Abstract
Saponin fractions of Quillaja saponaria Molina (QS) have cytotoxic activity against cancer cells in vitro, but are too toxic to be useful in the clinic. The toxic effect was abolished by converting QS fractions into stable nanoparticles through the binding of QS to cholesterol. Two fractions of QS were selected for particle formation, one with an acyl-chain (ASAP) was used to form killing and growth-inhibiting (KGI) particles, and the other without the acyl-chain (DSAP) was used to formulate blocking and balancing effect (BBE) particles. KGI showed significant growth inhibiting and cancer cell-killing activities in nine of 10 cell lines while BBE showed that on one cell line. The monoblastoid lymphoma cell line U937 was selected for analyzing the mode of action. Low concentrations of KGI (0.5 and 2 μg/mL) induced irreversible exit from the cell cycle, differentiation measured by cytokine production, and eventually programmed cell death (apoptosis). Compared to normal human monocytes, the U937 cells were 30-fold more sensitive to KGI. The nontoxic BBE blocked the cell killing effect of KGI in a concentration-dependent manner. In conclusion, the formulation of QS into nanoparticles has the potential of becoming a new class of anticancer agents.
Introduction
Pharmacological treatment of cancer makes continuous progress, but with some few exceptions, the new “targeted” drugs only provide marginal benefit as single drugs or in combination with established drugs.Citation1 Thus, there is an urgent need to identify and develop new anticancer drugs acting by new principles evading the drawbacks of currently available drugs. Saponins belong to a group of naturally derived compounds, which have demonstrated substantial cytotoxic activity through different mechanisms in vitro.Citation2–Citation4 Astragalus saponin (AST) inhibits cell proliferation through accumulation in S phase and G2/M arrest, with concomitant suppression of p21 expression and inhibition of cyclin-dependent kinase activity. Furthermore, it promotes apoptosis in HT-29 cells through caspase 3 activation and poly(ADP-ribose) polymerase cleavage.Citation5 Ginseng saponin induces apoptosis via p53 and p21 activation and down regulates cell cycle-related proteins.Citation6 Saponin extracts from the Quillaja saponaria Molina (QS) tree exhibit diverse biological activities such as hemolytic, anti-inflammatory, immune-stimulatory, antiviral, and cytotoxic activities.Citation7 QS is well studied and characterized, and has been fractionated into 21 or more fractions by reverse phase chromatography.Citation8,Citation9 The more hydrophobic fractions have an acyl-chain, ie, acyl-saponin (ASAP) corresponding to QS 21Citation8 or QHC,Citation10 rendering them highly cell lytic while the less hydrophobic fractions are lacking the acyl-chain, ie, desacyl-saponin (DSAP) corresponding to QS 7Citation8 or QHA,Citation10 having a low cell lytic effect.
The crude and ASAP fractions of QS cause side effects because of their hydrophobiclytic properties resulting in trapping at the site of administration, causing cell and tissue destruction leading to local and systemic adverse reactions.Citation11 Wang and colleagues used different QS fractions to show cancer cell-killing effects,Citation7 but they are too toxic to proceed to clinical testing. Thus, these free nonparticulate forms of QS demonstrate a low therapeutic index disqualifying them as candidate anticancer drugs. However, QS saponins formulated into 40 nm particles together with cholesterol and lipids have shown significantly reduced toxic effect on cells.Citation11 A similar particle, an immune-stimulating complex (ISCOM), is currently used as an adjuvant and delivery system commercialized by Isconova AB (Uppsala, Sweden) for animal vaccines and is also in human phase III studies.Citation11 The ASAP and DSAP have been formulated into particles similar to the ISCOMs. The chemical stability of ISCOM is of significant practical value and has a shelf-life of several years in aqueous solutions at +2 °C to +8 °C (compared to months for free saponins). The particles are also intact and stable both in vitro and in vivo. No release of the integrated components of the particles are observed (Dr Karin Lövgren-Bengtsson, Isconova AB).Citation11 In view of the well-known adjuvant effects of the two particles including stimulation and differentiation of immature and resting dendritic cells (manuscript in preparation), we decided to explore if the differentiation concept could be applied on cancer cell therapy.
In this study, we have elucidated the anticancer effects of the ASAP and DSAP particles. These particles were formulated using a similar technology as we applied for the formulation of ISCOMs.Citation12 Because of their different modes of anticancer cell effects, ASAP particles were denoted killing and growth-inhibiting (KGI) and DSAP particles were denoted blocking and balancing effect (BBE). The results demonstrate that ASAP formulated into KGI particles, selectively kills tumor cells by apoptosis at a 30-fold lower concentration than that required to kill normal cells, indicating their potential as anticancer drugs. In contrast, nonparticulate ASAP kills both normal and cancer cells by lytic and necrotic effects.
Materials and methods
Acyl- and desacyl-saponins and their particulate formulations
Purified QS fractions, the acyl-saponin (ASAP) and its particulate form KGI and the desacyl-saponin (DSAP) and its particulate form BBE (), were kindly supplied by Dr Karin Lövgren-Bengtsson (Isconova AB). These fractions are well characterized and used in commercial and experimental vaccine adjuvant formulations.Citation11,Citation13 The KGI and BBE particles were prepared as described previously.Citation13 Briefly, to 0.05 mL of lipid mixture containing 10 mg/mL each of cholesterol (C) and phosphatidylcholine (PC), 0.025 mL of ASAP/DSAP solution (100 mg/mL) and 0.05 mL of phosphate-buffered saline (PBS) was added, mixed well and incubated at room temperature overnight. Then the mixture was dialyzed against PBS at room temperature for three days with changing PBS every 12 hours. The particles were purified through a 10% sucrose cushion overnight at 50.000 rpm 10 °C in a SW50.1 rotor followed by re-suspension in PBS overnight.
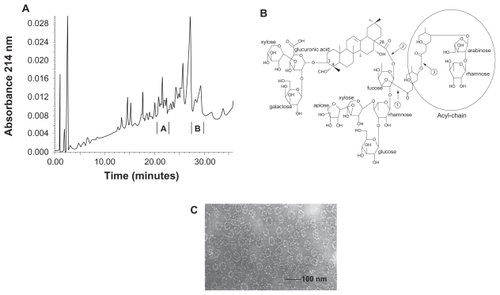
Figure 1 Selected Quillaja saponin (QS) fractions, and their particulate forms. A) Reversed phase chromatography profile of QS. Fraction A (desacyl-saponin, DSAP) is the least hydrophobic QS fraction being the active saponin component in BBE particles. Fraction B (acyl-saponin, ASAP) is the most hydrophobic QS fraction being the active component in KGI particles. B) The triterpenoid structure of QS. The core is a triterpenoid with two carbohydrate chains in the position 3 and 18 and an acyl chain terminated with arabinose and rhamnose monosaccharides demarked with a ring in the Figure. C) Electron microscopic picture of KGI. BBE shows the same morphology. The diameter of the spheres is about 40 nm.
Electron microscopy
Samples of KGI and BBE particles were dialyzed against PBS, applied to formvar-coated copper grids and contrasted with 2% ammonium molybdate, pH 7.0. Photographs of the specimens were taken in Phillips 300 electron microscope at 60 kV accelerating voltage.Citation14
Cells
The human monoblast cell lines THP-1, acute lymphoblastic leukemia Jurkat, and human histiocytic lymphoma cell lines U937 and U937-Vcr were kindly provided by Prof. Kenneth Nilsson (Rudbeck laboratory, Uppsala University, Uppsala, Sweden). The myeloma RPMI 8226/S cell line and its sublines 8226/Dox40 and 8226/LR-5 were kind gifts from Dr William Dalton (Department of Medicine, Arizona Cancer Center, University of Arizona, Tucson, AZ). The human acute myelocytic leukemia cell line MV-4-11 was obtained from American Type Culture Collection (Rockville, MD). The acute lymphoblastic leukemia cell lines CCRF-CEM and its subline CEM/VM-1 were kind gifts from William Beck (Department of Pharmacology, College of Medicine, University of Tennessee, Memphis, TN).
8226/Dox40 was selected for doxorubicin resistance and shows the classical multidrug resistance (MDR) phenotype with overexpression of P-glycoprotein 170.Citation15 8226/LR-5 was selected for melphalan resistance, proposed to be associated with increased levels of glutathione (GSH).Citation16 U937-Vcr was selected for vincristine resistance, proposed to be tubulin-associated. Citation17 CEM/VM-1, selected for teniposide resistance, expresses an atypical MDR, which is proposed to be topoisomerase II (topoII)-associated.Citation18
The cells were cultured in the recommended cell medium for the different cell lines. Normal human dendritic cells (DCs) and neutrophils were purchased from 3H Biomedical (Uppsala, Sweden).
For most experiments cell lines were grown in culture medium containing 10% fetal calf serum (FCS) under a humidified atmosphere with 5% CO2 at 37 °C in 96-well flat-bottomed micro-titer plates (Nunc, Roskilde, Denmark). For synchronization, cells were starved for 22 hours by lowering of the serum concentration in the cell culture medium to 0.5%. The medium was then changed to medium containing 10% FCS.
Hemolytic activity
Hemolytic activity was assessed in chicken red blood cells (RBC) as described by Rönnberg and colleagues.Citation10 The RBCs were washed with PBS and diluted to 0.5% in PBS. To 100 μl of this suspension, 100 μl of sample solution in PBS was added. The mixture was incubated at 37 °C for one hour and then centrifuged. Damaged cell membranes allow hemoglobin to leak out into the supernatant. The absorbance of the supernatant was then measured at 414 nm in a Labsystems Multiskan MCC/340 microtitre plate reader.
Cell viability
Trypan Blue (0.02%; National Veterinary Institute, Uppsala, Sweden) was used to analyze cell viability in nucleated cells. After exposure to saponin formulations, cells were stained with Trypan Blue. Stained dead and nonstained viable cells were counted in a microscope and the proportion of dead and live cells was calculated.
In some experiments the fluorometric microculture cytotoxicity assay (FMCA) was used for measurements of cell viability and cytotoxicity. Tumor cells were seeded in the presence or absence of the drug in 96-well micro-titer plates at a cell density of 5,000–20,000 cells/well. The FMCA is based on measurement of fluorescence generated from hydrolysis of fluorescein diacetate (FDA) to fluorescein by cells with intact plasma membranes and has been described in detail previously.Citation19 The plates were incubated at 37 °C in humidified atmosphere containing 5% CO2 for 72 hours. At the end of the incubation period the medium was removed by aspiration. After one wash in PBS, 50 μl/well of FDA dissolved in a physiological buffer (10 μg/mL) was added. The plates were incubated for 45 minutes and the generated fluorescence from each well was measured in a 96-well scanning fluorometer. The fluorescence is proportional to the number of intact cells in the well. Quality criteria for a successful analysis included a fluorescence signal in the control wells of more than five times the mean blank value, a mean coefficient of variation (CV) in the control wells of less than 30%.
Apoptosis and necrosis
Apoptosis was analyzed by fluorescent-activated cell sorting (FACS). Briefly, 100 μl cell suspension (~0.1–1 × 106 cells/mL) was transferred to a 5 mL culture tube. According essentially to the manufacturer’s instruction, after addition of 5 μl propidium iodide (PI; 50 μg/mL) and 2.5 μl annexin V-FITC (200 μg/mL) (BD Biosciences, Stockholm, Sweden) the samples were incubated for 15 minutes in the dark at room temperature. Then 400 μl of 1 × binding buffer was added to each tube and the samples were analyzed within one hour.
A multiparametric single-cell assay was also used for measurement of apoptosis.Citation20 Briefly, cells were plated in 96-well plates with flat optical bottom (Perkin-Elmer Inc., Wellesley, MA), and incubated overnight before addition of drugs. At indicated time points, fluorescent probes were added to stain apoptotic markers: FAM-DEVD-FMK to stain activated caspase-3, MitoTracker Red to evaluate mitochondrial membrane potential (MMP) and Hoechst 33342 to stain the nucleus. Analysis was performed in the ArrayScan® high content screening system (Cellomics Inc, Pittsburgh, PA) which is a computerized automated fluorescence imaging microscope that automatically identifies stained cells and reports the intensity and distribution of fluorescence in individual cells. Automatic focusing, image acquisition and analysis were performed to collect data on a user-defined number of cells. Images and data regarding intensity and texture of the fluorescence within the individual cells, as well as the average fluorescence of the cell population within a well were stored in a database for easy retrieval and analysis.
Measurement of cell metabolic activity
The Alamar Blue assay was used essentially according recommendations from the supplier (Serotec Ltd, Oxford, UK). Briefly, the cells were adjusted to 2 × 105 cells/mL in RPMI 1640 medium containing 10% FCS and 100 μg/mL kanamycin and 2 mM l-glutamine (GibcoBRL, Life Technologies AB, Täby, Sweden) and 100 μl/well cells cultured in 96-well plates (Nunc, Roskilde, Denmark). Then 100 μl/well of various QS formulations or medium alone (as control) was added in triplicates. Alamar Blue was added to a final concentration of 10% to the wells after specified time points. The cell metabolic activity was subsequently measured for up to eight hours. The OD values were read at 570 and 600 nm in a spectrophotometer (Labsystems Multiskan RC, type 351). Metabolic activity is expressed as percent of the cell control (survival index; SI) and IC50 is defined as the concentrations of the QS formulations resulting in 50% metabolic inhibition compared to control.
Thymidine kinase (TK) activity
The TK activity was determined with a kit obtained from Biovica (Uppsala, Sweden). Briefly 100 μl cell suspension at a concentration of 0.1–1 × 106 cells/mL was transferred to Eppendorf tubes and centrifuged at 200 g for 10 minutes. The cell pellet was re-suspended in 100 μl cold PBS and freeze/ thawed 2–3 times. After centrifugation at maximum speed for five minutes, the supernatant was collected. Ten μl of the samples were used according to the manufacturer’s protocol. The extracellular, ie, released TK activity was measured as above directly on the cell culture medium.
Cell cycle analysis
To investigate the effect of 0.25 and 1.00 μg/mL KGI and the effect of 10 and 100 μg/mL BBE on the cell cycle progression of tumor cells the lymphoma cell line, U937-GTB, was used. The U937-GTB cells were seeded in 12-well plates at 1 × 106 cells/mL and exposed to the different concentrations of KGI and BBE for six and 24 hours. At the end of the exposure time, cells were collected, rinsed twice with PBS and fixed in 70% ice-cold ethanol. The cells were stored at a concentration of 2 × 105 cells/mL at −20 °C until analysis. The fixed cells were then centrifuged, the cell pellet was rinsed with PBS and suspended after centrifugation in 1 mL of 0.02 mg/mL PI staining solution with 0.2 mg/mL DNase-free RNase and kept for ≥2 hours. DNA staining was then detected using the LSR II flow cytometer. ModFit LT software was used to analyze the percentage of the cells in different cell cycle phases.
Interleukin-8 production
Interleukin-8 (IL-8) was used for measurement of cellular cytokine production as a marker of tumor cell differentiation after exposure to QS formulations. IL-8 was measured using a commercial ELISA kit from Endogen (Pierce Endogen, Rockford, IL, USA) according to the manufacturer’s instruction.
Results
Chemical and physical structures of particulate KGI and BBE
illustrates QS separated by reversed phase chromatography as described previously.Citation8,Citation9 ASAP (see , fraction B) is highly hydrophobic owing to its acyl-chain () and is the active component in the KGI particle distinguishing it from the BBE particle. The DSAP fraction A (, fraction A) lacking an acyl-chain has low hydrophobicity () and is the active component in the BBE particle. KGI and BBE particles have equal morphology forming highly stable spherical particles with an average diameter of about 40 nm ().
Lytic and cytotoxic effects by free and nanoparticulate forms of ASAP and DSAP
The cell membrane lytic effect of the ASAP and DSAP or their particulate forms the KGI and BBE was measured on RBCs. Free ASAP lysed RBCs at a concentration of 5 μg/mL () whereas the KGI particles induced RBC lysis at a 20-fold higher concentration, ie, 100 μg/mL. Free DSAP did not cause lytic effects at concentrations up to 50 μg/mL as previously observed.Citation10 The BBE particle did not lyse RBCs even at 100 μg/mL ().
Figure 2 Free non-particulate ASAP causes early cell destruction but not the particulate form. A) Cell destruction measured by hemolytic effect. KGI and BBE are virtually non-lytic while free ASAP is highly lytic. B) Free ASAP induces early cell death in human normal monocytes like in cancer cells, but not the ASAP formulated into KGI particles measured by Trypan Blue staining. C) Free ASAP induces early cell death in U937 tumor cells within minutes at the concentrations indicated. KGI does not induce this rapid cell death. D) Cell membrane destruction measured by the release of the intracellular enzyme thymidine kinase (TK) to the extracellular medium. Free ASAP causes TK release, while KGI does not cause the leakage. E) Free ASAP causes cell necrosis but not KGI particles.
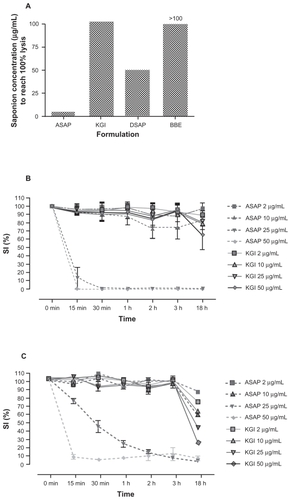
The cell membrane lytic effect of the free ASAP or the particulate form of the ASAP (KGI) was then measured in normal human monocytes (see Materials and methods) () and in the monoblast U937 tumor cells () after Trypan Blue staining. At 50 μg/mL, free ASAP induced cell death in both the normal monocytes and the U937 tumor cells within 30 minutes. At 25 μg/mL, free ASAP killed all the normal monocytes within 30 minutes compared to three hours for U937 tumor cells. Also 10 μg/mL of free ASAP killed a proportion of the normal monocytes after two to three hours. In contrast, the KGI particles did not induce cell death in the normal monocytes at any concentration tested during the first three hours but cell death was only apparent after 18–24 hours. KGI at all concentrations was more active against tumor cells compared to normal cells at 18 hours ().
Rapid leakage of the enzyme TK into the medium was observed from cells exposed to free ASAP, but not with KGI, corroborated the hemolytic activity results (). Necrotic changes, measured using PI staining, were noted within six and 12 hours in cells exposed to 25 μg/mL free ASAP (). The results indicate that the saponin in nanoparticulate form does not cause lytic reactions, cell membrane damage, and subsequent necrotic cell death.
KGI particles selectively kill monoblast U937 lymphoma cells
We selected the U937 cells to investigate the effects of KGI and BBE particles because of our background knowledge of tumor and normal cells of similar origin, ie, monocyte-derived immature dendritic cells (DCs). We compared the killing effects by KGI and BBE on cancer cells with that on the monocyte derived normal DCs. KGI killed U937 cells at a 30-fold lower concentration than was required to kill the normal DCs (). The effect on U937 cells was not reversible since the removal of the drug after three days did not allow cells to recover during the 12 days of culture. KGI at 2 μg/mL for three days reduced the cell number to 50% and eventually killed almost all tumor cells after 12 days of culture (), even 0.5 μg/mL of KGI caused continues repression of cancer cell growth during the 12 days of culture (data not shown). BBE did not induce cell death either in U937 or in normal DCs (). When a fixed concentration of 77 μg/mL KGI was mixed with increased concentrations of BBE, the killing effect of KGI was blocked in a concentration dependent manner. At a BBE:KGI ratio of 10:1, close to 100% blocking was observed ().
Figure 3 KGI kills preferentially cancer cells over normal cells with no recovery of residual cells while BBE blocks the killing effect of KGI. A) KGI induces cell death in U937 monoblast cancer cells at about 30-fold lower concentration than that required to kill normal monocytes derived immature DCs (mDC) measured by Alamar Blue method and expressed as IC50. B) The U937 cancer cells were synchronized in cell cycle and then cultured with 2 μg/mL of KGI for the first three days. The viable cells were counted at indicated time points. Even after a prolonged culture for 12 days there was no residual cancer cells treated with KGI reverting to replication. C) U937 cells were exposed to a fixed concentration of KGI (77 μg/mL) and increasing concentrations of BBE (X axis). As shown, close to 100% blocking by BBE was achieved when the ratio between BBE and KGI approaching 10 to 1.
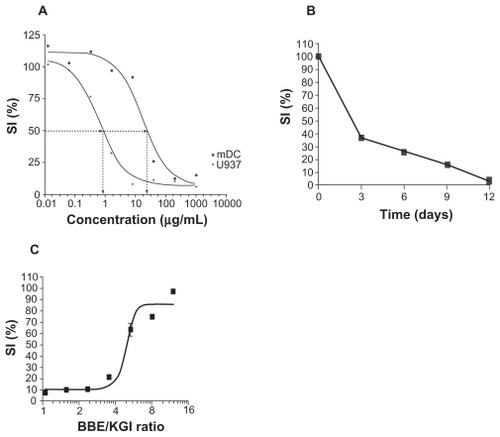
Table 2 Cytotoxic effect of KGI and BBE on different leukemia cell lines and normal cells measured after exposure for 24 hours expressed as IC50 (μg/mL)
KGI kills cancer cells by apoptosis
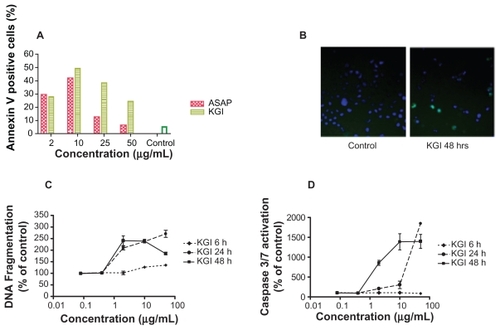
KGI dose-dependently induced apoptosis in U937 cells with a peak after 24 hours of exposure as judged by annexin V staining, depicts one representative of three experiments showing the same profile. At 24 and 48 hours an increase in DNA fragmentation as well as caspase 3/7 activity () was observed in the presence of intact plasma membranes, which is a hallmark of apoptosis.
Figure 4 KGI particles induce apoptosis in U937 cells demonstrated in various assays. A) Induction of apoptosis by free ASAP and KGI particles measured by Annexin V staining. B) Photomicrograph of control and KGI treated cells stained with apoptag and Hoechst 33342. C) Effect of KGI on DNA fragmentation. D) Caspase 3/7 activity using Arrayscan II as described in Material and Methods.
KGI and cell cycle
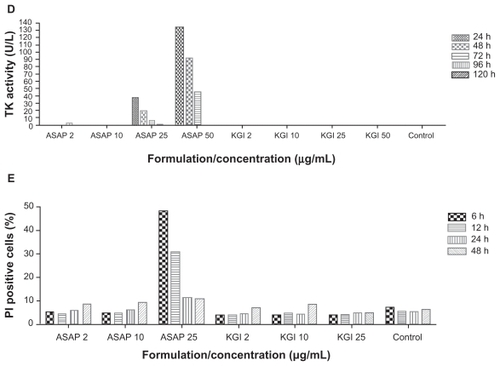
The effect of KGI on the cell cycle in U937 cells was analyzed by assessment of the TK-activity over time. The TK activity was correlated with the inhibition of cell-metabolism (recorded by the Alamar Blue assay), induction of cell death (measured by Trypan Blue exclusion staining) and also with apoptosis (detected by annexin V staining). In nonsynchronized cells, KGI at 2 μg/mL caused a marked reduction of TK activity after exposure of the tumor cells for 48–72 hours (). Reduction of the TK activity after exposure to 25 or 50 μg/mL of KGI was already evident at 24 hours. demonstrates that tumor cells synchronized in the cell cycle exposed to 2 μg/mL of KGI for 24 hours had reduced intracellular levels of TK compared to control cells coinciding with reduced number of viable cells () indicating that the KGI-treated cells did not enter a second cycle. Corroborating these results, cell cycle analysis with flow cytometry of KGI-exposed DNA-stained cells demonstrated accumulation of cells in G1 phase in parallel with depletion of cells in S-phase ().
Figure 5 ASAP in free form and formulated into KGI inhibit thymidine kinase (TK) activity. The decreased level of TK caused by KGI correlates with reduced cell growth. Both KGI and BBE can induce cytokine production but only KGI kills the cells. A) Very low or no intracellular levels of TK activity was recorded in U937 cancer cells treated with 25 and 50 μg/mL of free ASAP and KGI respectively. B) The intracellular levels of TK decreased close to 50% compared to control cells after exposure of cells synchronized in the cell cycle to KGI particles for 24 hrs. C) The number of viable cells synchronized in the cell cycle was reduced to ¼ of that of control cells after exposure to KGI for 24 hrs. D) KGI stimulates U937 cancer cells to produce IL-8 with a sharp increase at the concentrations of 0.5 to 1 μg/mL close to the IC50 of KGI while BBE stimulates production of IL-8 without killing the cells.
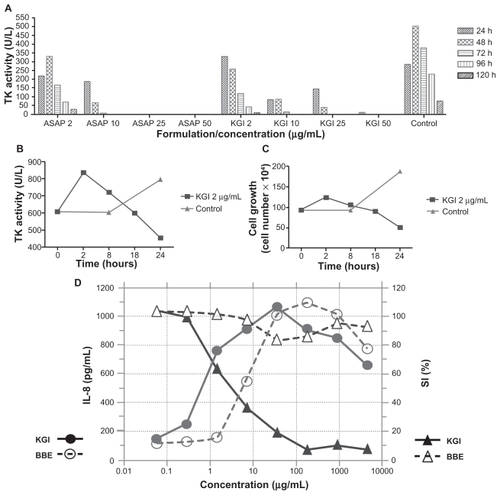
Table 1 KGI induces depletion of U937 cancer cells in the S-phase and accumulation in G1-phase of the cell cycle measured as proportion (%) of the total cell population
KGI and BBE trigger differentiation of U937 cells
The capacity of KGI and BBE to induce U937 cells to produce interleukin-8 (IL-8), as a marker for differentiation, was tested. The production of IL-8 was around 600 pg/mL at the IC50 for KGI (). BBE did not kill the cells at concentrations tested and at the same concentration as the IC50 for KGI, around 400 pg/mL of IL-8 was produced (). For KGI the effect on IL-8 production was paralleled by cytotoxicity whereas BBE was not cell toxic but induced cell differentiation ie, IL-8 production. KGI and BBE formulations could also activate immature human DCs to produce cytokines and to express the DC differentiation marker CD86 (data not shown).
Stand alone and synergistic effects of KGI and BBE on leukemic cell lines
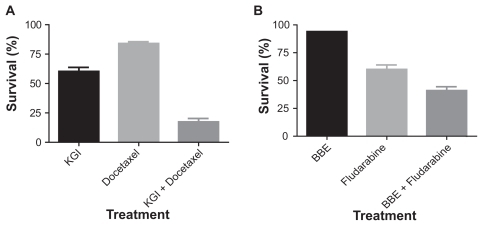
The cytotoxic effect of KGI and BBE was tested in several leukemia cell lines using the FMCA and Alamar Blue assays (). The results demonstrated that several other leukemic cell lines showed high sensitivity to KGI including the vincristine resistant subline U937-Vcr, acute lymphocytic leukemia CEM cells and acute myelocytic leukemia Mv-411 and THP-1 cells. In contrast, acute lymphocytic leukemia Jurkat- and etoposide-resistant CEM/R cells as well as the myeloma 8226 cell lines were less sensitive. None of the cell lines tested, apart from U937-Vcr, was sensitive to BBE. Moreover, as seen in , KGI and BBE exert strong synergistic effects with docetaxel and fludarabine on U937 cells, respectively.
Figure 6 The synergistic effect of KGI and BBE with other anticancer drugs on U937 cells A) KGI (10 μg/mL) exerts a strong synergistic effect with Docetaxel (0.0016 μM) and B) BBE (10 μg/mL) with Fludarabine (2 μM).
Discussion
Free nonparticulate saponins of various origin including QS fractions exert cytotoxic activity on cancer cells.Citation7 However, QS interacts with cholesterol present in the cell membranes on both normal and cancer cells, thereby trapping the free non-particulate saponin at the site of administration,Citation10 causing membrane damage, cell lysis, and subsequent necrosis resulting in a low therapeutic index. Possibly the free saponin binds more efficiently to cancer cells containing more cholesterol than normal cell, but that is not enough to give adequate therapeutic index to be of any clinical value.Citation21,Citation22
By formulating ASAP and DSAP with cholesterol into KGI and BBE nanoparticles respectively, the cell lytic toxicity is abolished. To note, no release of free ASAP or DSAP from the particles is recorded and the mode of action is connected to the particulate form. In contrast the concept for most particles intended for cancer treatment, eg, various polymer particles including PLG particles is to gradually release the free active components.Citation11,Citation13
The cancer cell-killing effect of ASAPCitation11,Citation13 is related to toxicity manifested by hemolysis, cell membrane damage causing leakage of TK, decreased cell metabolism, and necrosis. The release of intracellular compounds by cell membrane damage including both normal and cancer cells causes even severe toxic effects in vivo.Citation23 Thus, the therapeutic effect of free QA like ASAP is limited and out of the question for cancer treatment. Notably, free ASAP at 25 μg/mL induced a more rapid cell death of normal cells, ie, at 30 minutes vs one hour for U937 cells.
ASAP in the KGI particulate form caused U937 cancer cell death at low concentrations; 30-fold lower than what was required to kill the normal human monocytes or DCs, ie, a high therapeutic index was achieved. The high therapeutic index is explained by several factors. Firstly, fast replicating cancer cells are generally more sensitive than normal cells being the concept for cytostatic drugs. Secondly, by taking the cancer cells out of the cell cycle the essential recruitment/replacement of sensitive cancer cells is abolished. Thirdly, by guiding the cancer cells to differentiation manifested by, eg, IL-8, leads eventually to programmed cell death. All together, those are factors that result in the evasion of side effects. The different modes of retarding the cancer cell growth by KGI and BBE from that of cytostatic drugs predispose for synergistic effects as demonstrated between docetaxel/KGI and fludarabine/BBE (). KGI has in vivo studies shown high bioavailability after fast transport from the site of injection. In contrast the free form, being lytic, interacts with cell membranes causing local reactions and reduced bioavailability. KGI caused a late cell death by apoptosis requiring 12 hours or more to be observed. No reversal to cell replication was noticed during 12 days of culture even when KGI was removed after three days. The lytic effect of ASAP on red blood cells and nucleated cells was virtually abolished when formulated to KGI particles, and the cell death was confined to apoptosis. The apoptotic effect of KGI was preceded by exit from the cell cycle, G1 arrest (as shown in ) followed by activation and differentiation observed as production of IL-8. KGI as well as BBE cause maturation effects on immature normal human dendritic cells with downregulation of CD14, production of a number of pro-inflammatory cytokines, eg, IL-12, and expression of CD80, CD83, and CD86 (data not shown), which reinforces the concept that KGI and BBE stimulate cancer cells to apoptosis like they do with normal cells by cell differentiation rather than a cytostatic effect.
Different methods were used to measure apoptosis (annexin V staining, apoptag, and Hoechst 33342 staining, DNA fragmentation and caspase 3/7 activation). All methods clearly demonstrate that KGI induces apoptosis. The annexin V staining reveals that KGI induces apoptosis and no necrosis is observed in the early phase. Over time the number of necrotic cells increases as shown with PI double staining with annexin V. The explanation is that apoptotic cells in vitro over time undergo necrosis while in vivo phagocytosed. Higher concentrations of KGI induced apoptosis earlier than lower concentrations. Apoptotic cells are prone to necrosis and become, therefore, stained by PI over time (double stained with both PI and annexin V), leading to reduction of the proportion of annexin V single positive cells (). In addition, the killing activity of KGI was not confined to U937 cells but was also observed on other leukemia cell lines as reflected in .
DSAP formulated into BBE particles had no cytotoxic effect on the U937 cells, but blocked the cancer cell killing effect of KGI in a concentration dependent manner, approaching complete blocking in a BBE: KGI ratio of 10 to 1. This blocking indicates that BBE and KGI compete for one receptor and that KGI induced apoptosis probably requires a second receptor not targeted by DSAP and possibly related to the acyl-chain involving the terminal sugars arabinos and/or rhamnos. The indicated receptor activity has to be further elucidated in future studies.
KGI and BBE particles show high bioavailability and are virtually nontoxic, not causing local or systemic adverse effects in laboratory animals.Citation11 A product similar to the KGI particle, ie, the ISCOM particle has been tested in more than 1000 humans as an adjuvant for vaccines and is in human phase III clinical trials for this indication.Citation11 Thus, the combination of low toxicity, very high bioavailability, and high stability already observed in preclinical studies and in humans (Dr Karin Lövgren-Bengtsson, Isconova AB) indicates a favorable therapeutic index and the possibility of a rapid advancement to clinical testing in cancer.
In addition to the potential use of nanoparticulate QS as stand alone anticancer drug, there are prospects for synergy between KGI and BBE as well as a number of registered cancer drugs eg, docetaxel and fludarabine (). Besides direct pharmacodynamic interactions at the level of the tumor cell, the well documented adjuvant immunological effect of the QS particlesCitation11 might also augment the overall anticancer effect. Furthermore the KGI and BBE nanoparticles should also be explored as potential carriers for established anticancer drugs as they can be incorporated into the particles by various techniquesCitation24 as being successfully used commercially for delivery of vaccine antigens.Citation11
There are other potential clinical applications of the particulate saponins. The nanoparticles may be targeted to the tumor cells by inserting tumor-targeting molecules, potentially increasing the selectivity of drug delivery, hence increasing the therapeutic index. This principle is illustrated by incorporating envelope proteins from respiratory viruses into particles, rendering enhanced delivery to the common mucosal immune system after intranasal mode of administration as described by Hu and colleagues.Citation13,Citation25 ISCOMs supplied with the DD portion of protein A of Staphylococcus aureus are targeting B cells.Citation11 KGI supplied with the DD portion should target B cell lymphoma. The use of monoclonal anti-bodies attached to the particles recognizing specific surface structures on tumor cells is a device for targeting.
In conclusion, QS saponin formulated into nanoparticles represents a potential new mechanistic category of anticancer drugs fundamentally different from standard cytotoxic drugs by being considerably more selective and also by stimulating differentiation. Furthermore, as drug carriers, the particles with inborn cancer cell-killing and immune-stimulating properties create interesting prospects for synergism with integrated standard anticancer compounds.
Acknowledgments/disclosures
We are grateful to Prof. Jonas Blomberg, Department of Medical Sciences, Clinical Virology for providing us with the laboratory space. We also thank Dr Karin Lövgren, Isconova AB and Dr Simon Gronowitz, Biovica AB for laboratory assistance and valuable discussions. We also thank Einar Wanhainen, Duecom AB for his strong interest in the project and generous economic support. The authors report no conflicts or competing financial interests in this work.
References
- NygrenPLarssonROverview of the clinical efficacy of investigational anticancer drugsJ Intern Med20032531467512588538
- KerwinSMSoy saponins and the anticancer effects of soybeans and soy-based foodsCurr Med Chem Anticancer Agents20044326327215134504
- RaoAVSungMKSaponins as anticarcinogensJ Nutr19951253 Suppl717S724S7884557
- TianFZhangXTongYPE, a new sulfated saponin from sea cucumber, exhibits anti-angiogenic and anti-tumor activities in vitro andin vivoCancer Biol Ther20054887488216082187
- TinMMChoCHChanKJamesAEKoJKAstragalus saponins induce growth inhibition and apoptosis in human colon cancer cells and tumor xenograftCarcinogenesis20072861347135517148504
- HwangSJChaJYParkSGDiol- and triol-type ginseng saponins potentiate the apoptosis of NIH3T3 cells exposed to methyl methanesulfonateToxicol Appl Pharmacol2002181319220212079428
- WangZPSaponins as Anticancer Agent United States Patent 20050175623 A12005
- KensilCRPatelULennickMMarcianiDSeparation and characterization of saponins with adjuvant activity from Quillaja saponaria Molina cortexJ Immunol199114624314371987271
- KerstenGFSpiekstraABeuveryECCrommelinDJOn the structure of immune-stimulating saponin-lipid complexes (iscoms)Biochim Biophys Acta1991106221651712004105
- RonnbergBFekaduMMoreinBAdjuvant activity of non-toxic Quillaja saponaria Molina components for use in ISCOM matrixVaccine19951314137513828585296
- MoreinBHuKLovgrenKD’HondtESinghMANew ISCOMs meet unsettled vaccine demandsVaccine Adjuvants and Delivery SystemsHoboken, NJJohn Wiley & Sons, Inc2007191222
- LovgrenKMoreinBTheISCOMan antigen delivery system with built-in adjuvantMol Immunol19912832852862017197
- LovgrenKMoreinBThe ISCOM™ Technology. Vaccin adjuvants: Preparation methods and research protocolsMethods Mol Med200042239258
- MoreinBSundquistBHoglundSDalsgaardKOsterhausAISCOM, a novel structure for antigenic presentation of membrane proteins from enveloped virusesNature198430859584574606709052
- DaltonWDurieBAlbertsDGerlachJCressACharacterization of a new drug-resistant human myeloma cell line that expresses p-glycoproteinCancer Res198646512551302875788
- BellamyWDaltonWGleasonMGroganTTrentJDevelopment and characterization of a melphalan-resistant human multiple myeloma cell lineCancer Res19915199510021988143
- BotlingJLimingaGLarssonRNygrenPNilssonKDevelopment of vincristine resistance and increased sensitivity to cyclosporin A and verapamil in the human U-937 lymphoma cell line without overexpression of the 170 kDa p-glycoproteinInt J Cancer1994582692747913083
- DanksMSchmidtCCirtainMSuttleDBeckWAltered catalytic activity of and DNA cleavage by DNA topoisomerase II from human leukemic cells selected for resistance to VM-26Biochemistry198827886188692853972
- LarssonRKristensenJSandbergCNygrenPLaboratory determination of chemotherapeutic drug resistance in tumor cells from patients with leukemia using a fluorometric microculture cytotoxicity assay (FMCA)Int J Cancer1992501771851730510
- LovborgHNygrenPLarssonRMultiparametric evaluation of apoptosis: effects of standard cytotoxic agents and the cyanoguanidine CHS 828Mol Cancer Ther20043552152615141009
- HilfRGoldenbergHMichelIOrlandoRAArcherFLEnzymes, nucleic acids, and lipids in human breast cancer and normal breast tissueCancer Res1970306187418824318708
- PerkinsRGKummerowFAMajor lipid classes in plasma membrane isolated from liver of rats fed a hepatocarcinogenBiochim Biophys Acta197642434694801259971
- DumitriuIEBaruahPManfrediAABianchiMERovere-QueriniPHMGB1: guiding immunity from withinTrends Immunol200526738138715978523
- WikmanMFriedmanMPinitkiatisakulSGeneral strategies for efficient adjuvant incorporation of recombinant subunit immunogensVaccine200523171823312335
- HuKFLovgren-BengtssonKMoreinBImmunostimulating complexes (ISCOMs) for nasal vaccinationAdv Drug Deliv Rev2001511314915911516775