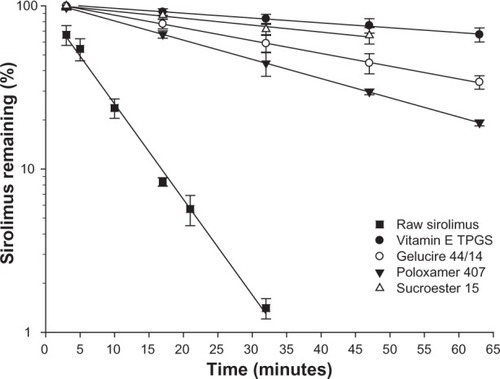
Sirolimus is very unstable, especially in acidic conditions. Therefore, stabilization in acidic conditions is the key factor in the development of sirolimus formulations. In our previous study, the screenings of excipients that enhanced the stability of sirolimus were performed for various excipients (polymer, surfactant, hydrotropic agent, organic acid/base, and sugar).Citation15 The results showed that surfactants stabilized sirolimus to a great degree. The results of the stability tests of the raw materials and a representative surfactant (vitamin E TPGS, Sucroester 15, Gelucire 44/14, and poloxamer 407) with a high stabilization effect are shown in . Without any surfactant, sirolimus was rapidly degraded in pH 1.2 simulated gastric fluids, and the remaining amount was less than 10% at 30 minutes. The semi-logarithmic plots of the remaining sirolimus versus time indicated pseudo first-order degradation behavior:
Figure 1 The effect of surfactants on the stability of sirolimus in pH 1.2 simulated gastric fluids.
Notes: Data are expressed as the mean ± standard deviation (n = 3). Vitamin E TPGS: Eastman Chemical Company(Kingsport, TN, USA), Gelucire 44/14 and Sucroester 15: Gattefosse (Lyon, France), Poloxamer 407: BASF Co, Ltd (Ludwigshafen, Germany)
where [C0] is the initial concentration and [C]t is the percentage remaining at time t, and kobs is the degradation pseudo-first-order rate constant calculated from the linear regression analysis. The half-life (t1/2) was calculated according to EquationEquation 2(2) :
The values of kobs and t1/2 are summarized in . These surfactants significantly enhanced the stability of sirolimus. The stabilization effect of the surfactants might be due to micelle formation. The most effective stabilizer was vitamin E TPGS, followed by Sucroester 15, Gelucire 44/14, and poloxamer 407. Among the surfactants, vitamin E TPGS and Gelucire 44/14 are semi-solid states that are easily mixed with oil and cosolvent in the melted state because of their low melting points of 37°C~41°C and 44°C, respectively. Thus, vitamin E TPGS and Gelucire 44/14 were the suitable surfactants used for SMEDDS formulation. From these results, vitamin E TPGS was selected as the surfactant for SMEDDS formulation, and Gelucire 44/14 was selected as a comparison.
Table 2 Effect of excipients on the stability of sirolimus in a dissolution medium of pH 1.2
Construction of the ternary phase diagram
The purpose of the construction of the ternary phase diagram is to determine the optimal formulation, which consists of clear homogeneous states, good self-emulsifying efficiency, and small droplet size when diluted with water. The visual test was performed to confirm the phase state of SMEDDS formulation. Since the surfactants used in these studies, vitamin E TPGS and Gelucire 44/14, are in semi-solid states, the SMEDDS formulation may experience phase separation. Therefore, the phase behavior of the SMEDDS formulation is important. In the cases of the SMEDDS formulation using vitamin E TPGS as a surfactant, 40 of the 63 formulations formed a homogeneous mixture and were marked in the ternary phase diagram. The maximum surfactant (vitamin E TPGS) weight ratio is 50%. All the formulations containing more than 50% surfactant precipitated or phase separated. In the case of Gelucire 44/14, among the 63 formulations, 31 formed a homogeneous mixture. The maximum surfactant (Gelucire 44/14) weight ratio is 30%.
The SMEDDS formed fine oil–water emulsions with only gentle agitation. Since the free energy to form an emulsion is relatively low, the formation of the emulsion is thermodynamically spontaneous.Citation24 The visual assessment of the SMEDDS formulation was performed to measure the apparent spontaneity of the emulsion formation. In cases of the SMEDDS formulation using vitamin E TPGS as a surfactant, 18 of the 40 homogenous formulations were judged as “good” (formed a clear or slightly bluish state) upon visual observation. The efficiency of the emulsification was good when the oil (Capryol™ PGMC) weight ratio was 20%~70%, cosolvent (glycofurol) weight ratio was 10%~70%, and surfactant (vitamin E TPGS) weight ratio was 10%~50%. In the case of Gelucire 44/14, among the 31 homogeneous formulations, eight were judged as “good.” The efficiency of the emulsification was good when the oil (Capryol™ PGMC) weight ratio was 10%~40%, cosolvent (glycofurol) weight ratio was 30%~80%, and surfactant (Gelucire 44/14) weight ratio was 10%~30%. It could be seen that the formulation using vitamin E TPGS as a surfactant exhibited a larger range of self-emulsification than Gelucire 44/14. The “good” self-emulsifying formulations were marked in the ternary phase diagram.
Droplet size seems to affect drug release. Smaller droplets resulted in better drug release by providing a large interfacial area over which the drug could diffuse into the gastrointestinal fluid and thus increase drug absorption.Citation25 The droplet sizes of the “good” self-emulsifying formulations were measured, and the results are summarized in . In this study, we established the “small” droplet size standard as 200 nm. In the case of the SMEDDS formulation using vitamin E TPGS as a surfactant, the ranges of the mean droplet size were 93.9~615.2 nm. Among the 18 “good” self-emulsifying formulations, 13 exhibited a mean droplet size of less than 200 nm. In the case of Gelucire 44/14, the ranges of the mean droplet size were 113.6~257.2 nm. Among the eight “good” self-emulsifying formulations, only three demonstrated a mean droplet size less than 200 nm.
Table 3 Composition of the SMEDDS formulations and the results of the droplet size analysis
When the SMEDDS formulation was introduced in water, the surfactants formed a layer around the oil droplets, reduced the interfacial energy, and provided a mechanical barrier to coalescence. However, the separation of the phases was merely delayed in these systems, which were still considered unstable from a thermodynamic point of view.Citation24 To evaluate the stability of the emulsion formation, the droplet sizes after 24 hours of dilution in water were measured and the results are presented in . In this study, we established the standard of emulsion stability after 24 hours as the droplet size of 300 nm. In the case of the SMEDDS formulation using vitamin E TPGS as a surfactant, the mean droplet sizes after 24 hours increased 0.86~2.93-fold from the initial droplet size. Among the 13 formulations that had a less than 200 nm initial droplet size, ten exhibited a less than 300 nm droplet size after 24 hours. In the case of Gelucire 44/14, the mean droplet sizes after 24 hours showed no significant increase. All formulations in which the initial size was less than 200 nm were smaller than 300 nm after 24 hours. The increasing ratio was 0.98~1.02 and suggests that the initial droplet sizes were smaller in the vitamin E TPGS system than in the Gelucire 44/14 system. The Gelucire 44/14 system maintained the initial droplet size over a longer period of time than the vitamin E TPGS system.
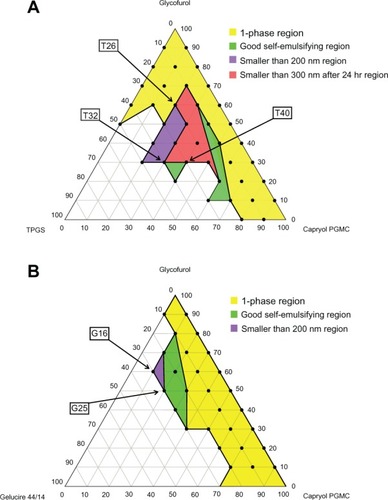
The results of the phase separation test, self-emulsification test, initial droplet size test, and the after 24 hours droplet size test were marked on the ternary phase diagrams and all the diagrams were combined into one overall diagram. shows the ternary phase diagram of the vitamin E TPGS and Gelucire 44/14 systems.
Figure 2 The pseudo ternary phase diagram indicating the 1-phase region (yellow), good self-emulsifying region (green), smaller than 200 nm region (purple), and smaller than 300 nm after 24 hours region (red). (A) Capryol™ PGMC–glycofurol–vitamin E TPGS system; (B) Capryol™ PGMC–glycofurol–Gelucire 44/14.
Note: Capryol™ PGMC and Gelucire 44/14: Gattefossé (Lyon, France).
Determination of optimal formulation
To determine the optimal formulation, each of the ternary phase diagrams was overlaid and the optimal liquid SMEDDS formulation was selected in the formulation that included all four regions (1-phase region, good self-emulsifying region, smaller than 200 nm region, and smaller than 300 nm after 24 hours region). In the Capryol™ PGMC-glycofurol-vitamin E TPGS system, the formulation T26 exhibited the smallest droplet size (93.9 ± 2.3 nm) with a 20% oil weight ratio. When the total SMEDDS weight was 101 mg (1 mg sirolimus + 100 mg liquid component), the weight of the oil was 20 mg. The sirolimus concentration in the oil was 50 mg/g and almost reached saturated solubility (57.81 mg/g in Capryol™ PGMC) and could have led to precipitation. Therefore, the formulation T32 which showed the second smallest droplet size (108.2 ± 11.4 nm) was selected as an optimal formulation. The formulation T40, which had a relatively higher oil weight ratio than T32 (droplet size: 172.0 ± 2.6 nm), was selected as a comparison. In the Capryol™ PGMC-glycofurol-Gelucire 44/14 system, the formulation G16, which demonstrated the smallest droplet size (113.6 ± 6.6 nm), had too low of an oil weight ratio (10%). Therefore, the formulation G25, which had a 20% oil weight ratio (droplet size 169.0 ± 4.1 nm), was selected as a comparison with the T32 formulation. T32 (Capryol™ PGMC:glycofurol:vitamin E TPGS = 30:30:40 weight ratio) was determined to be the optimal formulation, and T40 (Capryol™ PGMC:glycofurol:vitamin E TPGS = 40:30:30 weight ratio) and G25 (Capryol™ PGMC:glycofurol:Gelucire 44/14 = 20:50:30 weight ratio) were selected to compare the dissolution profile and bioavailability, respectively.
Droplet size of the reconstituted microemulsions
In the case of sirolimus, low stability in acidic conditions may be a major obstacle in its formulation. To maintain a stable state in pH 1.2 dissolution conditions, a high concentration of surfactant was required. Rapamune® oral solution, the commercial product of sirolimus, with 1 mL of other excipients was used to deliver 1 mg sirolimus. In the SMEDDS formulation of this study, the weight of the other excipients was 100 mg. To stabilize the SMEDDS formulation, more surfactants were necessary. Another surfactant, Sucroester 15, which demonstrated a stabilization effect of sirolimus in pH 1.2 simulated gastric fluids, was added in the liquid SMEDDS formulation. Generally, SMEDDS is solidified using high surface-area colloidal inorganic adsorbent substances such as silica, silicates, magnesium trisilicate, magnesium hydroxide, and talcum.Citation26 However, in our preliminary study, sirolimus was found to have extremely low stability when it was in contact with inorganic metals such as aluminum and magnesium (data not shown). Therefore, we decided to use mannitol as an absorbent. To evaluate the reconstitution property of the solid SMEDDS formulation, the droplet size of the reconstituted microemulsion was measured. The mean droplet size of the T32 solid SMEDDS formulation was 94.1 ± 8.7 nm. When mannitol and the Sucroester 15 mixture without the SMEDDS formulation were treated in the same manner, no emulsion droplets were detected. The droplet size of the microemulsion from the solid SMEDDS was slightly decreased, but the difference was not statistically significant compared to the liquid SMEDDS (unpaired t-test, P = 0.164). The decreased droplet size of the solid SMEDDS was probably attributed to the addition of more surfactant (Sucroester 15). From these results, the adsorption of the liquid SMEDDS in mannitol and Sucroester 15 mixture did not seem to have a remarkable effect on droplet size.
In vitro dissolution study
In vitro dissolution studies were performed for raw sirolimus powder, liquid SMEDDS, and solid SMEDDS. The dissolution profiles for each formulation in distilled water are shown in . The release amount of sirolimus from the SMEDDS formulation was significantly higher than that of raw sirolimus powder. The release amount of sirolimus from the liquid SMEDDS formulation rapidly reached levels greater than 90% within 10 minutes. This could suggest that the drug, which completely dissolved in the SMEDDS formulation, could be released due to its small droplet size, permitting a faster rate of release into the aqueous phase compared to raw sirolimus powder. The release amount of sirolimus from the solid SMEDDS formulation slowly reached 90% compared to the liquid SMEDDS formulation. This was due to the delayed release caused by Sucroester 15 in the solid SMEDDS formulation. The dissolution profiles of each formulation in pH 1.2 simulated gastric fluids are shown in . In the liquid SMEDDS formulation, the released sirolimus in pH 1.2 simulated gastric fluids was rapidly degraded, and the release amount of sirolimus was less than 20% within 30 minutes. The release amount of sirolimus from the solid SMEDDS formulation slowly increased to 50% at 2 hours. However, in the solid SMEDDS without sucroester, the released sirolimus was rapidly degraded, similar to the liquid SMEDDS formulation. Mannitol did not enhance the stability of sirolimus. Thus, the enhanced stability of the solid SMEDDS formulation might be due to the slow release rate and the stabilization effect of Sucroester 15.
Figure 3 Dissolution profiles of sirolimus in (A) distilled water and (B) pH 1.2 simulated gastric fluid.
Note: Data are expressed as the mean ± standard deviation (n = 3).
Abbreviation: SMEDDS, self-microemulsifying drug delivery system.
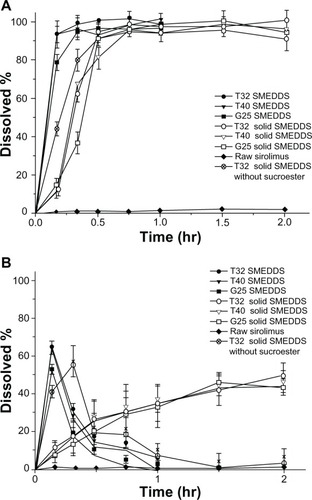
As a result of the addition of Sucroester 15 in the solid SMEDDS formulation, sirolimus maintained a more stable state than the liquid SMEDDS formulation in pH 1.2 simulated gastric fluids. These increased release profiles and stability in acidic conditions could affect the bioavailability.
In vivo pharmacokinetic study in rats
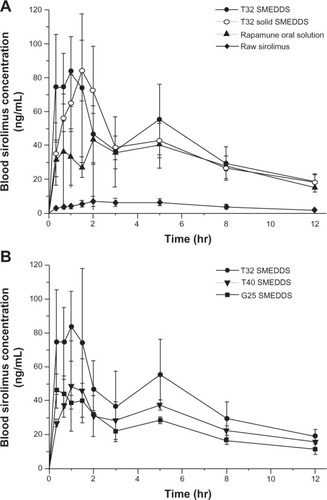
In order to evaluate the bioavailability of the sirolimus SMEDDS formulation, an in vivo pharmacokinetic study was performed in rats. shows the time courses of sirolimus blood concentration after the oral administration. The pharmacokinetic parameters are presented in . The absorption of drug from the T32 liquid SMEDDS formulation was significantly improved compared with the raw material and other formulations. For the raw sirolimus powder, the AUC0→12 h, Cmax, and Tmax were 56.3 ± 7.9 ng hr/mL, 8.8 ± 2.4 ng/mL, and 3.75 ± 1.5 hours, respectively. After the administration of the T32 liquid SMEDDS formulation, the AUC0→12 h, Cmax, and Tmax were 483.93 ± 120.3 ng hr/mL, 108.9 ± 25.1 ng/mL, and 1.0 ± 0.5 hours, respectively. The T32 liquid SMEDDS formulation exhibited a higher bioavailability than the raw sirolimus powder, with an approximately 8.6-fold and 12.3-fold increase in AUC0→12 h, and Cmax, respectively. Based on the one-way analysis of variance of the AUC00→12 h, values, there were significant differences (P < 0.05) between the samples. All liquid SMEDDS and T32 solid SMEDDS formulations showed significantly increased AUC0→12 h, over raw sirolimus powder. In addition, the T32 liquid SMEDDS formulation showed a significantly increased AUC0→12 h over commercial Rapamune® oral solution. The higher bioavailability of the T32 SMEDDS formulation was probably due to the protection of oil droplets containing sirolimus in the gastrointestinal tract by the surfactant. The surfactant enhanced the absorption of the drug by disturbing the cell membrane.Citation27 As shown in , the absorption of sirolimus increased with the surfactant concentration. In the T32 SMEDDS formulation, the AUC0→12 h and Cmax values of the liquid and solid formulation had no significant differences (P = 0.372 and P = 0.146, respectively). In the T32 solid SMEDDS formulation, the Tmax value was slightly delayed but the difference was not statistically significant. It may be that the delayed Tmax is due to the slow release rate of sucroester. This result suggests that SMEDDS may be a useful tool for enhancing the bioavailability of sirolimus.
Table 4 Pharmacokinetic parameters of sirolimus in rats after the oral administration of raw sirolimus powder or SMEDDS formulation
Figure 4 Blood concentration–time profile of sirolimus in rats after the oral administration of (A) raw sirolimus powder, T32 liquid, and solid SMEDDS formulations and Rapamune® oral solution; (B) liquid SMEDDS formulations at a dose equivalent to 5 mg sirolimus/kg of body weight.
Notes: Data are expressed as the mean ± standard deviation (n = 4). Rapamune® (Wyeth, now Pfizer Inc, New York, NY, USA).
Abbreviation: SMEDDS, self-microemulsifying drug delivery system.
Conclusion
To develop SMEDDS for enhancing the bioavailability of poorly water-soluble sirolimus, the SMEDDS formulation composed of oil, cosolvent, and surfactant was established. Based on the results of the solubility test in various vehicles and a stability test in pH 1.2 simulated gastric fluids, the oil, cosolvent, and surfactant were selected. Through the construction of a ternary phase diagram, we determined that the optimal combination of SMEDDS formulation was as follows: 30% of Capryol™ PGMC as the oil, 30% of glycofurol as the cosolvent, and 40% of vitamin E TPGS as the surfactant (weight ratio). To improve the stability and overcome the drawback of liquid formulation, the solid SMEDDS were prepared using Sucroester 15 and mannitol. In vitro dissolution studies revealed that the release amount of sirolimus from the SMEDDS formulation was higher than raw sirolimus powder, and the drug stability of solid SMEDDS formulation improved in pH 1.2 simulated gastric fluids. Also, in vivo studies in rats demonstrated that the SMEDDS formulation exhibited a significantly increased absorption than raw sirolimus powder. Our studies suggest the potential use of SMEDDS formulation for the delivery of poorly water-soluble drugs, such as sirolimus, through oral administration.
Acknowledgment
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MEST) (No 2008-0060608).
Disclosure
The authors report no conflicts of interest in this work.
References
- VézinaCKudelskiASehgalSNRapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principleJ Antibiot (Tokyo)197528107217261102508
- MartelRRKliciusJGaletSInhibition of the immune response by rapamycin, a new antifungal antibioticCan J Physiol Pharmacol19775514851843990
- SimamoraPAlvarezJMYalkowskySHSolubilization of rapamycinInt J Pharm20012131–2252911165091
- RoufMABilensoyEVuralIHincalAADetermination of stability of rapamycin following exposure to different conditionsEur J Pharm Sci2007321S46S46
- LuengoJIKonialianALHoltDAStudies on the chemistry of rapamycin: novel transformations under Lewis-acid catalysisTetrahedron Lett1993346991994
- SteffanRJKearneyRMHuDCBase catalyzed degradations of rapamycinTetrahedron Lett1993342336993702
- YohannesDMyersCDDanishefskySJDegradation of rapamycin: synthesis of a rapamycin derived fragment containing the tricarbonyl and triene sectorsTetrahedron Lett1993341320752078
- VasquezEMSirolimus: a new agent for prevention of renal allograft rejectionAm J Health Syst Pharm200057543744810711524
- RosenHAbribatTThe rise and rise of drug deliveryNat Rev Drug Discov20054538138515864267
- ShenLJWuFLLNanomedicines in renal transplant rejection – focus on sirolimusInt J Nanomedicine200721253217722509
- AlemdarAYSadiDMcAlisterVCMendezILiposomal formulations of tacrolimus and rapamycin increase graft survival and fiber outgrowth of dopaminergic graftsCell Transplant200413326327115191164
- RoufMBilensoyEVuralIHincalAAInclusion complexation of rapamycin with beta-cyclodextrin to improve solubility and stability of the drugEur J Pharm Sci2007321S46S47
- BuechGBertelmannEPleyerUSiebenbrodtIBorchertHHFormulation of sirolimus eye drops and corneal permeation studiesJ Ocul Pharmacol Ther200723329230317593014
- PreethamACSatishCSFormulation of a poorly water-soluble drug sirolimus in solid dispersions to improve dissolutionJ Dispers Sci Technol2011326778783
- KimMSKimJSParkHJChoWKChaKHHwangSJEnhanced bioavailability of sirolimus via preparation of solid dispersion nanoparticles using a supercritical antisolvent processInt J Nanomedicine201162997300922162657
- BalakrishnanPLeeBJOhDHEnhanced oral bioavailability of Coenzyme Q10 by self-emulsifying drug delivery systemsInt J Pharm20093741–2667219446761
- ShahNHCarvajalMTPatelCIInfeldMHMalickAWSelf-emulsifying drug delivery systems (SEDDS) with polyglycolyzed glycerides for improving in vitro dissolution and oral absorption of lipophilic drugsInt J Pharm199410611523
- KimCKChoYJGaoZGPreparation and evaluation of biphenyl dimethyl dicarboxylate microemulsions for oral deliveryJ Control Release2001701–214915511166415
- PrajapatiBGPatelMMConventional and alternative pharmaceutical methods to improve oral bioavailability of lipophilic drugsAsian J Pharm20071118
- SunMSiLZhaiXThe influence of co-solvents on the stability and bioavailability of rapamycin formulated in self-microemulsifying drug delivery systemsDrug Dev Ind Pharm201137898699421417621
- HuXLinCChenDSirolimus solid self-microemulsifying pellets: formulation development, characterization and bioavailability evaluationInt J Pharm20124381–212313322850296
- KhooS-MHumberstoneAJPorterCJHEdwardsGACharmanWNFormulation design and bioavailability assessment of lipidic self-emulsifying formulations of halofantrineInt J Pharm19981671–2155164
- KommuruTRGurleyBKhanMAReddyIKSelf-emulsifying drug delivery systems (SEDDS) of coenzyme Q10: formulation development and bioavailability assessmentInt J Pharm2001212223324611165081
- CraigDQMBarkerSABanningDBoothSWAn investigation into the mechanisms of self-emulsification using particle-size analysis and low-frequency dielectric-spectroscopyInt J Pharm19951141103110
- ConstantinidesPPLipid microemulsions for improving drug dissolution and oral absorption: physical and biopharmaceutical aspectsPharm Res19951211156115728592652
- CarliFChielliniERemediaSRL inventors assigneePharmaceutical composition comprising a water/oil/water double microemulsion incorporated in a solid support WO patent WO 2003-0134212202003
- SwensonESCuratoloWJIntestinal permeability enhancement for proteins, peptides and other polar drugs – mechanisms and potential toxicity 2Adv Drug Deliv Rev1992813992