 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
For the purposes of obtaining a hepatocyte-selective drug delivery system, a novel tetravalent galactosylated diethylenetriaminepentaacetic acid-distearoyl phosphatidylethanolamine (4Gal-DTPA-DSPE) was synthesized. The chemical structure of 4Gal-DTPA-DSPE was confirmed by proton nuclear magnetic resonance and mass spectrometry. The four galactose-modified liposomes (4Gal-liposomes) were prepared by thin-film hydration method, then doxorubicin (DOX) was encapsulated into liposomes using an ammonium sulfate gradient loading method. The liposomal formulations with 4Gal-DTPA-DSPE were characterized by laser confocal scanning microscopy and flow cytometry analysis, and the results demonstrated that the 4Gal-liposomes facilitated the intracellular uptake of DOX into HepG2 cells via asialoglycoprotein receptor-mediated endocytosis. Cytotoxicity assay showed that the cell proliferation inhibition effect of 4Gal-liposomes was higher than that of the conventional liposomes without the galactose. Additionally, pharmacokinetic experiments in rats revealed that the 4Gal-liposomes displayed slower clearance from the systemic circulation compared with conventional liposomes. The organ distributions in mice and the study on frozen sections of liver implied that the 4Gal-liposomes enhanced the intracellular uptake of DOX into hepatocytes and prolonged the circulation. Taken together, these results indicate that liposomes containing 4Gal-DTPA-DSPE have great potential as drug delivery carriers for hepatocyte-selective targeting.
Introduction
Liver diseases, including virus infections, liver cirrhosis, and hepatocellular carcinoma, have become a significant health challenge around the world, due to the lack of curative treatment options besides liver resections and transplantation.Citation1
Hepatocytes, the liver parenchymal cells, constitute 60%–80% of the mass of the liver tissue, and liver diseases mainly develop from the hepatocytes.Citation2 Although hepatocytes are the main functional cells of the liver, high uptake of compounds into other liver cell types, such as Kupffer cells, often leads to serious degradation of them,Citation3 which in some cases destroys their therapeutic activity. However, hepatocyte targeting is often equated with liver targeting, and total liver uptake of a compound is measured without proper identification of the cell type. This has induced the necessity of the development of cell-specific delivery carriers, through surface modification, which are usually transferred via a receptor-mediated endocytosis system. Asialoglycoprotein receptors (ASGP-Rs) are exclusively expressed on the membranes of hepatocytes, providing active membrane-bound sites, and have been used as the target receptors for drug delivery to the hepatocytes.Citation4,Citation5 ASGP-Rs contain 1–5 × 105 binding sites per cell, and their main function is to recognize, bind, and internalize ASGPs that contain terminal galactose (Gal) or N-acetylgalactosamine (GalNAc) residues.Citation6,Citation7
Many studies have proved that both natural and synthetic carbohydrates can establish the structure–affinity relationship for the ASGP-R. Baenziger and MaynardCitation8 and Baenziger and FieteCitation9 have shown that the human receptor exhibits specificity for terminal Gal and GalNAc on desialylated glycoproteins. Lee et alCitation10 have also demonstrated that the affinity and specificity of the ASGP-R is a consequence of oligovalent interactions with its physiological ligands, a process termed “cluster glycoside effect.” Synthetic oligosaccharides tested on rabbit hepatocytes by Lee et al further strengthened the binding hierarchy of polyvalent ligands: tetra-antennary > triantennary >> biantennary >> monoantennary as a cluster glycoside effect.
Hepatocyte-selective targeting can be achieved through introduction of cells recognizing ligands on the liposomal surface. As many studies have proved that Gal-modified liposomes can be recognized by the ASGP-R on the liver parenchymal cells and incorporated into the cells by endocytosis, Gal was used as a liver-targeting moiety. Many studies have verified that liposomes modified with galactosylated lipid achieves effective targets to hepatocytes.Citation11–Citation14 Moreover, the half maximal inhibitory concentration values for mono-, bi-, tri-, and tetra-antennary oligosaccharides were found to be approximately 1 × 10−3, 1 × 10−6, 5 × 10−9, and 10−9 M, respectively. In other words, although the number of Gal residues/mol of ligand increased only four-fold, the inhibitory potency increased 1,000,000-fold.Citation15
Most studies have focused on cholesterol (Chol) as a lipophilic anchor moiety, because galactosylated Chol derivatives can be easily synthesized, where Chol and Gal ligands are linked by an ether bond.Citation16 However, it is very easy for Chol to fall out from the liposome membrane if the hydrophilic head group is too large, whereas distearoylphosphatidylethanolamine (DSPE) anchor may be located deeper in the liposome membrane with its two long aliphatic chains (2 × 17-CH2–), thus steadily inserting into the walls of lipid bilayer structures.Citation17,Citation18 In addition, YeagleCitation19 reported that red cell membrane sodium–potassium adenosine triphosphatase activity gradually decreased with elevated Chol levels. Furthermore, the proportion of Chol in the cell membrane limited the amount of Chol in liposomes,Citation20 thus limiting the quantity of ligands in liposomes. In contrast, DSPE is a natural body component with good biocompatibility, and the maximum amount of phospholipid in liposomes can reach 80%.Citation21 Therefore, the quantity of ligands in liposome can be greatly increased when DSPE serves as a lipophilic anchor moiety. Hence, DSPE was employed to connect Gal ligands in our study. Although multivalent Gal ligands have been previously reported,Citation22 few articles describe ligands beyond three Gal units. As we mentioned, targeting efficiency increases from monoantennary to tetra-antennary as a cluster glycoside effect. Therefore, in our study, four Gals were firstly connected to a DSPE simultaneously to improve the targeting efficiency.
In the present study, we designed and synthesized a novel multifunctional liposomal material, tetravalent galactosylated diethylenetriaminepentaacetic acid-distearoylphosphatidylethanolamine (4Gal-DTPA-DSPE), containing (1) a lipophilic anchor moiety (DSPE) for stable incorporation into liposomes, (2) a DTPA for connection of DSPE and ligands, and (3) four Gal moieties for the cell surface receptors in hepatocytes.
Doxorubicin (DOX) was selected as a model drug, as it can be efficiently encapsulated in liposomes via transmembrane sulfate ammonium gradients and form a stable drug–sulfate gel in the liposome interior, which results in a greater stability of DOX liposomes in plasma and during storage. Additionally, DOX is a cancer chemotherapeutic agent, and its fluorescence allows it to be identified within tissues and cells.
This study aimed to develop a Gal-modified liposomal formulation for DOX delivery and evaluate its effect of targeting to the liver. 4Gal-liposomes were composed of 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), Chol, and 4Gal-DTPA-DSPE. To evaluate the liver-targeting delivery property of 4Gal-liposomes, in vitro cellular uptake of DOX-loaded 4Gal-liposomes was visualized by confocal scanning microscopy and measured by flow cytometry. The cytotoxicity study was conducted to evaluate the safety of 4Gal-liposomes by 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Furthermore, pharmacokinetics of 4Gal-liposomes studied in rat and tissue distribution was carried out by in vivo imaging. Finally, the analysis of frozen sections of liver was carried out in order to study the mechanism of the targeting ability of 4Gal-liposomes to liver tissue.
The results suggest that the compound described in this work could serve as a valuable tool for studying hepatic endocytosis, and is a suitable carrier for site-specific drug delivery to the liver.
Materials and methods
Materials
DTPA was purchased from Aladdin Chemistry Co Ltd (Shanghai, People’s Republic of China). DSPE and DSPC were purchased from Genzyme Corporation (Cambridge, MA, USA). Anhydrous pyridine was purchased from Sigma Chemical Co (St Louis, MO, USA). 2,3,4,6-Tetra-O-acetyl-β-D-galactopyranosyl bromide was purchased from J&K Scientific Co Ltd (Shanghai, People’s Republic of China). HepG2 cells and Hela cells were purchased from the Laboratory Animal Center of Sun Yat-sen University (Guangzhou, People’s Republic of China). Cells were cultured in Dulbecco’s Modified Eagle’s medium supplemented with 10% fetal bovine serum and antibiotics (streptomycin 100 U/mL, penicillin 100 U/mL) at 37°C in humidified air with 2% carbon dioxide. All other chemicals were of reagent grade.
Experimental animals
Male Kunming mice (18–20 g) and male Sprague Dawley rats (220–250 g) were purchased from the Laboratory Animal Center of Sun Yat-sen University. All experimental procedures were approved and supervised by the Institutional Animal Care and Use Committee of Sun Yat-sen University.
Synthesis of 4Gal-DTPA-DSPE conjugates
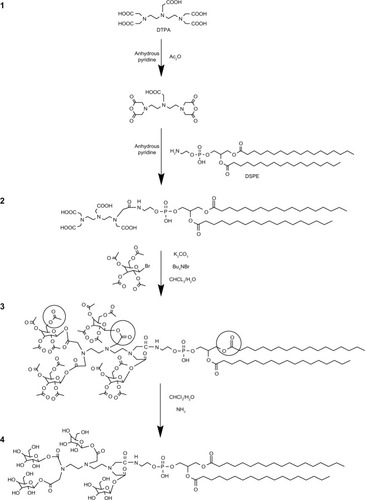
4Gal-DTPA-DSPE was synthesized by the following procedure (shown in ): (1) activation of DTPA, (2) connection of DTPA and DSPE, (3) galactosylation of DTPA-DSPE, and (4) removal of protection from hydroxyl groups. In the synthetic process, the carboxyl groups of DTPA were firstly activated by the acetic anhydride dissolved in anhydrous pyridine.Citation23 Then the amino group of DSPE was covalently linked to a carboxyl group of DTPA.Citation17 The next step was to connect the remaining carboxyl groups of DTPA and 1-hydroxyl group of Gals (other hydroxyl groups were protected by acetylation).Citation24 Finally the protecting groups of hydroxyl groups were removed selectivelyCitation25 The detailed synthetic routes of the compound are depicted in Supplementary material. The structure of 4Gal-DTPA-DSPE and intermediate products was characterized by 1H-NMR (1H-Nuclear Magnetic Resonance) and mass spectrometry (–).
Figure 1 The synthetic routes of tetravalent galactosylated diethylenetriaminepentaacetic acid-distearoylphosphatidylethanolamine (4Gal-DTPA-DSPE) conjugates.
Preparation and characterization of liposomes
DSPC, Chol, and 4Gal-DTPA-DSPE (molar ratio is shown in ) were dissolved in CHCl3 and dried under an N2 stream. A trace amount of CHCl3 was removed by keeping the lipid film under a vacuum. The lipid film was hydrated with 250 mM (NH4)2SO4 to obtain a blank liposome suspension. The liposome suspension was then sequentially extruded through polycarbonate membranes (Avanti® Mini-Extruder; Avanti Polar Lipids, Inc, Alabaster, AL, USA) with a pore size of 200 nm and 100 nm. The resulting liposomes were dialyzed (molecular weight cutoff size 14,000) against phosphate-buffered saline (PBS) (pH 7.4) at 37°C. For drug loading, DOX was dissolved in a small volume of deionized water and added to the liposomes to achieve a drug:lipid ratio of 1:10 (mol/mol). The loading process was carried out at 65°C for 30 —minutes, and DOX liposomes were obtained. The particle size and zeta potential of the DOX liposomes were analyzed using a Malvern Zetasizer Nano ZS90 (Malvern Instruments Ltd Malvern, UK). DOX-loaded 4Gal-liposomes were stained with phosphotungstic acid and observed by transmission electron microscopy (FEI Tecnai G2 F30, FEI company, Hillsboro, OR, USA). To determine the encapsulation efficiency (EE), unencapsulated DOX was separated from liposomes by size exclusion chromatography using a Sephadex G-50 column (Pharmacia Company, Uppsala, Sweden). PBS (pH 7.4) was used as the eluent. The eluted liposomes were collected and lysed with Triton X-100 (1%, v/v). The DOX concentration was determined by ultraviolet spectrophotometry (233 nm). The EE of DOX was calculated based on the ratio of liposomal drug to total drug.
Table 1 Composition and physicochemical properties of the prepared liposomes
Cellular internalization
Confocal laser scanning microscopy
HepG2 cells and Hela cells were used for the cell internalization study. HepG2 cells expressing ASGP-Rs were derived from a human hepatocellular carcinoma. Hela cells without ASGP-Rs served as the control.Citation26–Citation32 Cells were seeded on a cover glass in a 24-well culture plate at a density of 7 × 104 cells per well. The cells were incubated for 24 hours to 50% confluence and then treated with free DOX and a variety of liposomal DOX formulations for 2 hours. All groups were given a DOX equivalent dose of 30 μg/mL. The cells were washed three times with cold PBS, fixed with 4% paraformaldehyde at room temperature, and permeabilized with 0.5% Triton X-100 in PBS. The cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) (1 μg/mL) in order to visualize the nuclei. A Zeiss LSM710 laser scanning confocal microscope (Carl Zeiss Meditec AG, Jena, Germany) was used to investigate the intracellular uptake and subcellular distribution of DOX (excitation/emission wavelength: 488 nm/560 nm).
Flow cytometry analysis
Cell suspension (8 × 105 cells/well) was seeded in a 24-well culture plate and incubated for 24 hours until 80% confluence. The cells were then treated with free DOX and a variety of liposomal DOX formulations for 2 hours. All groups were given a DOX equivalent dose of 30 μg/mL. The cells were harvested and washed three times with cold PBS. The drug-free cells served as a reference sample. The cellular uptake of DOX was measured by using a flow cytometer EPICS XL (Beckman Coulter, Inc, Fullerton, CA, USA). The intracellular DOX was excited with an argon laser at a wavelength of 488 nm, and the fluorescence was detected at 575 nm. Data were analyzed with FlowJo software (Tree Star, Inc, Ashland OR, USA).
Competition assay
Free Gal was used as a competitive inhibitor to study whether the cellular uptake of the 4Gal-liposomes was via ASGP-Rs. HepG2 cells and Hela cells were seeded in 24-well plates at a density of 7 × 104 cells (in 500 μL culture media) per well and incubated for 24 hours until 50% confluence, to which 200 μL of Gal solution (100 mM) was added and then 37 μL of 4Gal-liposomes (DOX concentration of 600 μg/mL) was added to incubate for 2 hours. The total volume of culture media was approximately 700 μL. The treatment samples were the same as those in “Confocal laser scanning microscopy.”
Cell cytotoxicity assay
The cytotoxicity of free DOX and various liposomes (conventional liposomes, 4Gal-liposomes, and blank 4Gal-liposomes) on HepG2 cells and Hela cells was examined via MTT assay. Briefly, cells were seeded in 96-well plates at a density of 1 × 104 cells per well and incubated for 24 hours. Then the cells were treated with serial concentrations of free DOX or a variety of liposomal DOX formulations. The drug-free cells served as a reference sample, and the cell-free culture medium served as a blank control. After 24 hours incubation, 10 μL of MTT solution (5 mg/mL) was added to each well and incubated for a further 4 hours. Finally, the medium was replaced with 150 μL dimethyl sulfoxide (DMSO), and the optical density (OD) was determined with a microplate reader at a wavelength of 570 nm in triplicate. Relative inhibition was calculated by the following formula. Experiments were repeated three times, and data were presented as mean ± standard deviation.
Pharmacokinetic studies in rats
To obtain preliminary parameters about the pharmacokinetic properties of the 4Gal-liposomes, 15 Sprague Dawley rats were divided into three groups at random and treated with free DOX, conventional liposomes, and 4Gal-liposomes (10%), respectively. All groups were given a DOX equivalent dose of 10 mg/kg, and blood samples were collected at 10 minutes, 30 minutes, 1 hour, 2 hours, 4 hours, 6 hours, and 8 hours after drug administration from the jugular vein. Then the plasma was obtained by centrifuging immediately at 5,000 rpm for 10 minutes. A total of 20 μL of internal standard (salicylic acid 10.4 μg/mL in methanol) was added to 100 μL of plasma and mixed for 30 seconds. After adding 25 μL of perchloric acid (17.5%, v/v) and eddying for 1 minute, the plasma samples were centrifuged at 13,000 rpm for 10 minutes. Then an aliquot of 20 μL of the supernatant solution was injected into the high performance liquid chromatograph (HPLC) (Hitachi, Tokyo, Japan). Samples were separated by Luna-C18 column (150 × 4.6 mm, 5 μm; Phenomenex, Torrance, CA, USA). The mobile phase consisting of NH4H2PO4 (0.01 mol/L)-acetonitrile-acetic acid (76:24:0.4, v/v/v) was pumped at a flow rate of 1.0 mL/min. The column eluent was monitored at 233 nm at 40°C.
In vivo biodistribution study
For the purpose of investigating the targeting ability of 4Gal-liposomes to liver, Kunming mice received a single intravenous injection of free DOX and a variety of DOX liposomes at a DOX equivalent dose of 5 mg/kg. At 3 hours postadministration, the mice were sacrificed and major organs such as hearts, livers, spleens, lungs, and kidneys were excised. The distribution of DOX (excitation/emission wavelength: 488 nm/560 nm) was detected using an in vivo imaging system (Night OWL II LB983; Berthold Technologies GmbH and Co KG, Bad Wildbad Germany).
Study on frozen sections of liver
Free DOX and a variety of liposomal DOX formulations were injected intravenously into the tail vein of the mice at a DOX equivalent dose of 5 mg/kg. Mice were sacrificed at 3 hours postinjection. The liver was excised and frozen rapidly in dry ice, allowing the generation of 10 μm-thick cryosections. The tissue sections were fixed in cold acetone for 10 minutes, washed with PBS, blocked with bovine serum albumin for 1 hour, stained with fluorescein isothiocyanate-phalloidin (Sigma Chemical Co), and mounted with the DAPI-containing medium (VECTASHIELD® with DAPI; Vector Laboratories, Inc, Burlingame, CA, USA). Images were captured using a Zeiss LSM710 laser scanning confocal microscope.
Statistical analysis
Pharmacokinetic analysis was carried out by a two-compartment model method using the 3P97 practical pharmacokinetic program (edited by The Chinese Society of Mathematical Pharmacology New Edition 1997.11). Data were expressed as mean ± standard deviation, and the statistical differences between the groups were determined by one-way analysis of variance using SPSS 13.0 software (IBM Corporation, Armonk, NY, USA). Data were considered significantly different at the level of P < 0.05 and very significantly different at the level of P < 0.01.
Results
Preparation and characteristics of liposomes
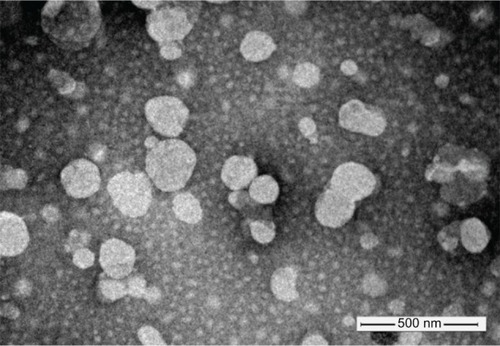
The characterization results of liposomes are listed in , and the transmission electron microscopy image of 4Gal-liposomes is shown in . The liposomes had a mean diameter of approximately 160 nm and relatively narrow distribution. The liposomes with or without Gal modification showed similar vesicle sizes, polydispersity indexes, and zeta potentials, indicating that the incorporation of 4Gal-DTPA-DSPE into lipid membrane had no influence on the physical properties of liposomes. DOX proved to be an excellent tool compound for target validation studies of liposomes. It could be conveniently encapsulated into liposomes at high concentration. EE of DOX into liposomes was >90% at a drug:lipid ratio of 1:10.
Figure 2 Negative stain (phosphotungstic acid) transmission electron microscopy image of four galactose-modified liposomes.
Cellular internalization
The results of cellular uptake were displayed qualitatively by confocal images and quantitatively by flow cytometry analysis (shown in and ). Strong DOX fluorescence intensity was observed in the nuclei of HepG2 cells treated with Gal-modified liposomes (), which indicated that 4Gal-liposomes were internalized more efficiently by HepG2 cells than conventional liposomes (). shows that the uptake could be blocked by 100 mM free Gal, indicating that Gal-modified liposomes were internalized by HepG2 cells via the ASGP-R, which was frequently expressed on the surface of hepatocytes. Similarly, flow cytometry results showed that the cellular uptake of Gal-modified liposomes was higher than that of unmodified liposomes and could be blocked by free Gal (shown in ).
Figure 3 Confocal scanning microscopy images of HepG2 cells (A) and Hela cells (B) incubated with blank medium (A1 and A2), free doxorubicin (B1 and B2), conventional liposomes (C1 and C2), four galactose-modified liposomes (4Gal-liposomes) (5%) (D1 and D2), 4Gal-liposomes (10%) (E1 and E2), and 100 mM galactose + 4Gal-liposomes (10%) (F1 and F2) for 2 hours at 37°C. Cells were fixed and then treated with 4′,6-diamidino-2-phenylindole for nuclei staining. Red: fluorescence of doxorubicin. Blue: fluorescence of 4′,6-diamidino-2-phenylindole. Pink: the merger fluorescence of blue and red.
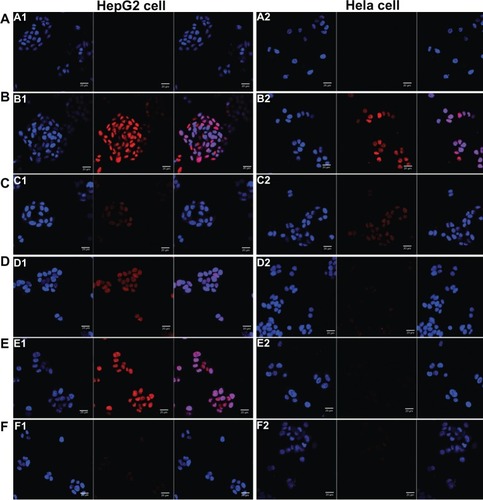
Figure 4 Flow cytometry analysis (A) of HepG2 cells (a) and Hela cells (b) after incubating with free doxorubicin (DOX) and DOX liposomes for 2 hours with 10% fetal bovine serum medium. The relative fluorescence intensity (B) of free DOX and DOX liposomes in HepG2 cells (a) and Hela cells (b) after incubating with 10% fetal bovine serum medium for 2 hours using flow cytometry analysis (n = 3), **P < 0.05.
Abbreviation: 4Gal-liposomes, four galactose-modified liposomes.
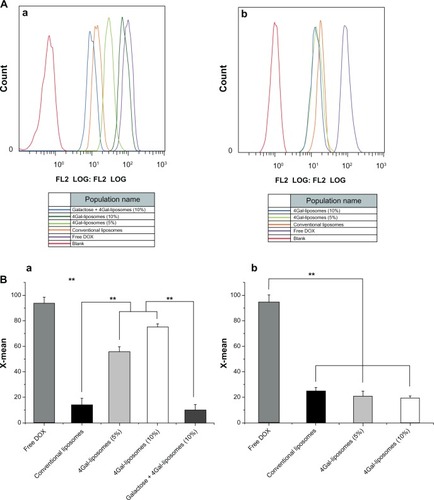
Hela cells, which lack ASGP-Rs, were selected to investigate whether the cellular uptake of Gal-modified liposomes was via the ASGP-R interaction. show that Gal-modified liposomes had a minor tendency to be internalized by Hela cells, and there was no significant difference between conventional liposomes () and Gal-modified liposomes. The fluorescence intensity of Gal-modified liposomes in Hela cells was weaker than that in HepG2 cells, and the results of flow cytometry (shown in ) were in accordance with the confocal images.
Taken together, these results indicate that the liposomes that contained 4Gal-DTPA-DSPE could effectively target the HepG2 cells via the ASGP-R.
Cell cytotoxicity assay (MTT)
The cytotoxicity of free DOX and DOX liposomes at various concentrations is shown in . We found that the cytotoxicity in HepG2 cells increased with increasing DOX and DOX liposome concentration shown in . Compared with unmodified liposomes, the cellular uptake of Gal-modified liposomes was greater because of the Gal-mediated endocytosis process, resulting in a higher cytotoxicity.
Figure 5 Relative inhibition of free doxorubicin (DOX) and DOX liposomes incubated in HepG2 cells (A) and Hela cells (B) with serum for 24 hours (n = 3). **P < 0.05, ***P < 0.01.
Abbreviation: 4Gal-liposomes, four galactose-modified liposomes.
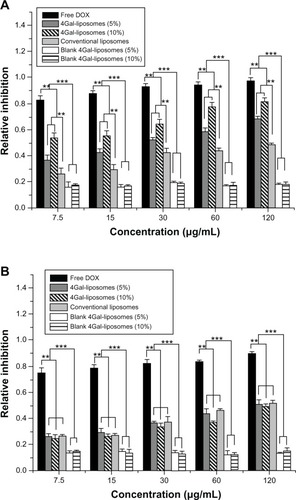
The cytotoxicity of free DOX and DOX liposomes in Hela cells is shown in . No significant difference in the cytotoxicity of Hela cells was shown between unmodified and Gal-modified liposomes, because there was no ASGP-R on the surface of Hela cells. Moreover, blank 4Gal-liposomes did not induce a visible cytotoxicity effect, indicating that the 4Gal-DTPA-DSPE possessed good biocompatibility.
Pharmacokinetics of 4Gal-liposomes
To investigate the pharmacokinetics process in vivo, free DOX, conventional liposomes, and 4Gal-liposomes (10%) were administrated into three groups of rats. Then blood samples were collected at the designated time points, and DOX concentrations were measured by high-performance liquid chromatography with ultraviolet detection. The plasma clearance curves of free DOX, conventional liposomes, and 4Gal-liposomes (10%) in rats are shown in . Clearance of free DOX from the blood circulation was very rapid, and the DOX concentration decreased to 0.18 μg/mL at 4 hours. Compared with free DOX, conventional liposomes and 4Gal-liposomes displayed slower clearance from the circulating system in vivo. The plasma concentrations of DOX in the conventional liposomes and 4Gal-liposomes groups were 0.76 μg/mL and 1.21 μg/mL at 4 hours postinjection, respectively. However, elimination rates in the plasma of the rats treated with 4Gal-liposomes were even slower than conventional liposomes. It was assumed that the circulation time of 4Gal-liposomes was prolonged with the high density of hydrophilic Gals on the surface.
Figure 6 Plasma concentrations of doxorubicin (DOX) in normal mice after intravenous injection of free DOX, conventional liposomes, and four galactose-modified liposomes (4Gal-liposomes) (10%). All groups were given a DOX equivalent dose of 10 mg/kg.
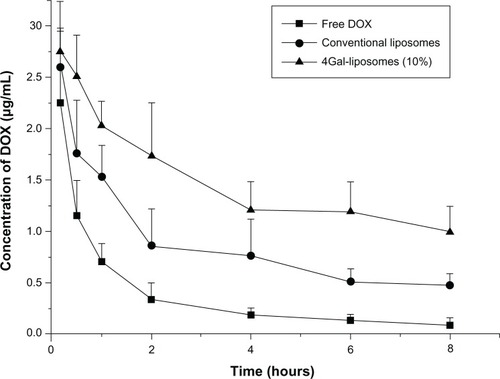
The key pharmacokinetic parameters are summarized in . The elimination half-life of 4Gal-liposomes was increased by 4.9-fold and 2.1-fold in comparison with that of free DOX and conventional liposomes, respectively. In addition, the value of the area under the concentration curve was found to be significantly increased for 4Gal-liposomes.
Table 2 DOX pharmacokinetics (mean ± SD) in plasma after intravenous injection of free DOX, conventional liposomes and 4Gal-liposomes (10%)
Tissue distribution in vivo of 4Gal-liposomes
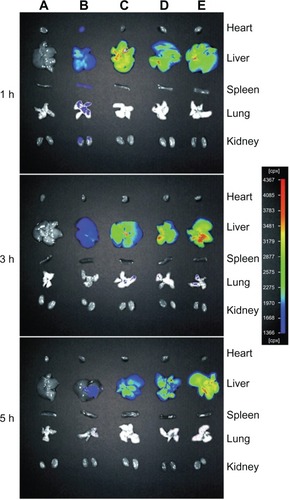
To investigate the dynamic biodistribution of 4Gal-liposomes in mice, the fluorescence images of various organs at different time points were recorded by the in vivo imaging system. Representative fluorescence images of mice after administration of free DOX and DOX liposomes are shown in . The fluorescence of free DOX quickly decreased in liver (Group B), and the fluorescence was also observed in the heart, spleen, and kidney, which indicated the toxicity of free DOX to other organs. Fluorescence of Group D (4Gal-liposomes 5%) and Group E (4Gal-liposomes 10%) exhibited significantly enhanced accumulation of 4Gal-liposomes in liver in comparison with those injected with conventional liposomes (Group C) at 3 hours and 5 hours, confirming the in vivo targeting ability of 4Gal-liposomes toward liver tissue. We could assume that the fluorescence of 4Gal-liposomes increased after 3 hours because of the high density of aqueous layer on the surface of liposomes, which extended the mean residence time. For conventional liposomes, the fluorescence accumulated in liver might be attributed to the well-known passive effect of targeting. As shown in Group D and Group E, almost no fluorescence was observed in other tissues, indicating few liposomes entering these organs. The organ distributions implied that the liver-targeting ability of DOX might be enhanced by the liver-targeting delivery system of 4Gal-liposomes.
Figure 7 The fluorescence images of various organs of Kunming mice sacrificed at 1 hour (h), 3 hours, and 5 hours after injection with phosphate-buffered saline (A), free doxorubicin (B), conventional liposomes (C), four galactose-modified liposomes (5%) (D), and four galactose-modified liposomes (10%) (E) in vivo imaging system.
Study on frozen sections of liver
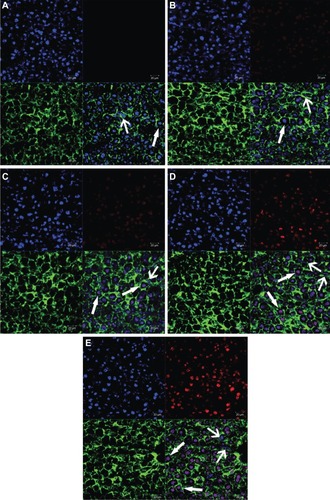
The analysis of frozen sections of liver was carried out to study the mechanism of the targeting ability of 4Gal-liposomes to liver tissue. The fluorescence intensity images from DOX are shown in . The figure reveals that some labeled nuclei were large and round (presumed hepatocyte) and brightly stained, whereas other nuclei were oblong, oval (presumed nonparenchymal), or, in some cases, indented.Citation33,Citation34 Thus, the nonparenchymal cells and hepatocytes could be distinguished by their distinct morphologies, as indicated by the arrow → (parenchymal cells) and arrow ← (nonparenchymal cells). Distribution of relatively strong DOX fluorescence could be observed in the hepatocytes treated with Gal-modified liposomes, indicating that the liposomes incorporated with the 4Gal-DTPA-DSPE showed a remarkably specific effect of targeting to the hepatocytes.
Figure 8 Confocal scanning microscopy images of liver sections of free doxorubicin (DOX) and DOX liposomes, blank (A), free DOX (B), conventional liposomes (C), four galactose-modified liposomes (5%) (D), and four galactose-modified liposomes (10%) (E). Nuclei were stained blue with 4′,6-diamidino-2-phenylindole, fluorescein isothiocyanate was shown as green fluorescence, DOX was shown as red fluorescence, and the merger image is on the bottom right.
Notes: Arrows with triangle head point to hepatocytes, and the others point to non-parenchymal cells.
Discussion
Synthesis and characterization of 4Gal-DTPA-DSPE conjugates
In this study, we focused on the potential ligands with higher affinity than monoantennary galactosides. DSPE as a lipophilic moiety was incorporated into the membrane of liposomes, and the amino group of DSPE was linked to the carboxyl group of DTPA. DTPA was employed to connect DSPE and Gals with its five modifiable carboxyl groups.
In the synthetic process (shown in ), DTPA was firstly activated by the acetic anhydride to form DTPA anhydride. The amino group of DSPE was then covalently linked to the free carboxyl group of DTPA anhydride. Coupling the carboxyl group of DTPA anhydride with the amino group of DSPE was performed by mixing a 10-fold molar excess (a 20-fold equivalent excess) of DTPA anhydride with the DSPE in anhydrous pyridine. The lipid solution should be dropwise added into the vigorously stirred DTPA anhydride solution. In this way, only one hydroxyl group of DTPA participated in the reaction, preventing multisubstituted products. The remaining carboxyl groups could be further coupled to the galactosyl groups. Pyridine was used as a solvent and catalyst. It was important to ensure that the pyridine was absolutely anhydrous, because DTPA anhydride would be hydrolyzed when encountering even a trace amount of water.
The next step was to connect the carboxyl groups of DTPA and 1-hydroxyl group of Gals. Three methods have been studied. Firstly, thionyl chloride (SOCl2) was used to activate the carboxyl group of DTPA. However, DSPE was found to be unstable in the strong acidic environment of SOCl2. We presumed that the ester bond of DSPE was unstable under this condition. Secondly, dicyclohexylcarbodiimide was utilized as an activator, and 4-dimethylaminopyridine acted as a catalyst to attach Gals to the carboxyl group of DTPA by covalent binding. However, the target compound still could not be achieved by this strategy. Thirdly, we therefore tried to activate the hydroxyl groups of Gals instead of carboxyl groups of DTPA. Under the optimized phase-transfer-catalyzed conditions (K2CO3, Bu4 NBr, CH2Cl2-H2O, reflux), DSPE-DTPA was coupled with 2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl bromide, generating the desired product.
The final step was the deacetylation of the hydroxyl groups of galactosides. As two types of ester bonds, namely galactosylated ester bond and lecithin ester bond, should not be hydrolyzed (shown in ), it was very crucial to selectively break the ester bond of acetyl. Firstly, triethylamine was used to give a base solution to hydrolyze the ester bond of acetyl. However, a side product always existed through thin layer chromatography (TLC) analysis. We believed that in a strong base solution, the glycosidic bond was easily broken, leading to reaction with CH3OH to form the side product. Hence, dry gaseous ammonia was used in an ice water bath to form a mild base environment. We found that the reaction temperature had a significant influence on the ratio of the desired product to the side product. When the reaction temperature was 0°C approximately (in an ice water bath), the ratio was appropriate. Under these mild conditions, the reaction time was monitored by TLC and we obtained the desired compound.
The surface hydration modification of 4Gal-liposomes
Surface modification has been achieved by incorporating hydrophilic moieties, such as polyethylene glycol (PEG), which were chemically conjugated to lipids in order to reduce immune recognition and rapid clearance.Citation35 The surface of the liposomal membrane was modified with dendritic hydrophilic Gals to reduce aggregation and avoid recognition by the reticuloendothelial system (RES). This strategy was similar to liposome PEGylation and is often referred to as surface hydration modification. In this work, four galactose were conjugated to the carboxyl groups of DTPA, which were linked to the terminal amino group of DSPE. This led to the presence of hydrophilic groups on the surface of the liposomal membrane, and a dense aqueous layer might be formed around the liposomes by interaction between the dendritic hydrophilic hydroxyl groups of Gals and water molecules, thus avoiding the RES uptake and prolonging circulation time.
Intracellular uptake of liposomes
DOX is a potent anticancer drug that is known to readily intercalate into DNA strands,Citation36 and many studies have shown that DOX preferentially accumulates into the nuclear compartment of cells.Citation37,Citation38 Free DOX is mainly located in the nucleus and shows the most intense intracellular fluorescence (shown in ) as the positive control in vitro, attributed to its direct and rapid partition into the membrane without release from liposomes and its highly nucleophilic nature.Citation39 However, free DOX presents serious cardiotoxicity, which limits clinical application.Citation40 The administration of DOX in liposome-encapsulated form has been advocated as a means of changing the distribution of DOX in vivo and reducing the cardiac damage induced by DOX.Citation41–Citation44 Preclinical experiments with liposome-encapsulated DOX indicate that this form of delivery may be effective in decreasing the cardiotoxic effect of the drug. In addition, drastic changes in the clinical pharmacokinetics of DOX have been observed using liposomal delivery.Citation45,Citation46 Currently, PEGylated liposomal DOX (Doxil®; Janssen Products, LP, Horsham, PA, USA) is a US Food and Drug Administration-approved marketed DOX formulation.Citation47,Citation48 However, liposomal DOX is less effective than free DOX.Citation49,Citation50 Therefore, our study aimed to develop a Gal-modified liposomal formulation for DOX delivery in order to reduce its cardiotoxicity and enhance its effect of targeting to hepatocyte by ASGP-R-mediated endocytosis.
To demonstrate the specific cell binding and internalization of 4Gal-liposomes, ASGP-R-positive HepG2 cells were chosen as target cells, whereas ASGP-R-negative Hela cells were applied as negative cells. The confocal microscopy images and flow cytometry data demonstrated that 4Gal-liposomes resulted in significantly higher cell association by ASGP-R-positive HepG2 cells compared with the negative control. But similar cellular behavior was found with the two liposomal formulations when they were incubated in ASGP-R-negative Hela cells. In the competition study, the HepG2 cells’ association of 4Gal-liposomes was suppressed to a lower level by the presence of excess free Gal, whereas no significant changes were found in Hela cells. All these phenomena suggest that 4Gal-liposomes could enhance specific cell binding and cellular uptake in HepG2 cells due to the mediating of Gal, and depending on the ASGP-R expression level on the cell surface as well.
Liposome uptake by liver in vivo
As hepatocytes represent most hepatic cells and liver diseases mainly develop from hepatocytes, it was very important to confirm that the drugs were not only concentrated in nonparenchymal cells but also internalized by hepatocytes. The frozen sections of liver that stained green (the cell membrane), blue (the nuclei of the cells), and red (the DOX) could distinguish the hepatocytes from nonparenchymal cells. and show that there was significant difference of distribution among free DOX and liposomal formulations, and Gal-modified liposomes showed a remarkably specific effect of targeting to the liver tissue after 3 hours.
The pharmacokinetic experiments and biodistribution studies revealed that the inclusion of 4Gal-DTPA-DSPE in the liposomal bilayer extended systemic circulation. There was a general consensus that serum proteins adsorbed on to the surface of conventional liposomes could mediate recognition of the liposomes by macrophages of the RES, and facilitate clearance of liposomes from the circulation. Coating liposomes with 4Gal-DTPA-DSPE decreased the blood clearance considerably, most likely due to reduced protein adsorption and liposome aggregation. We assumed that with 4Gal-DTPA-DSPE modification of the liposomal surface, a dense aqueous layer was formed around the liposomes, thus avoiding the attraction of opsonins. As a result, 4Gal-liposomes that escaped trapping by the cells of the RES had a prolonged circulation time and accumulated in the liver by active targeting.
Conclusion
In the present study, a hepatocyte-targeting drug delivery system was successfully constructed by incorporating synthetic 4Gal-DTPA-DSPE (5% and 10%, mol/mol) into liposomes, where Gal was used for active targeting to the liver and applying for prolonged circulation. DOX, as a drug model, was effectively encapsulated into the liposomes. The cellular uptake and cell cytotoxicity tests indicated that 4Gal-liposomes had a significant targeting function toward human hepatoma cells and could deliver DOX into HepG2 cells effectively. Furthermore, the results of pharmacokinetic and biodistribution experiments provided evidence that 4Gal-liposomes possessed an enhanced plasma half-life and higher liver accumulation in vivo. Finally, the study of frozen sections of liver confirmed that the drugs were internalized by hepatocytes rather than concentrated in nonparenchymal cells. These results suggest that liposomes containing 4Gal-DTPA-DSPE could be a potential drug carrier system for hepatocyte-selective targeting.
Future direction
The purpose of this study was to investigate whether content delivery of DOX could be targeted to the normal liver. The next step of this study is to explore the targeted delivery characteristics of this formulation in liver tumors of animal models.
Supplementary materials
Synthesis of 4Gal-DTPA-DSPE conjugates
Synthesis of DTPA anhydride (1)
Diethylenetriaminepentaacetic acid (DTPA) (19.75 g, 0.05 mol) and acetic anhydride (37.8 mL, 0.4 mol) were added to anhydrous pyridine (25 mL), and the mixture was stirred for 24 hours at 65°C ± 5°C. Then the mixture was filtered and the solid was washed with acetic anhydride (20 mL) and anhydrous ether (20 mL) three times, respectively, and the solvent was removed in a vacuum. Compound (1) was obtained as a white powder (87% yield). Proton nuclear magnetic resonance (1H-NMR) (400 MHz, dimethyl sulfoxide), δ/ppm: 3.71 (s, 8H), 3.31 (s, 2H), 2.75 (t, 4H), 2.60 (t, 4H).
Synthesis of DSPE-DTPA (2)
DTPA anhydride (5 g, 13.99 mmol) was added to anhydrous pyridine (100 mL). The mixture was heated until the lipid was dissolved to give a clear solution. In a separate flask, distearoylphosphatidylethanolamine (DSPE) (1 g, 1.34 mmol) was warmed with anhydrous pyridine (100 mL) until dissolved. The lipid solution was added to the vigorously stirred DTPA anhydride solution. After addition, the solution was heated to reflux for 70 minutes, during which time an orange tint appeared but the solution remained clear. H2O (50 mL) was then added to the reaction mixture, and refluxing was continued for 70 minutes to hydrolyze remaining anhydride linkages. The mixture was cooled to room temperature and rotary evaporated to dryness. The residue was purified by column chromatography on a silica gel eluted with CHCl3:CH3OH:H2O: formic acid (65:25:4:1). Compound (2) was obtained as a yellow powder (48% yield). Citation1H-NMR (400 MHz,CDCl3), δ/ppm: 5.15 (s, 2H), 4.36 (d 1H), 4.09 (d 3H), 3.95 (s, 2H), 3.70 (s, 8H), 3.44 (s, 2H), 3.29 (s, 2H), 2.73 (t, 4H), 2.58 (t, 4H), 2.12 (t, 3H), 1.61 (s, 8H), 1.27 (s, 52H), 0.89 (t, 6H). MS (ESI+) m/z: [M+H]+ calcd for C55H103 N4O17P [M+H]+1123.4, found 1123.90.
Synthesis of 4-Ac-galactose-DTPA-DSPE (3)
K2CO3 (199 mg, 1.2 equivalent), H2O (11.7 mL), and Bu4NBr (38.7 mg, 0.1 equiv) were added to a solution of DSPE-DTPA (333.6 mg, 0.3 mmol) and 2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl bromide (639.6 mg, 5.2 equiv) in CH2Cl2 (18 mL). The resulting mixture was refluxed for 6 hours at 40°C and was then diluted with CH2 Cl2(36 mL). The organic phase, after being washed with water and brine, respectively, was dried over Na2SO4 and then concentrated in a vacuum. The residue was purified by column chromatography on a silica gel eluted with CHCl3:CH3OH (9:1). Compound (3) was obtained as yellow oil (59% yield). 1H-NMR (400 MHz,CDCl3), δ/ppm: 6.15 (d 4H), 5.52 (d 4H), 5.41 (t, 4H), 5.07 (t, 4H), 4.83 (t, 1H), 4.75 (t, 3H), 4.49 (t, 4H), 4.16 (s, 8H), 3.70 (s, 8H), 3.30 (s, 2H), 2.74 (t, 4H), 2.59 (t, 4H), 2.15 (s, 12H), 2.11 (s, 13H), 2.06 (s, 12H), 2.01 (s, 12H), 1.60 (s, 7H), 1.28 (s, 55H), 0.88 (t, 6H). MS (ESI+) m/z: [M+H]+ calcd for C111H175 N4O53P [M+H]+2444.55, found 2444.13.
Synthesis of 4Gal-DTPA-DSPE (4)
Compound (3) (253 mg, 0.1 mmol) was dissolved in a mixture of anhydrous CHCl3 (3 mL) and anhydrous CH3OH (8 mL). Then the mixture was stirred for 5 minutes to form a homogenous phase, and dry gaseous ammonia was introduced for 1 hour into the flask, which was kept in an ice–salt bath. Next, the solution was stirred in an ice water bath for 3–4 hours (the reaction time was determined by TLC). After removal of the solvent, sticky yellow oil was recovered. The oil was purified by column chromatography on a silica gel eluted with CHCl3:CH3OH (2:1). Compound (4) was obtained as yellow oil (63% yield). 1H-NMR (400 MHz,CDCl3), δ/ppm: 6.10 (d 4H), 5.21 (s, 1H), 4.92 (t, 4H), 4.47 (t, 4H), 4.41 (d 4H), 4.22 (d 8H), 3.70 (s, 8H), 3.29 (s, 2H), 2.75 (t, 4H), 2.60 (t, 4H), 2.31 (d 4H), 1.88 (s, 6H), 1.58 (s, 4H), 1.26 (s, 53H), 0.88 (t, 6H). MS (ESI+) m/z: [M+H]+ calcd for C79H143N4O37P [M+H]+1770.92, found 1770.94.
Figure S1 Proton nuclear magnetic resonance (400 MHz) spectrum of diethylenetriaminepentaacetic acid anhydride.
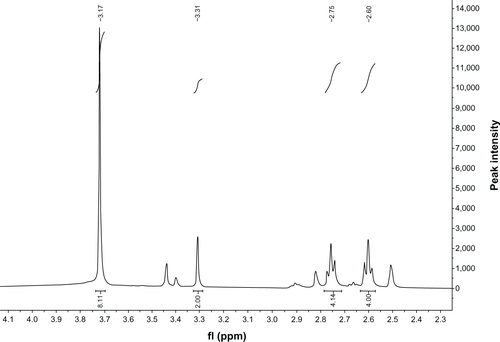
Figure S2 Proton nuclear magnetic resonance (400 MHz) spectrum of diethylenetriaminepentaacetic acid-distearoylphosphatidylethanolamine.
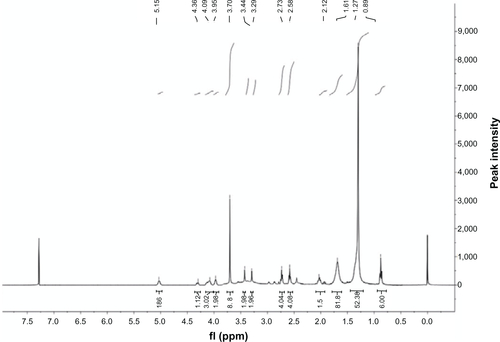
Figure S3 Proton nuclear magnetic resonance (400 MHz) spectrum of tetravalent galactosylated diethylenetriaminepentaacetic acid-distearoylphosphatidylethanolamine.
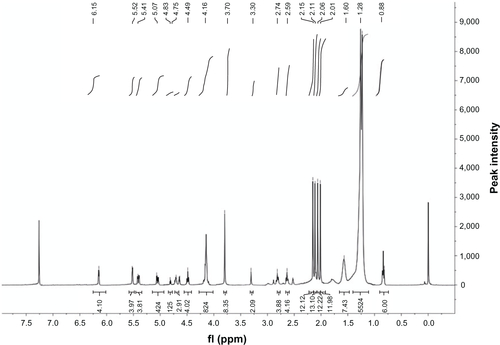
Figure S4 Proton nuclear magnetic resonance (400 MHz) spectrum (A) and mass spectrum (B) of galactosylated diethylenetriaminepentaacetic acid-distearoylphosphatidylethanolamine.
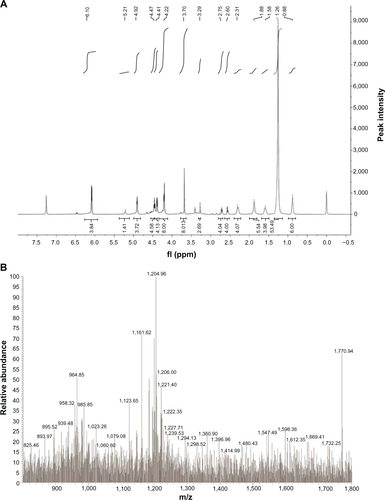
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No 81173003/h3008).
Disclosure
The authors report no conflicts of interest in this work.
References
- LiLWangHOngZYPolymer-and lipid-based nanoparticle therapeutics for the treatment of liver diseasesNano Today201054296312
- WangSXuHXuJSustained liver targeting and improved antiproliferative effect of doxorubicin liposomes modified with galactosylated lipid and PEG-LipidAAPS PharmSciTech201011287087720490957
- SmithJSXuZByrnesAPA quantitative assay for measuring clearance of adenovirus vectors by Kupffer cellsJ Virol Methods20081471546017850893
- StockertRJThe asialoglycoprotein receptor: relationships between structure, function, and expressionPhysiol Rev19957535916097624395
- PoelstraKPrakashJBeljaarsLDrug targeting to the diseased liverJ Control Release2012161218819722370583
- SpiessMThe asialoglycoprotein receptor: a model for endocytic transport receptorsBiochemistry1990294310009100182125488
- GeffenISpiessMAsialoglycoprotein receptorInt Rev Cytol B1992137181219
- BaenzigerJMaynardYHuman hepatic lectin. Physiochemical properties and specificityJ Biol Chem198025510460746137372599
- BaenzigerJFieteDGalactose and N-acetylgalactosamine-specific endocytosis of glycopeptides by isolated rat hepatocytesCell1980222 Pt 26116207448875
- LeeYCTownsendRRHardyMRBinding of synthetic oligosaccharides to the hepatic Gal/GalNAc lectin. Dependence on fine structural featuresJ Biol Chem198325811992026848494
- SatoATakagiMShimamotoAKawakamiSHashidaMSmall interfering RNA delivery to the liver by intravenous administration of galactosylated cationic liposomes in miceBiomaterials20072871434144217141864
- MandalADasSBasuMChakrabartiRDasNHepatoprotective activity of liposomal flavonoid against arsenite-induced liver fibrosisJ Pharmacol Exp Ther20073203994100117138861
- ManagitCKawakamiSYamashitaFHashidaMEffect of galactose density on asialoglycoprotein receptor-mediated uptake of galactosylated liposomesJ Pharm Sci200594102266227516136555
- MuraoANishikawaMManagitCTargeting efficiency of galactosylated liposomes to hepatocytes in vivo: effect of lipid compositionPharm Res200219121808181412523658
- MammenMChoiSKWhitesidesGMPolyvalente Wechselwirkungen in biologischen Systemen: Auswirkungen auf das Design und die Verwendung multivalenter Liganden und Inhibitoren. [Poly valent interactions in biological systems: implications for the design and use of multivalent ligands and inhibitors]Angewandte Chemie19991102029082953 German
- KawakamiSMunakataCFumotoSYamashitaFHashidaMTargeted delivery of prostaglandin E1 to hepatocytes using galactosylated liposomesJ Drug Target20008313714210938523
- GrantCWMKarlikSFlorioEA liposomal MRI contrast agent: phosphatidylethanolamine-DTPAMagn Reson Med19891122362432779414
- IshiwataHVertut-DoïAHiroseTMiyajimaKPhysical-chemistry characteristics and biodistribution of poly(ethylene glycol)-coated liposomes using poly(oxyethylene) cholesteryl etherChem Pharm Bull. (Tokyo)1995436100510117641302
- YeaglePLCholesterol and the cell membraneBiochimica et Biophysica Acta19858223–42672873904832
- GleasonMMMedowMSTulenkoTNExcess membrane cholesterol alters calcium movements, cytosolic calcium levels, and membrane fluidity in arterial smooth muscle cellsCirc Res19916912162272054935
- KirbyCClarkeJGregoriadisGEffect of the cholesterol content of small unilamellar liposomes on their stability in vivo and in vitroBiochem J198018625915987378067
- KhorevOStokmaierDSchwardtOCuttingBErnstBTrivalent, Gal/GalNAc-containing ligands designed for the asialoglycoprotein receptorBiorg Med Chem200816952165231
- FengLZhangLYanZGuBWangCLuWExperimental study on liposomal drug delivery system for lymphatic targetingActa Biophysica Sinica201026735749
- ZhuSLiYYuBSynthesis of betavulgaroside III, a representative triterpene seco-glycosideJ Org Chem200873134978498518529027
- ZhaoHLiXLiuHLiYWuCSynthesis and characterization of ferrocenylmethylaminobenzoic acids and their glucosyl estersSynth React Inorg Met Org Chem2000303473483
- ParkIYKimTHKimYKChoiYJNahJWChoCSGalactosylated chitosan/carbonate apatite nanohybridization for cell specificity and high transfection efficiency as a DNA carrierKey Eng Mater2007342437440
- ParkI-KJiangH-LCookS-EGalactosylated chitosan (GC)-graft-poly (vinyl pyrrolidone)(PVP) as hepatocyte-targeting DNA carrier: in vitro transfectionArch Pharm Res200427121284128915648108
- ZhangX-QWangX-LZhangP-CGalactosylated ternary DNA/polyphosphoramidate nanoparticles mediate high gene transfection efficiency in hepatocytesJ Control Release2005102374915681095
- JereDJiangHAroteRDegradable polyethylenimines as DNA and small interfering RNA carriersExpert Opin Drug Deliv20096882783419558333
- ParkIKimTKimSVisualization of transfection of hepatocytes by galactosylated chitosan-graft-poly (ethylene glycol)/DNA complexes by confocal laser scanning microscopyInt J Pharm2003257110311012711166
- JiangH-LKwonJ-TKimE-MGalactosylated poly (ethylene glycol)-chitosan-graft-polyethylenimine as a gene carrier for hepatocyte-targetingJ Control Release2008131215015718706946
- DíezSMiguélizIDe IlarduyaCTTargeted cationic poly (D, L-lactic-co-glycolic acid) nanoparticles for gene delivery to cultured cellsCell Mol Biol Lett200914234736219194666
- LongmuirKJHaynesSMBarattaJLKasabwallaNRobertsonRTLiposomal delivery of doxorubicin to hepatocytes in vivo by targeting heparan sulfateInt J Pharm2009382122223319664697
- BarattaJLNgoALopezBKasabwallaNLongmuirKJRobertsonRTCellular organization of normal mouse liver: a histological, quantitative immunocytochemical, and fine structural analysisHistochem Cell Biol2009131671372619255771
- LianTHoRJYTrends and developments in liposome drug delivery systemsJ Pharm Sci200190666768011357170
- GabbayEGrierDFingerleRInteraction specificity of the anthracyclines with deoxyribonucleic acidBiochemistry197615102062776212
- ColeyHAmosWTwentymanPWorkmanPExamination by laser scanning confocal fluorescence imaging microscopy of the subcellular localisation of anthracyclines in parent and multidrug resistant cell linesBr J Cancer199367613168099807
- MarquardtDCenterMSDrug transport mechanisms in HL60 cells isolated for resistance to adriamycin: evidence for nuclear drug accumulation and redistribution in resistant cellsCancer Res19925211315731631591727
- DüzgüneşNNirSMechanisms and kinetics of liposome–cell interactionsAdv Drug Del Rev1999401318
- LiuXChuaCCGaoJPifithrin-α protects against doxorubicin-induced apoptosis and acute cardiotoxicity in miceScience Signaling20042863H933
- GabizonAASelective tumor localization and improved therapeutic index of anthracyclines encapsulated in long-circulating liposomesCancer Res19925248918961737351
- GabizonAIsacsonRLibsonEClinical studies of liposome-encapsulated doxorubicinActa Oncol19943377797867993646
- GabizonAMeshorerABarenholzYComparative long-term study of the toxicities of free and liposome-associated doxorubicin in mice after intravenous administrationJ Natl Cancer Inst19867724594693461207
- ParkJWLiposome-based drug delivery in breast cancer treatmentBreast Cancer Res2002439512052251
- GabizonAALiposomal anthracyclinesHematol Oncol Clin North Am1994824318040147
- OlsonFMayhewEMaslowDRustumYSzokaFCharacterization, toxicity and therapeutic efficacy of adriamycin encapsulated in liposomesEur J Cancer Clin Oncol19821821671767201392
- ChenZGSmall-molecule delivery by nanoparticles for anticancer therapyTrends Mol Med2010161259460220846905
- WangRBillonePSMullettWMNanomedicine in action: an overview of cancer nanomedicine on the market and in clinical trialsJ Nanomater201310.1155/2013/629681.
- ChandaroyPSenAAlexandridisPHuiSWUtilizing temperature-sensitive association of Pluronic F-127 with lipid bilayers to control liposome–cell adhesionBiochimica et Biophysica Acta200215591324211825586
- XiangYLiangLWangXWangJZhangXZhangQChloride channel-mediated brain glioma targeting of chlorotoxin-modified doxorubicine-loaded liposomesJ Control Release2011152340241021435361