 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The goal of the present study was to compare the drug release properties and stability of the nanoporous silica with different pore architectures as a matrix for improved delivery of poorly soluble drugs. For this purpose, three dimensional ordered macroporous (3DOM) silica with 3D continuous and interconnected macropores of different sizes (200 nm and 500 nm) and classic mesoporous silica (ie, Mobil Composition of Matter [MCM]-41 and Santa Barbara Amorphous [SBA]-15) with well-ordered two dimensional (2D) cylindrical mesopores were successfully fabricated and then loaded with the model drug indomethacin (IMC) via the solvent deposition method. Scanning electron microscopy (SEM), N2 adsorption, differential scanning calorimetry (DSC), and X-ray diffraction (XRD) were applied to systematically characterize all IMC-loaded nanoporous silica formulations, evidencing the successful inclusion of IMC into nanopores, the reduced crystallinity, and finally accelerated dissolution of IMC. It was worth mentioning that, in comparison to 2D mesoporous silica, 3DOM silica displayed a more rapid release profile, which may be ascribed to the 3D interconnected pore networks and the highly accessible surface areas. The results obtained from the stability test indicated that the amorphous state of IMC entrapped in the 2D mesoporous silica (SBA-15 and MCM-41) has a better physical stability than in that of 3DOM silica. Moreover, the dissolution rate and stability of IMC loaded in 3DOM silica was closely related to the pore size of macroporous silica. The colorimetric 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and Cell Counting Kit (CCK)-8 assays in combination with direct morphology observations demonstrated the good biocompatibility of nanoporous silica, especially for 3DOM silica and SBA-15. The present work encourages further study of the drug release properties and stability of drug entrapped in different pore architecture of silica in order to realize their potential in oral drug delivery.
Introduction
Oral delivery is commonly recognized as the most convenient and preferred route for drug administration. However, the large majority of the newly discovered chemical entities and many existing drug substances suffer from poor aqueous solubility, insufficient dissolution throughout the gastrointestinal tract, and, consequently, low bioavailability when administered orally.Citation1 Developing strategies to improve the drug dissolution rate and to enable the effective oral delivery of water-insoluble compounds is currently one of the greatest challenges to formulation scientists in the pharmaceutical field.Citation2,Citation3
Various approaches have been employed to tackle the formulation challenges of poorly water-soluble drugs, such as nanocrystal or nanosizing techniques,Citation4,Citation5 lipid-based formulation,Citation6,Citation7 and solid dispersions,Citation8,Citation9 and so on. Among these methods, adsorption onto porous substrates with a high surface area is a well-known and widely used technique to enhance dissolution of poorly soluble drugs.Citation10–Citation14 Porous silicate is a porous material that has been commonly used as a pharmaceutical excipient. Several grades of porous silicate having different characteristics such as particle size, pore size, and specific surface area are commercially available and have been widely employed to encapsulate poorly soluble drugs, such as Aerosil®, Sylysia®, Florite®, Neusilin® and aerogel.Citation15–Citation18
Since the discovery of mesoporous (2 nm <pore size <50 nm) silica materials in the 1990s, the synthesis and application of mesoporous silica have received substantial attention due to their high surface area, well defined and tunable pore size, uniform porous structure, and easily modified surface.Citation19–Citation22 Among these mesoporous silica, Mobil Composition of Matter (MCM)-41 and Santa Barbara Amorphous (SBA)-15, with a two dimensionally ordered hexagonal arrangement of cylindrical pores of uniform size (typically 2–10 nm) disposed parallel to each other (), are probably the most investigated materials.Citation23–Citation25
Figure 1 Schematic illustration of the pore structures of (A) 2D mesoporous silica, (B) 3DOM silica, and (C) molecular structure of indomethacin.
Notes: 2D mesoporous silica contains cylindrical mesopores arranged parallel to each other, while 3DOM silica contains spherical macropores connected by windows.
Abbreviation: 3DOM, three dimensional ordered macroporous.
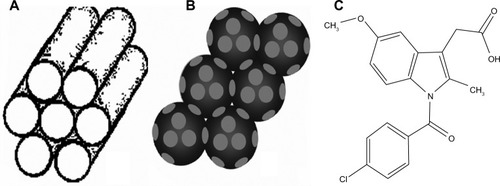
In contrast to mesoporous materials templated by surfactant micelles, three dimensionally ordered macroporous (3DOM) materials with pore sizes in the sub-micrometer range are generally templated by colloidal crystals, allowing the introduction of uniform spherical voids with controlled size, typically hundreds of nanometers in diameterCitation26 (). The low specific surface areas, relative to mesoporous materials, combined with the three dimensionally interconnected pore networks, allowed ready accessibility to guest molecules and enhanced mass transport.Citation27,Citation28 On account of these unique structural advantages, 3DOM silica is expected to be very promising for drug delivery applications, especially for poorly water-soluble compound. Taking the colloidal crystal method commonly used for 3DOM materials into account, the pore size of 3DOM silica can be tailored simply by adjusting the diameter of the colloidal crystals.Citation29,Citation30 In view of this characteristic, it is highly desirable to synthesize 3DOM silica with different pore sizes and then to explore the influence of pore size of 3DOM silica on the drug loading and release properties of poorly soluble drugs. Moreover, it would also be very interesting to compare the drug loading and release characteristics between 3D macroporous silica and widely investigated 2D mesoporous silica SBA-15 and MCM-41, especially as, to our knowledge, the comparison between these two types of materials has not yet been reported.
Amorphous or molecular forms of drugs often show better solubility compared to their crystalline counterparts.Citation31 However, the amorphous state of drug will spontaneously turn into the stable crystallization under certain conditions. Pure amorphous indomethacin (IMC) recrystallizes typically in days if stored at ambient conditions.Citation32 Therefore, stability is one of the important factors to be considered when silica is used as a drug carrier. Although many articles have reported porous silica as a drug delivery system for poorly water-soluble drugs, to our knowledge, the stability tests about macroporous silica as drug carriers have never been reported to date. Here, we have compared properties of 3DOM silica with 2D cylindrical mesoporous silica with respect to stability of IMC loaded in the drug delivery system.
Over the last decade, porous silica with many appealing properties has been widely used in biomedical applications and drug delivery.Citation33 In addition, there is growing concern over its toxicity and biocompatibility after administration in vivo.Citation34–Citation36 Hence, to evaluate the potential of 3DOM silica and 2-D mesoporous silica for oral application, the cytotoxicity test is imperative before they can be given by oral administration. It is worth mentioning that despite the significant progress in the utilization of mesoporous silica for drug delivery applications, there have been only limited studies on the biocompatible evaluation of mesoporous silica.Citation37,Citation38 Particularly, the cytotoxicity of 3DOM silica has also never been studied. In this work, human colon carcinoma (Caco-2) cells, a widely used in vitro model of intestinal epithelial cells, were chosen to investigate the cytotoxicity of this nanoporous silica due to the similarities with intestinal epithelial cells in morphology and physiology.Citation37 The pore architecture of silica particles greatly influences their biocompatibility and therefore should be carefully designed.Citation39
For this purpose, 3DOM silica with different pore sizes and classic mesoporous silica (ie, MCM-41 and SBA-15) were successfully fabricated. IMC (), a non-steroidal anti-inflammatory drug, was used as a Class II of Biopharmaceutical Classification System (BCS-II) model drug and loaded into all types of nanoporous silica via the solvent deposition method at the same drug-silica ratio.Citation40,Citation41 Various technical tools, such as scanning electron microscopy (SEM), N2 adsorption, differential scanning calorimetry (DSC), and X-ray diffraction (XRD) were applied to systematically characterize all IMC-loaded nanoporous silica formulations, aiming to explore the possible mechanism for drug dissolution improvement and the stability of different kinds of silica.
Materials
Tetraethoxysilane (TEOS), styrene, methyl methacrylate (MMA), and cetyltrimethylammonium bromide (CTAB) were obtained from Tianjin Bodi Chemical Holding Co, Ltd (Tianjin, People’s Republic of China). Pluronic P123 EO20-PO70-EO20 was obtained from Sigma Chemicals, Perth, WA, Australia. IMC (purity >99.0%) was purchased from Shijiazhuang Pharmaceutical Group (Huasheng Pharm Co, Ltd, Hebei, People’s Republic of China). All other chemicals were of analytical grade as required and used without further purification.
Methods
Preparation of nanoporous silica
Preparation of 2D mesoporous silica MCM-41 and SBA-15
2D ordered mesoporous silica MCM-41 microparticles were synthesized by using cationic surfactant CTAB as a template at room temperature.Citation42 Briefly, 0.2 g of CTAB was dissolved in a mixture of 20.5 mL of NH4OH and 27.0 mL of distilled water with constant stirring. When the solution became homogenous, 1.0 mL of TEOS was introduced, giving rise to white slurry. The mixture was stirred at 60°C for 3 hours. Then, the obtained suspension was homogenized by an ATS AH110D homogenizer at 600 bars for 10 cycles to reduce the aggregation of particles (ATS Engineer Inc., Shanghai, People’s Republic of China). Finally, the resulting product was filtered, washed with distilled water, dried, and calcined at 600°C for 4 hours.
2D ordered mesoporous silica SBA-15 microparticles were synthesized according to the procedure reported by Zhao et al with some modifications.Citation43 Briefly, 2.0 g P123 was dissolved in a mixture of 15 mL water and 60 mL 2 M hydrochloric acid solution with constant stirring at 40°C. Then, 4.6 mL TEOS was added to that solution with stirring for 24 hours at 40°C. The mixture was aged at 100°C overnight without stirring. The solid product was filtered, washed with deionized water, air dried, and calcined at 600°C for 5 hours to remove the Pluronic 123.
Preparation of 3DOM silica
3DOM silica with different pore sizes were fabricated by using noncross-linked monodisperse poly-(methyl methacrylate) (PMMA) and polystyrene (PS) latex spheres as templates, which were synthesized using an emulsifier-free emulsion polymerization techniqueCitation44 (see details in Supplementary materials). The latex spheres were then close-packed into highly ordered colloidal crystal templates by centrifugation for 4 hours (4000 rpm), then allowed to air-dry. The detailed procedure for the creation of 3DOM structure was the same as in our recent publication.Citation45 The prepared 3DOM silica using PMMA or PS as a template was milled and sieved to obtain microparticles with nominal size of <100 micrometers, and denoted as M1 and M2, respectively.
Drug loading procedure via solvent deposition method
The solvent deposition method,Citation46,Citation47 which involved a combination of soak equilibrium and subsequent solvent evaporation, was employed in the present study to load IMC into both 3DOM and 2D mesoporous silica. Firstly, IMC was dissolved in acetone to obtain a yellow transparent solution (50 mg/mL). Then an aliquot of this solution was mixed with a certain amount of silica to obtain samples with a theoretical drug-silica ratio of 1:3 (weight [w]/w). After gentle stirring for 12 hours, the solvent was allowed to evaporate under reduced pressure and then dried at room temperature. The residue was placed in a vacuum desiccator with blue silica gel for 12 hours. The obtained nanoparticles preparations were labeled as IMC-M1 and IMC-M2 for 3DOM materials, IMC-MCM for MCM-41, and IMC-SBA for SBA-15 materials, respectively.
Field emission-SEM and transmission electron microscopy
SEM images were obtained with a field emission scanning electron microscope (JEOL-6700, JEOL Ltd, Tokyo, Japan). Prior to examination, samples were mounted onto metal stubs using double-sided adhesive tape and sputtered with a thin layer of gold under vacuum. Transmission electron microscopy (TEM) was performed with a Tecnai G2 F30 microscope instrument (FEI, Hillsboro, OR, USA) with an acceleration voltage of 200 kV. The powder samples were suspended in ethanol, dropped on copper grid, then allowed to dry overnight before TEM analysis.
Nitrogen adsorption
Nitrogen adsorption measurements were performed at −196°C on a SA3100 surface area analyzer (Beckman coulter, Brea, CA, USA). All drug-loaded samples were outgassed for 12 hours at 50°C before measurement. The temperature was kept low to avoid drug degradation. The adsorption-desorption isotherms were recorded and the specific surface areas were calculated by applying the Brunauer–Emmett–Teller (BET) method in the relative pressure range between 0.05 and 0.2. The pore size distribution was calculated from the adsorption branch of N2 adsorption– desorption isotherms using the conventional Barrett–Joyner–Halenda (BJH) method. The total pore volume was estimated from the amount adsorbed at a relative pressure of 0.9814.
Solid state characterization of drug-loaded porous silica
The solid state of IMC in 3DOM and 2D mesoporous silica formulations was evaluated by DSC and XRD. DSC was performed on a differential scanning calorimeter (DSC 60, Shimadzu Co, Kyoto, Japan). Samples were heated at 10°C/minute in aluminum pans under a nitrogen flow. XRD was performed using a Rigaku Geigerflex powder X-ray diffractometer (Rigaku Denki, Tokyo, Japan) with a copper anode (Cu Kα radiation, λ =0.15405 nm, 30 kV, 30 mA). XRD patterns were recorded over the 2θ range from 5° to 50° with a step size of 0.02° and a scan speed of 4°/minute.
In vitro dissolution testing
Dissolution tests were carried out with USP dissolution apparatus using the paddle method (KC-8D, Tianjin Guoming Medical Equipment Co, Ltd, Tianjin, People’s Republic of China). The dissolution medium consisted of 900 mL phosphate buffer (pH 6.8) prepared using KH2PO4 and NaOH. Dissolution studies were carried out at 37°C ± 0.5°C with a paddle speed of 100 ± 1 rpm. A sample corresponding to 25 mg IMC was introduced onto the surface of dissolution medium at time zero. Aliquots (5 mL) of the dissolution medium were withdrawn and passed through a 0.45 μm membrane filter at appropriate intervals (5, 10, 15, 20, 30, 45, and 60 minutes). In addition, an equal volume of fresh dissolution medium was immediately added to keep the volume constant. An ultraviolet (UV) detection wavelength of 320 nm was used to determine the amount of IMC dissolved. All measurements were carried out in triplicate.
Stability test
The IMC loaded samples were stored in a desiccator under 75% relative humidity (RH; saturated solution of NaCl) at a constant temperature (40°C ± 1°C). The samples were protected from light. The crystalline form of IMC in loaded samples was measured by XRD and DSC after a storage period of three months to evaluate the solid state stability. The stressed samples were designated as M1-ST and M2-ST for 3DOM materials, MCM-ST for MCM-41 and SBA-ST for SBA-15 materials.
Cell culture and exposure to silica carriers
Caco-2 cells were cultured in 25 cm2 culture flasks (Corning Incorporated, Corning, NY, USA) using Dulbecco’s Modified Eagle’s Medium (DMEM; Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS), 1% non-essential amino acids, 100 units/mL penicillin and 100 μg/mL streptomycin. The culture was maintained at 37°C in an atmosphere of 5% CO2 and 95% relative humidity.
For all experiments, cells were trypsinized, harvested and then seeded at a density of 105 per well in 96-well plates. The cell suspensions were incubated and allowed to attach for 24 hours, and then treated with various concentrations of silica carriers (20, 50, 100, 200, 500 μg/mL) for another 24 hours. Prior to use, silica carriers were sterilized in an autoclave and then dispersed in phosphate buffered saline (PBS) solution by vortexing and ultrasonification.
In vitro cytotoxicity studies by MTT and Cell Counting Kit-8 assays
Firstly, in vitro cytotoxicity of various silica carriers against Caco-2 cells was assessed by the classic 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay. After 24 hour of treatment with various silica carriers at different concentrations, 20 μL of MTT (5 mg/mL in PBS solution) was added to each well and incubated for another 4 hours, allowing the live cells to reduce the yellow MTT to dark-blue formazan crystals. The medium was then replaced with 100 μL of dimethyl sulfoxide (DMSO) to dissolve the blue formazan crystals. The absorbance was read on a microplate reader (KHB ST-360, JingGong Industrial Co., Ltd., Shanghai, People’s Republic of China) at a wavelength of 492 nm.
Secondly, in vitro cytotoxicity of various silica carriers against Caco-2 cells was assessed by using the Cell Counting Kit (CCK)-8 assay (Beyotime Biotechnology, Jiangsu, People’s Republic of China). According to the manufacturer’s instruction, 10 μL of CCK-8 solution was added into each well after 24 hour of exposure to silica carriers, and then incubated for an additional 1 hour. The absorbance was monitored at 450 nm.
For both MTT and CCK-8 assays, the relative cell viability (%) related to control wells containing cell culture medium without silica samples was calculated by
where Abs(test) is the absorbance of test samples, and Abs(reference) is the absorbance of reference sample.
All data from MTT and CCK-8 assays were shown as the mean ± standard deviation (SD; n=3). Statistical comparisons were made with Student’s t-test. P-values <0.05 were considered statistically significant.
External morphology observation by phase contrast inverted microscopy
To qualitatively observe the external morphology of Caco-2 cells after incubation with various types of silica, cells were visualized by phase contrast inverted microscope. Firstly, cells were exposed to different types of porous silica at the concentration of 500 μg/mL for 24 hours. After the completion of exposure period, cells (control and exposed) were washed with PBS and observed by phase contrast inverted microscopy at 400× magnification.
Results and discussion
Preparation and characterization of nanoporous silica
Colloidal crystal templating was an effective, simple, and flexible method for the creation of 3DOM materials.Citation29,Citation30 In general, this procedure involved the assembly of latex spheres, the vacuum infiltration of latex spheres by a sol-gel precursor, and the final removal of the template by calcinations. In the present experiment, uniform PMMA and PS latex spheres were obtained by soap-free emulsion polymerization. Under selected conditions, highly monodisperse PMMA and PS latex spheres with narrow size distributions and mean particle size of 280 nm and 680 nm were obtained (see details in Supplementary materials). The average particle size and morphology of the latex spheres were characterized by dynamic light scattering and SEM, respectively ( and in Supplementary materials). Subsequently, highly ordered colloidal crystal arrays assembled through close packing of latex spheres into well-arranged colloidal crystals were employed for the preparation of 3DOM silica. Both SEM () and TEM () images revealed the formation of well-defined three-dimensionally ordered macroporous structures of 3DOM silica with PMMA and PS spheres as colloidal crystal templates, respectively. The macropores were interconnected through windows which were formed as a result of the contact between the template spheres prior to infiltration of the precursor solution. Based on SEM and TEM observations, the pore sizes of 3DOM silica were estimated to be approximately 200 nm and 500 nm for M1 and M2, respectively. In comparison to the original colloidal spheres, the pore sizes were slightly reduced due to calcinations shrinkage.
Figure 2 SEM images of (A) M-1, (B) M-2, (C) SBA-15, and (D) MCM-41.
Abbreviations: M, macroporous; MCM, Mobil Composition of Matter; SBA, Santa Barbara Amorphous; SEM, scanning electron microscopy.

Figure 3 TEM images of (A) M-1, (B) M-2, (C) SBA-15, and (D) MCM-41.
Abbreviations: M, macroporous; MCM, Mobil Composition of Matter; SBA, Santa Barbara Amorphous; TEM, transmission electron microscopy.

MCM-41 and SBA-15 with 2D cylindrical pores were the most widely studied mesoporous material-based drug carriers and were often used as models to compare with other materials due to the simplicity and ease in preparation.Citation47 In our work, MCM-41 possessing well-defined morphology was synthesized under basic conditions in a straightforward way by self-assembly of silica species and cationic CTAB, while SBA-15 was hydrothermally synthesized with a block copolymer P123 as a template under acidic conditions. SEM images () revealed that the as synthesized MCM-41 represented grape-like morphology with relatively uniform sizes of 0.5–1 μm, while the as synthesized SBA-15 sample consisted of many rope-like fibers with relatively uniform sizes of 1 μm aggregated into wheat-like macrostructures, which were in good agreement with previous reports.Citation43 TEM images () revealed that the inner pores of both MCM-41 and SBA-15 were unidirectional and arranged in a honeycomb structure.
Drug loading and characterization
In this experiment, the solvent deposition method was applied instead of the solvent adsorption method generally adopted for mesoporous silica in order to obtain a much higher drug loading amount usually desirable in oral drug delivery application. This method included the immersion of porous silica carrier into concentrated solution of IMC and then the rapid evaporation of acetone after complete adsorption equilibrium between drug solutions and carriers. Crude IMC powder was found to be irregular-shaped particles with a relatively wide particle size distribution (mean particle size of several micrometers, see in Supplementary materials). However, after incorporation into all nanoporous silica by the solvent deposition method, large crystals of IMC disappeared, suggesting the successful entrapment of IMC into porous silica (). For both 3DOM silica formulations (), the outer pores of 3DOM silica were blocked by IMC and became ambiguous except that a few needle-like crystals could be observed on the surface of the 3DOM silica. In the case of 2D mesoporous silica formulations (), it was observed that the morphology of IMC-loaded MCM-41 and SBA-15 was not changed compared to pure silica, demonstrating that most IMC was filled into the pore channels of MCM-41 and SBA-15.
Figure 4 SEM images of (A) IMC-loaded M-1, (B) IMC-loaded M-2, (C) IMC-loaded SBA-15, and (D) IMC-loaded MCM-41.
Abbreviations: IMC, indomethacin; M, macroporous; MCM, Mobil Composition of Matter; SBA, Santa Barbara Amorphous; SEM, scanning electron microscopy.

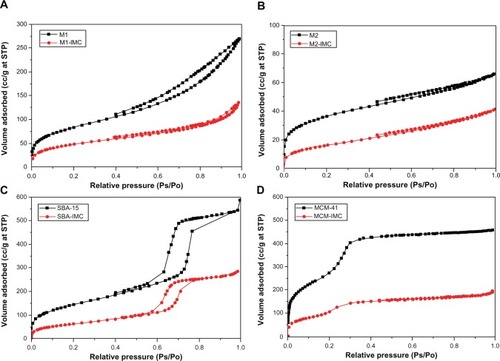
Furthermore, the drug loading characteristics of 3DOM silica and mesoporous silica samples were confirmed by the results of N2 adsorption-desorption measurements. The N2 adsorption-desorption isotherms of all nanoporous silica before and after drug loading are presented in . For both pure 3DOM silica M1 and M2, Type II nitrogen isotherms with some adsorption hysteresis was observed (), typical for a nonporous or macroporous adsorbent with no significant mesoporosity.Citation48 For both pure SBA-15 and MCM-41, however, typical type IV sorption isotherm was observed () and the sharpness of the adsorption branches was indicative of a narrow mesopore size distribution. The isotherm of SBA-15 exhibited a sharp inflection in P/P0 range from 0.60 to 0.80, while the isotherm of MCM-41 displayed the inflection over a narrow range between 0.2 and 0.3. After drug loading, the nitrogen sorption isotherms of both 3DOM and mesoporous silica showed obvious decrease in the total volume of adsorbed N2, indicating the substantial pore filling. summarizes the physical parameters, such as the BET surface area, total pore volume and BJH average pore diameter for nanoporous silica before and after drug loading. After drug loading, the decrease in BET surface area, pore volume, and pore size were observed and these changes can be attributed to the filling of IMC into pores of nanoporous silica thereby blocking the adsorption of nitrogen molecules.
Table 1 Detailed textural parameters of samples by N2 adsorption measurements
Figure 5 The N2 adsorption-desorption isotherms of (A) M1 and M1-IMC, (B) M2 and M2-IMC, (C) SBA-15 and SBA-IMC and (D) MCM-41 and MCM-IMC.
Abbreviations: M1-IMC, IMC-loaded M1; M2-IMC, IMC-loaded M2; SBA-IMC, IMC-loaded SBA-15; MCM-IMC, IMC-loaded MCM-41; IMC, indomethacin; M, macroporous; MCM, Mobil Composition of Matter; Ps, measured pressure; Po, saturated vapor pressure of N2; SBA, Santa Barbara Amorphous; STP, standard temperature and pressure.
Solid state characterization by XRD and DSC
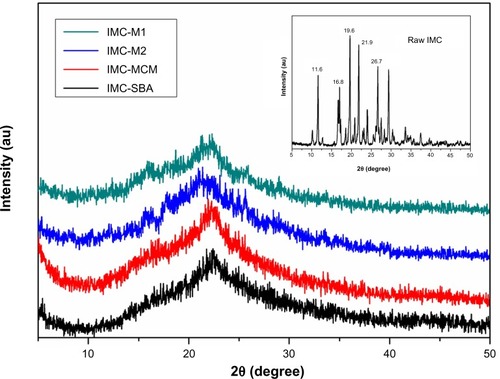
The crystal property of pure IMC and IMC-loaded silica formulations was evaluated by XRD analysis. As shown in the XRD patterns (), pure IMC showed intense and characteristic diffraction peaks at 2θ =11.6°, 16.8°, 19.6°, 21.9°, and 26.7°, which corresponded to the stable γ-form of IMC. After incorporation into nanoporous silica, a significant loss of IMC crystallinity was observed, irrespective of the types of silica. For MCM-41 and SBA-15 samples, the characteristic peak of IMC disappeared completely, implying noncrystalline state of the confined drugs. However, in the case of 3DOM silica preparations, very weak peaks could still be detected, suggesting the presence of traces of crystalline drugs. It was assumed that mesoporous silica with a smaller pore size might be more effective to form less perfect crystal due to space confinement. When confined to the narrow pores, the drug molecules are prevented from arranging themselves into a crystal lattice.
Figure 6 XRD patterns for raw IMC powders (insert) and IMC-loaded nanoporous silica samples.
Abbreviations: au, arbitrary unit; IMC, indomethacin; M, macroporous; MCM, Mobil Composition of Matter; SBA, Santa Barbara Amorphous; XRD, X-ray diffraction.
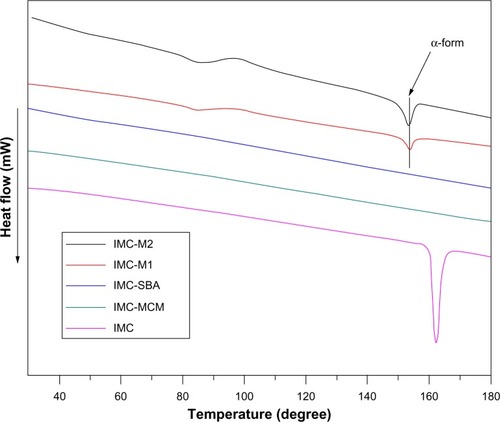
To further confirm the physical state of IMC, DSC measurement was used. shows the DSC curves of pure IMC and IMC-loaded silica formulations. For the crystalline IMC, a single sharp endothermic melting peak at 161°C was observed, in agreement with the melting point of γ-crystalline IMC. However, when loaded into nanoporous silica, IMC suffered a marked reduction in crystallinity reflected in the disappearance of the melting peak at 161°C. In the case of 2D mesoporous silica formulations, no trace of an endothermic peak was observed in the DSC curves. In combination with XRD results, IMC was proved to be in a non-crystalline state when confined into MCM-41 and SBA-15, which was consistent with previous reports.Citation49 Nevertheless, in the DSC curves of both 3DOM silica formulations, an endothermic peak at 155°C was corresponding to the meta-stable α-form. In combination with XRD analysis, the coexistence of both amorphous and crystalline IMC in 3DOM silica was proved. Moreover, for the 3DOM M2 carrier with larger pore diameter at about 500 nm, the crystal diffraction peak of IMC encapsulated in M2 was stronger at 2θ =16°–18° in XRD pattern and the endothermic peak was more obvious at 155°C in DSC curve compared with that in M1 which had a pore diameter of 200 nm, indicating that in the scope of macropores the larger pore diameter the carrier had, the easier the formation of drug crystal could be. Various reports have revealed the hydrogen interaction between drug molecules and porous silica. In addition to the space restriction of nanoporous silica, the formation of hydrogen bonding between the silanol groups present on the surface of silica and carboxyl group in IMC might be one responsibility for the observed loss of crystallinity.Citation50,Citation51
Figure 7 DSC thermograms for pure IMC powders and IMC-loaded nanoporous silica samples.
Abbreviations: DSC, differential scanning calorimetry; IMC, indomethacin; M, macroporous; MCM, Mobil Composition of Matter; SBA, Santa Barbara Amorphous.
In vitro dissolution
The in vitro dissolution behaviors of nanoporous silica formulations, including 3DOM silica, MCM-41, and SBA-15, were compared to original IMC crystals. As presented in , for pure IMC crystals, less than 70% of IMC was released within 60 minutes, indicating the poor dissolution of the pure drug. However, the dissolution of IMC from all nanoporous silica formulations, irrespective of the silica type, was significantly higher than from pure IMC. The enhanced dissolution rate of IMC by inclusion into nanoporous silica was supposed to improve its absorption in the digestive tract and thereby improve the efficacy.Citation16,Citation17 Hence, improving the dissolution rate and solubility are essential to increase the bioavailability of poorly water soluble drugs.
Figure 8 In vitro dissolution profiles for pure IMC powders and IMC-loaded nanoporous silica samples.
Note: Each data point represents the mean ± standard deviation of three determinations.
Abbreviations: IMC, indomethacin; M, macroporous; MCM, Mobil Composition of Matter; min, minutes; SBA, Santa Barbara Amorphous.
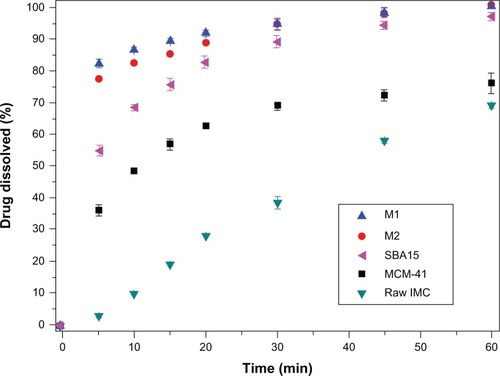
Several factors may contribute to the dissolution enhancement: lack of crystalline form and increased surface area of the drug when after confined into pores of nanoporous silica as well as the hydrophilic surface of silica carriers. It is well known that drug nanosizing is an effective way to increase the dissolution velocity, because nanosized drug crystals can increase the effective surface area available for dissolution according to the Noyes–Whitney equation.Citation4,Citation5 Since the pore size of both 3DOM and 2D mesoporous silica was within the nanometers range, drugs incorporated into pores of silica may not be able to grow to large crystals. On the other hand, the formation of the highly ordered crystalline form may be restricted by the confined space of the nanopores, thus retaining its noncrystalline or amorphous form.Citation19,Citation52 The formation of the less ordered or amorphous form, compared to the crystalline form, is well known to dramatically increase the apparent solubility and dissolution rate of poorly water-soluble drugs.Citation53
Comparing the dissolution profiles of IMC from 3DOM silica and 2D mesoporous silica formulations, the dissolution rate of IMC from 3DOM silica was faster than from MCM-41 and SBA-15. The difference in matrix architecture, including the pore size and pore geometry, may be mainly responsible for the difference in the drug dissolution profile. Drug release from the pores of the carrier into the dissolution medium requires two main processes: dissolving of the entrapped drug is the first step and diffusion of the dissolved drug through the pore channel into the dissolution medium is the second. The particle size of IMC entrapped in 2D mesoporous silica (2–50 nm) MCM-41 and SBA-15 is much smaller than in 3D macroporous silica M1 and M2, which could make drugs dissolve quickly. However, it is the diffusion process throughout the elongated mesopores that is the rate-limiting step that decides the dissolution rate of IMC. For 3DOM carriers, a large pore diameter and interconnecting 3D pore networks might facilitate fast diffusional transport of dissolved drugs and shorten the diffusion distance from the inner pores to the outer dissolution medium owing to the reduced pore restriction and good accessibility, then leading to fast drug diffusion of 3DOM carriers. Therefore, the dissolving of drug entrapped in the 3DOM silica is the rate-limiting process of the dissolution. The particle sizes of IMC entrapped in these two kinds of carriers (2D mesoporous silica and 3D macroporous silica) with nanopores are all in the nanometer range, which could be dissolved rapidly when the carriers contacted the bulk medium. Therefore, the reason that the dissolution velocity of IMC from 3DOM silica was faster than from MCM-41 and SBA-15 might be due to the dissolved velocity of IMC in 3DOM silica which is the rate-limiting step of dissolution and is faster than the diffusion process of IMC entrapped in the 2D mesoporous silica MCM-41 and SBA-15.
For 3DOM silica with different pore sizes, it was observed that IMC displayed a much faster dissolution rate from M1 with a smaller pore size of 200 nm than from M2 with a larger pore size of 500 nm. This may be due to the fact that drug particles entrapped in M1 had a much smaller particle size than in M2, resulting in a higher surface area for dissolution according to the Noyes–Whitney equation. The dissolution of the drug entrapped in M1 is faster than in M2, and the dissolution process is the rate-limiting step for 3DOM silica. Consequently, in comparison to a large pore space, the drug particle size might play a more pronounced role in the drug release behavior in the case of macroporous materials within the sub-micrometer range.
Stability test
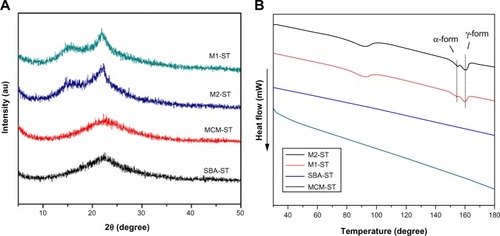
The solid-state samples were characterized by DSC and XRD and compared to the original state to evaluate stability. As seen in , the emergence of a drug melting peak of IMC at 161°C in the DSC curves of M1-ST and M2-ST, as well as the characteristic crystalline diffraction peaks at 2θ =15° in the XRD pattern, prove that some amorphous IMC has changed to the stable crystalline state during storage in the accelerated test for 3 months. But there is still some amorphous drug according to the result of XRD. On the contrary, the DSC curves and XRD patterns of MCM-ST and SBA-ST indicated that the physical state of IMC in 2D mesoporous silica MCM-41 and SBA-15 showed no significant change compared to the initial samples. This study further proved that mesoporous material has a strong recrystallization-inhibition effect, and it was still able to effectively inhibit drug crystallization even if drugs were kept in storage for 3 months under accelerated conditions. These results may be mainly due to the space confinement effect. The rigidity of mesoporous carriers can effectively inhibit drugs from mutually aggregating, thus preventing the formation of crystals. However, for 3DOM silica, the pore size was in the sub-micron range and interconnected with each other, thus weakening the inhibition of mutual aggregation. Therefore, the drug loaded in M1 and M2 aggregated with each other, and transformed into the crystalline state in the accelerated tests. In addition, the diffraction peak intensity of M2-ST at 2θ =17° is stronger than that of M1-ST, which could indicate that with the increase of the carrier pore, the inhibition of drug crystallization weakens gradually. These results indicate that the amorphous state of IMC in 2D mesoporous silica has a better physical stability than the 3DOM M1 and M2 silica.
Figure 9 (A) XRD curves and (B) DSC patterns of physical stability of IMC-loaded porous silica systems after storage at a relative humidity of 75% and 40°C.
Abbreviations: au, arbitrary unit; DSC, differential scanning calorimetry; IMC, indomethacin; M, macroporous; MCM, Mobil Composition of Matter; SBA, Santa Barbara Amorphous; XRD, X-ray diffraction.
In vitro cell cytotoxicity evaluations
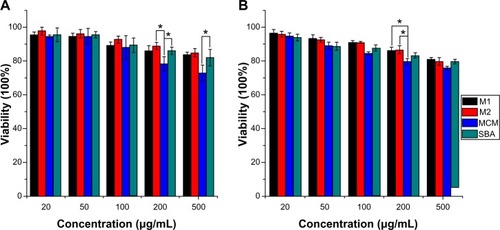
A number of methods have been developed to study cell viability and proliferation in cell populations. Among these methods, the colorimetric assay was a convenient and widely used tool for the study of the metabolic activity of viable cells, which is usually performed in a microplate format which allows many samples to be analyzed rapidly and simultaneously.Citation54 In principle, the cells were incubated with a colorimetric substrate (eg, MTT or 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium [WST]-8), which is reduced to a colored formazan exclusively by metabolically active cells. In the MTT assay, MTT can be reduced by viable cells to a colored, water-insoluble formazan salt, which can then be solubilized and easily quantified. However, in the CCK-8 assay, which contains WST-8, viable cells can convert WST-8 to a water-soluble formazan, thus avoiding the solubilization step and enabling the simplification of assay performance.Citation55 In our preset study, both the MTT and CCK-8 assay were used and combined in order to measure the cytotoxicity of nanoporous silica against Caco-2 cells. The results of MTT and CCK-8 assays in indicate that both 3DOM silica and SBA-15 silica exhibited negligible cytotoxicity on Caco-2 cells at tested concentrations. Even at the highest concentration of 500 μg/mL, cell viability was higher than 80%. However, MCM-41 silica was slightly toxic at high concentrations with cell viabilities lower than 80% and showed more toxicity at the same concentration than 3DOM silica and SBA-15. This result was probably due to the smaller diameter of MCM-41 nanoparticles when compared to other materials, which is therefore able to enter the cells through endocytosis.
Figure 10 Effect of nanoporous silica on cell viability of Caco-2 cells by (A) MTT and (B) CCK-8 assays.
Notes: Experiments were carried out in triplicate and error bars indicate standard deviation; *P<0.05.
Abbreviations: CCK, Cell Counting Kit; M, macroporous; MCM, Mobil Composition of Matter; MTT, 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; SBA, Santa Barbara Amorphous.
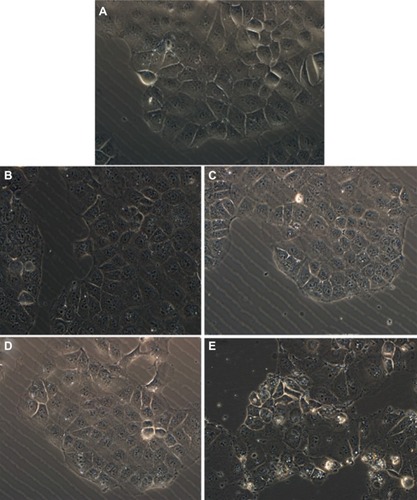
Variation of the shape and morphology of cells upon their exposure to toxic materials could be observed by optical measurements. In our work, cell morphologic changes were observed by phase-contrast microscopy and photographs of adherent cells were taken after removal of the medium containing any floating cells. shows the general external morphology of control cells and nanoporous silica-exposed cells. Except for the slight shrinkage and irregular shape observed for cells treated with MCM-41 silica, no obvious differences were found between cells untreated and treated with other nanoporous silica, demonstrating their low toxic effect and good biocompatibility. These results were in agreement with those obtained from the MTT and CCK-8 assays. The slight cell cytotoxicity of spherical MCM-41 against Caco-2 cells might due to its relatively small particle size, nearly spherical morphology, and large surface areas, resulting in the increased cellular associations and thus enhanced intrinsic toxicity.Citation56
Figure 11 Phase contrast inverted microscope image of Caco-2 cells untreated (control) and treated with various silica carriers at the same concentration. (A) Control, (B) M1, (C) M2, (D) SBA-15, and (E) MCM-41.
Abbreviations: M, macroporous; MCM, Mobil Composition of Matter; SBA, Santa Barbara Amorphous.
Conclusion
The feasibility of two types of nanoporous silica materials, including mesoporous silica (MCM-41 and SBA-15 with 2D hexagonal pores) and 3D macroporous silica (3DOM silica), as matrices for the dissolution enhancement of the poorly soluble drug IMC was studied in this work. Moreover, the influence of pore geometry and size of the silica host on the loading and release properties of IMC was studied systematically. IMC was loaded into porous silica via a soaking/solvent evaporation method to obtain a drug-silica ratio of 1:3 (w/w). A significant loss of crystallinity for IMC was observed after entrapment into both 3DOM silica and 2D mesoporous silica, probably due to spatial confinement and hydrogen bonds formation between silica surface and IMC molecule. All nanoporous silica exhibited dissolution-enhancing effects for IMC. Several factors seem to play roles, including both the lack of crystalline form and extremely large surface area for dissolution as well as the hydrophilic surface of porous silica. Compared to 2D cylindrical mesopores, 3DOM silica could reduce the diffusion resistance significantly and shorten the diffusion distance from the inner pores to the outer dissolution medium, thus leading to faster drug release. However, 3DOM silica can’t effectively inhibit drugs from mutually aggregating. Therefore, the amorphous drug could partly turn into a crystalline form after accelerated testing after 3 months. Therefore, the stability of amorphous drug entrapped in 3D macroporous silica is a significant factor to be considered. The cytotoxicity results obtained from MTT, CCK-8, and phase contrast microscopy clearly showed that the presence of 3DOM silica and SBA-15 had no significant effect on the cell proliferation and cell viability at tested concentrations, demonstrating its good biocompatibility. We hope that our research encourages further research on the application of silica with different pore architecture as drug delivery systems.
Supplementary materials
Synthesis of monodisperse PMMA and PS microspheres
For a typical synthesis, a three-necked round-bottomed flask, which was equipped with a condenser, a thermometer, and a pipe for nitrogen introduction, was filled with 200 mL of deionized water and heated to 70°C before a certain amount of monomer was added. Prior to use, the monomers (styrene and MMA) were washed in a separatory funnel four times with 5% NaOH and then four times with distilled water. Under constant stirring (300 rpm) and with N2 bubbling for 30 minutes, a solution of potassium persulfate (KPS) initiator (0.01 g/mL) preheated to 70°C was added to initiate polymerization reaction. The temperature of the reaction system was kept at 70°C for 2 hours (PMMA spheres) or 24 hours (PS spheres). Dynamic light scattering (also known as photon correlation spectroscopy; PCS) was performed by using the Submicron Particle Sizer NICOMP 380 (NICOMP, Santa Barbara, California, USA) to monitor the mean particle size of the resulting latex spheres. The particle size distribution of PMMA and PS spheres is NICOMP distribution, and the angle of measurements is 90°. Prior to measurement, the latex spheres were filtered to remove any large agglomerates. By varying the reaction conditions listed in , mono-disperse PS spheres with average diameters of 780 nm and PMMA spheres with average diameters of 280 nm were synthesized as shown in .
Preparation and characterization of highly ordered colloidal templates
PMMA and PS colloidal templates were obtained by centrifugation and then characterized by SEM (). presents typical SEM images of PMMA and PS nanospheres in a dry monolithic form. It was found that both PMMA and PS colloidal spheres were closely packed into a highly ordered 3D arrangement with uniform particle size of 250 nm and 550 nm, respectively. In comparison with results from dynamic light scattering analysis (280 nm and 680 nm), a size reduction was observed, probably due to the drying and assembly process. These obtained well-arranged 3D colloidal crystal arrays could be available templates for the creation of 3DOM silica materials.
Figure S1 The intensity-weighted particle size distributions for (A) PMMA and (B) PS microspheres based on dynamic light scattering.
Abbreviations: PMMA, poly-(methyl methacrylate); PS, polystyrene; Intens-WT, intensity-weighted; REL, relative; Diam, diameter.
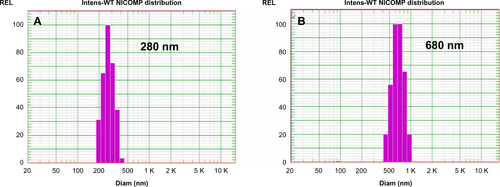
Figure S2 SEM images for colloidal templates of (A) PMMA and (B) PS spheres.
Abbreviations: PMMA, poly-(methyl methacrylate); PS, polystyrene; SEM, scanning electron microscopy.

Figure S3 SEM image of crude IMC powders.
Abbreviations: IMC, indomethacin; SEM, scanning electron microscopy.

Table S1 Reaction conditions for the synthesis of microspheres by soap-free emulsion polymerization
Acknowledgments
This work was supported by National Basic Research Program of China (973 Program) (NO 2009CB930300), National Natural Science Foundation of China (NO 81072605) and major national platform for innovative pharmaceuticals (2009ZX09301-012).
Disclosure
The authors report no conflicts of interest in this work.
References
- LipinskiCALombardoFDominyBWFeeneyPJExperimental and computational approaches to estimate solubility and permeability in drug discovery and development settingsAdv Drug Deliv Rev2001461–332611259830
- FahrALiuXDrug delivery strategies for poorly water-soluble drugsExpert Opin Drug Deliv20074440341617683253
- DressmanJChristosRDrug solubility: how to measure it, how to improve itAdv Drug Del Rev2007597531532
- Merisko-LiversidgeELiversidgeGGCooperERNanosizing: a formulation approach for poorly-water-soluble compoundsEur J Pharm Sci200318211312012594003
- MüllerRHJacobsCKayserONanosuspensions as particulate drug formulations in therapy. Rationale for development and what we can expect for the futureAdv Drug Deliv Rev200147131911251242
- HaussDJOral lipid-based formulationsAdv Drug Deliv Rev200759766767617618704
- ColeETCadéDBenameurHChallenges and opportunities in the encapsulation of liquid and semi-solid formulations into capsules for oral administrationAdv Drug Deliv Rev200860674775618096270
- LeunerCDressmanJImproving drug solubility for oral delivery using solid dispersionsEur J Pharm Biopharm2000501476010840192
- VasconcelosTSarmentoBCostaPSolid dispersions as strategy to improve oral bioavailability of poor water soluble drugsDrug Discov Today20071223–241068107518061887
- SharmaSSherPBadveSPawarAPAdsorption of meloxicam on porous calcium silicate: characterization and tablet formulationAAPS PharmSciTech200564E618E62516408863
- FriedrichHFussneggerBKolterKBodmeierRDissolution rate improvement of poorly water-soluble drugs obtained by adsorbing solutions of drugs in hydrophilic solvents onto high surface area carriersEur J Pharm Biopharm200662217117716275049
- SanganwarGPGuptaRBDissolution-rate enhancement of fenofibrate by adsorption onto silica using supercritical carbon dioxideInt J Pharm20083601–221321818550302
- KinoshitaMBabaKNagayasuAImprovement of solubility and oral bioavailability of a poorly water-soluble drug, TAS-301, by its melt-adsorption on a porous calcium silicateJ Pharm Sci200291236237011835196
- SmirnovaIMamicJArltWAdsorption of drugs on silica aerogelsLangmuir2003192085218525
- TanASimovicSDaveyAKRadesTPrestidgeCASilica-lipid hybrid (SLH) microcapsules: a novel oral delivery system for poorly soluble drugsJ Control Release20091341627019013488
- BahlDBognerRHAmorphization of Indomethacin by Co-Grinding with Neusilin US2: amorphization kinetics, physical stability and mechanismPharm Res200623102317232516927179
- SmirnovaISuttiruengwongSSeilerMArltWDissolution rate enhancement by adsorption of poorly soluble drugs on hydrophilic silica aerogelsPharm Dev Technol20049444345215581080
- Quintanar-GuerreroDGanem-QuintanarANava-ArzaluzMGPiñón-SegundoESilica xerogels as pharmaceutical drug carriersExpert Opin Drug Deliv20096548549819413456
- SlowingIIVivero-EscotoJLWuCWLinVSMesoporous silica nanoparticles as controlled release drug delivery and gene transfection carriersAdv Drug Deliv Rev200860111278128818514969
- ColillaMManzanoMVallet-RegíMRecent advances in ceramic implants as drug delivery systems for biomedical applicationsInt J Nanomedicine20083440341419337409
- KresgeCLeonowiczMRothWVartuliJBeckJOrdered mesoporous molecular sieves synthesized by a liquid-crystal template mechanismNature19923596397710712
- ChenBWangZQuanGIn vitro and in vivo evaluation of ordered mesoporous silica as a novel adsorbent in liquisolid formulationInt J Nanomedicine2012719920922275835
- Vallet-RegíMBalasFColillaMManzanoMDrug confinement and delivery in ceramic implantsDrug Metab Lett200711374019356016
- HeikkiläTSalonenJTuuraJMesoporous silica material TUD-1 as a drug delivery systemInt J Pharm2007331113313817046183
- Vallet-RegíMDoadrioJCDoadrioALIzquierdo-BarbaIPérez-ParienteJHexagonal ordered mesoporous material as a matrix for the controlled release of amoxicillinSolid State Ionics20041721–4435439
- HollandBTBlanfordCFSteinASynthesis of macroporous minerals with highly ordered three-dimensional arrays of spheroidal voidsScience199828153765385409677191
- LytleJCYanHErgangNSSmyrlWHSteinAStructural and electrochemical properties of three-dimensionally ordered macroporous tin(IV) oxide filmsJ Mater Chem20041416161622
- LeeKLytleJErgangNOhSSteinASynthesis and rate performance of monolithic macroporous carbon electrodes for lithium-ion secondary batteriesAdv Funct Mater2005154547556
- VelevODLenhoffAMColloidal crystals as templates for porous materialsCurr Opin Colloid In200051–25663
- SteinASchrodenRCColloidal crystal templating of three-dimensionally ordered macroporous solids: materials for photonics and beyondCurr Opin Solid St Mater Sci200156553564
- HancockBCDisordered drug delivery: destiny, dynamics and the Deborah numberJ Pharm Pharmacol200254673774612078989
- YoshiokaMHancockBCZografiGCrystallization of indomethacin from the amorphous state below and above its glass transition temperatureJ Pharm Sci19948312170017057891297
- WilliamsACTimminsPLuMForbesRTDisorder and dissolution enhancement: deposition of ibuprofen on to insoluble polymersEur J Pharm Sci2005263–428829416087323
- TangFLiLChenDMesoporous silica nanoparticles: synthesis, biocompatibility and drug deliveryAdv Mater Weinheim201224121504153422378538
- FuCLiuTLiLLiuHChenDTangFThe absorption, distribution, excretion and toxicity of mesoporous silica nanoparticles in mice following different exposure routesBiomaterials201334102565257523332175
- GuzmánELiggieriLSantiniEInfluence of silica nanoparticles on dilational rheology of DPPC–palmitic acid Langmuir monolayersSoft Matter2012839383948
- HeikkiläTSantosHAKumarNCytotoxicity study of ordered mesoporous silica MCM-41 and SBA-15 microparticles on Caco-2 cellsEur J Pharm Biopharm201074348349420025968
- Di PasquaAJSharmaKKShiYLCytotoxicity of mesoporous silica nanomaterialsJ Inorg Biochem200810271416142318279965
- LeeSYunHSKimSHThe comparative effects of mesoporous silica nanoparticles and colloidal silica on inflammation and apoptosisBiomaterials201132359434944321889200
- WangFHuiHBarnesTJBarnettCPrestidgeCAOxidized mesoporous silicon microparticles for improved oral delivery of poorly soluble drugsMol Pharm20107122723619874003
- HuYWangJZhiZJiangTWangSFacile synthesis of 3D cubic mesoporous silica microspheres with a controllable pore size and their application for improved delivery of a water-insoluble drugJ Colloid Interface Sci2011363141041721820127
- CaiQLinWYXiaoFSPangWQChenXHZouBSThe preparation of highly ordered MCM-41 with extremely low surfactant concentrationMicropor Mesopor Mater1999321–2115
- ZhaoDFengJHuoQTriblock copolymer syntheses of mesoporous silica with periodic 50 to 300 angstrom poresScience199827953505485529438845
- ZhangJJinYLiCCreation of three-dimensionally ordered macroporous Au/CeO2 catalysts with controlled pore sizes and their enhanced catalytic performance for formaldehyde oxidationAppl Catal B: Environmental2009911–21120
- HuYZhiZWangTJiangTWangSIncorporation of indomethacin nanoparticles into 3-D ordered macroporous silica for enhanced dissolution and reduced gastric irritancyEur J Pharm Biopharm201179354455121767643
- Barzegar-JalaliMMalekiNGarjaniAEnhancement of dissolution rate and anti-inflammatory effects of piroxicam using solvent deposition techniqueDrug Dev Ind Pharm200228668168612149960
- HuYZhiZZhaoQ3D cubic mesoporous silica microsphere as a carrier for poorly soluble drug carvedilolMicropor Mesopor Mater2012147194101
- SingKEverettDHaulRReporting physisorption data for gas/solid systems - with special reference to the determination of surface area and porosityPure Appl Chem1985574603619
- MellaertsRMolsRJammaerJAIncreasing the oral bioavailability of the poorly water soluble drug itraconazole with ordered mesoporous silicaEur J Pharm Biopharm200869122323018164930
- JiCBarrettAPoole-WarrenLAFosterNRDehghaniFThe development of a dense gas solvent exchange process for the impregnation of pharmaceuticals into porous chitosanInt J Pharm20103911–218719620214968
- MadiehSSimoneMWilsonWMehraDAugsburgerLInvestigation of drug-porous adsorbent interactions in drug mixtures with selected porous adsorbentsJ Pharm Sci200796485186317221849
- BlagdenNde MatasMGavanPTYorkPCrystal engineering of active pharmaceutical ingredients to improve solubility and dissolution ratesAdv Drug Deliv Rev200759761763017597252
- HancockBCParksMWhat is the true solubility advantage for amorphous pharmaceuticals?Pharm Res200017439740410870982
- MosmannTRapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assaysJ Immunol Methods1983651–255636606682
- LiYSunLJinMSize-dependent cytotoxicity of amorphous silica nanoparticles in human hepatoma HepG2 cellsToxicol In Vitro20112571343135221575712
- Di PasquaAJSharmaKKShiYLCytotoxicity of mesoporous silica nanomaterialsJ Inorg Biochem200810271416142318279965