
Abstract
The objective of the present study was to demonstrate that a novel hydroxypropyl-β-cyclodextrin functionalized calcium carbonate (HP-β-CD/CC) based amorphous solid dispersion (ASD) can be used to increase the solubility and oral bioavailability of water-insoluble drugs. Irbesartan (IRB) was selected as a model compound and loaded into the nanoporous HP-β-CD/CC matrix using an immersion method. The IRB-loaded HP-β-CD/CC formulation was characterized by various analytical techniques, such as specific surface area analysis, scanning electron microscopy (SEM), dynamic light scattering (DLS), powder X-ray diffraction (PXRD), and differential scanning calorimetry (DSC). Analyses with PXRD and DSC confirmed that IRB was fully converted into the amorphous form in the nanopores of HP-β-CD/CC. From the solubility and dissolution tests, it was observed that the aqueous solubility and dissolution rate of IRB-loaded HP-β-CD/CC were increased significantly compared with those of pure IRB and IRB-loaded mesoporous silica. Likewise, the IRB-loaded HP-β-CD/CC formulation exhibited better absorption compared with that of the commercially available IRB capsules in beagle dogs. The mean peak plasma concentration (Cmax) and the area under the mean plasma concentration–time curve (AUC[0→48]) of IRB-loaded HP-β-CD/CC were 1.56- and 1.52-fold higher than that of the commercial product, respectively. Furthermore, the IRB-loaded HP-β-CD/CC formulation exhibited excellent stability against re-crystallization. These results clearly demonstrate that HP-β-CD/CC based porous ASD is a promising formulation approach to improve the aqueous solubility and the in vivo absorption performance of a water-insoluble compound like IRB.
Introduction
It is estimated that more than two-thirds of the compounds currently emerging from the drug discovery pipeline have a solubility <100 μg/mL and thus, according to the definition of the United States Pharmacopeia, they may be considered poorly soluble or insoluble in aqueous media.Citation1,Citation2 The poor aqueous solubility of an active pharmaceutical ingredient (API) presents a major challenge for the drug development industry when developing dosage forms for API belonging to biopharmaceutical classification system (BCS) classes II (poor solubility and high permeability) and IV (poor solubility and poor permeability).Citation3–Citation5 Water-insoluble drugs exhibit low oral bioavailability and are ineffective therapeutically due to their low dissolution profile in physiological fluids.Citation6,Citation7
In recent years, there has been considerable interest in developing mesoporous inorganic materials as prospective oral drug carriers to improve the aqueous solubility and dissolution properties of hydrophobic drugs.Citation8,Citation9 Mesoporous inorganic materials offer several advantages for drug delivery, including a large inner surface area and pore volume to highly disperse the drug molecules, a high drug loading capacity, excellent thermal stability, and chemical inertness.Citation9,Citation10 After adsorption to the nanopores of inorganic carriers, the drug remains entrapped in an amorphous state possessing much higher internal energy relative to the crystalline form and leading to enhanced in vitro dissolution rate.Citation11,Citation12 Furthermore, hydrophobic drugs loaded in hydrophilic porous carriers can improve wettability and increase porosity.Citation13 It has been found that several types of mesoporous silica nanoparticles are able to improve the aqueous solubility and dissolution properties of poorly water-soluble drugs. However, the mesoporous silica industry struggles with high manufacturing costs due to expensive surfactants and silica sources used in the fabrication, as well as environmental concerns related to the often toxic surfactants needed as pore-forming templates. Similar to mesoporous silica, mesoporous calcium carbonate (CaCO3) is also a promising oral drug carrier due to its porous structure, relatively high surface area, ability to protect encapsulated components, excellent biocompatibility, and biodegradability.Citation14–Citation16 In addition, chemical decomposition of calcium carbonate is fast under acidic conditions in the stomach by the liberation of carbon dioxide. Furthermore, the synthesis of mesoporous calcium carbonate is cost-effective and does not need the use of any toxic surfactants. However, compared with other inorganic drug carriers, mesoporous calcium carbonate has been less studied. More recently, a novel hybrid β-cyclodextrin/calcium carbonate (β-CD/CC) matrix has been fabricated by combining calcium chloride and sodium carbonate in the presence of β-CD.Citation17 As is well-known, β-CD has been mainly used as a complexing agent to increase the solubility, stability, and bioavailability of water-insoluble drugs. Combining these unique properties of β-CD with mesoporous calcium carbonate, a hybrid β-CD/CC matrix has potential applications in the pharmaceutical and biomedical areas. However, to the best of our knowledge, no study has explored its potential in improving the solubility, dissolution, and bioavailability of water-insoluble drugs following oral administration.
Herein we report, for the first time, the application of a uniform hydroxypropyl-β-cyclodextrin functionalized calcium carbonate (HP-β-CD/CC) matrix as a drug delivery vehicle. Irbesartan (IRB), an angiotensin II receptor blocker, was chosen as a model drug as it is a lipophilic compound with negligible water solubility and belongs to BCS class II drugs.Citation18,Citation19 Therefore, improving the solubility and dissolution rate of such a drug is expected to enhance its bioavailability and, hence, its therapeutic potency. In this work, specific surface area analysis, SEM, PXRD, DSC, in vitro solubility, and dissolution tests were conducted to characterize the IRB-loaded HP-β-CD/CC formulation. For comparison, IRB loaded into the conventional mesoporous silica matrix was also investigated. Furthermore, the in vivo oral absorption of IRB from the IRB-loaded HP-β-CD/CC formulation was evaluated using beagle dogs. In addition, the cytotoxicity of HP-β-CD/CC on HT-29 human colon carcinoma cells was also evaluated. We believe that the new information produced from our research will help promote the use of hybrid HP-β-CD and mesoporous calcium carbonate materials in pharmaceutical applications.
Materials and methods
Materials
HP-β-CD (C63H112O42) was obtained from Sinopharm (Beijing, People’s Republic of China). Poloxamer 407 (EO106PO70EO106) was generously provided by BASF (Ludwigshafen, Germany). Crude IRB (C25H28N6O, purity more than 99%) was provided by Huahai Pharma Ltd., (Zhejiang, People’s Republic of China) and used as-received. High performance liquid chromatography (HPLC)-grade acetonitrile (C2H3N) and methanol (CH4O) were purchased from Concord (Tianjin, People’s Republic of China). PrestoBlue® was purchased from Life Technologies (Carlsbad, CA, USA). Dulbecco’s Modified Eagle’s Medium (DMEM) was purchased from Solarbio (Beijing, People’s Republic of China). Purified water was used throughout this study. All the other reagents and chemicals used were of analytical grade or better.
Preparation of drug-loaded microparticles
Synthesis of nanoporous HP-β-CD/CC microparticles
Nanoporous HP-β-CD/CC microparticles were synthesized using the co-precipitation method.Citation17 First, 3.6 g calcium chloride (CaCl2) and 1.2 g HP-β-CD were dissolved in 100 mL purified water with constant stirring. Next, the calcium chloride solution obtained was transferred to a water bath set at a temperature of 20°C and, after 15 minutes, a sodium carbonate (Na2CO3) solution (100 mL, 0.33 M) was added to the calcium chloride solution under vigorous magnetic agitation (600 rpm). The co-precipitation reaction was ceased after 15 minutes and the mixture was centrifuged in Eppendorf tubes at 6,000× g for 5 minutes to recover the formed particles. Finally, the resulting particles were washed thoroughly with ethanol, followed by drying at 60°C. The obtained powder was designated as HP-β-CD/CC.
Synthesis of mesoporous silica microparticles
For comparison, a classic mesoporous silica matrice, Santa Barbara amorphous materials (SBA)-16, was synthesized using a procedure similar to that previously published.Citation20 Typically, 2.5 g Poloxamer 407 was dissolved with stirring in 180 mL HCl solution (1.6 M). After a homogeneous solution was obtained, 9.0 mL tetramethyl orthosilicate (C4H12O4Si) was slowly added to the surfactant solution. The mixture was stirred for 20 hours at 35°C. Next, the resulting mixture was treated hydrothermally at 80°C for 48 hours without stirring. Finally, the obtained precipitate was collected, washed with ethanol, and calcined at 600°C for 5 hours.
Preparation of IRB-loaded HP-β-CD/CC and IRB-loaded mesoporous silica
The model drug IRB was loaded into two different porous matrices using the immersion procedure, which infuses the drug into the nanopores by means of capillary forces. Briefly, 0.4 g IRB was dissolved in 20 mL ethanol solution. Next, 1.6 g porous matrices (HP-β-CD/CC and SBA-16, respectively) were immersed in 20 mL IRB solution and ultrasonicated for 15 minutes. After stirring for 4 hours, the mixture was transferred to a flask and the solvent was removed under reduced pressure in a rotary evaporator at 50°C. Finally, the dried IRB-loaded particles (labeled as IRB-HP-β-CD/CC and IRB-SBA-16, respectively) were micronized by grinding and passed through a 100-mesh sieve.
Particle characterization
SEM images of the prepared HP-β-CD/CC and SBA-16 samples were taken with S-4800 type microscope (Hitachi, Japan) in high-vacuum mode. Transmission electron microscopy (TEM) images of the particles were recorded on a Tecnai G2 microscope (FEI, the Netherlands) operated at an accelerating voltage of 200 kV. The particle size distribution was measured by DLS using a Zetasizer Nano sizer (Malvern, UK). Nitrogen sorption isotherms and pore characterization of the prepared samples were recorded at −196°C using an ASAP 2020 volumetric adsorption analyzer (Micromeritics, USA). The prepared HP-β-CD/CC and SBA-16 samples were degassed at 100°C for 6 hours prior to measurement, while the samples loaded with IRB and pure IRB powder were degassed at 40°C overnight in the degas port to avoid possible thermal degradation. The specific surface area was calculated by the standard Brunauer–Emmett–Teller (BET) model. The pore size distribution was determined according to the Barrett–Joyner–Halenda (BJH) method.
DSC measurement
Thermal response profiles of each sample were evaluated using a Q2000 differential scanning calorimeter (TA Instruments, Belgium). The instrument was calibrated with indium as a reference and maintained under an inert atmosphere with nitrogen purging. An accurately weighed sample (3–6 mg) was loaded into an aluminum crucible and crimped with a perforated lid. The crucible was then heated from 30°C to 250°C at 10°C/min.
PXRD measurement
The PXRD patterns of each sample were recorded on an AXS D8 Advance diffractometer (Bruker AXS GmbH, Germany) equipped with a Cu-Ka radiation point source. A sample approximately 0.5 mm thick was gently consolidated in an aluminum holder, placed in the diffractometer, and scanned at a current of 30 mA and a voltage of 40 kV. The scan range was 5°–40° (2θ), with a step size of 0.02° and a scan speed of 0.03° per second.
HPLC analysis
A Waters 2695 HPLC instrument, consisting of a 1525 binary pump and a 2487 dual wavelength absorbance detector (Waters Corporation, USA) was used to determine the concentration of IRB. The analysis was carried out using a Waters XBridge™ C18 150×4.6 mm, 5 μm. The mobile phase consisted of 0.02 M phosphate buffer and acetonitrile (60%/40%, v/v) and was pumped isocratically at a flow rate of 1.0 mL/min. A sample aliquot of 10 μL was injected and the effluent monitored at 245 nm.
Drug loading measurement
The drug content of the HP-β-CD/CC based IRB formulation was determined by dissolving an accurately weighed amount of IRB formulation (about 5 mg) in 100 mL methanol. These suspensions were ultrasonicated for 15 minutes and subsequently placed in a rotary mixer for 12 hours. Then, the porous matrix was separated from the IRB solution by centrifugation (4,000× g, 15 minutes). The supernatant layer was taken, suitably diluted with mobile phase, and the drug concentration was determined by HPLC. The drug loading was calculated as: weight of drug in samples ×100/weight of samples.
Solubility measurement
The aqueous solubilities of the raw drug and the drug-loaded in the formulations were determined in purified water, enzyme-free simulated gastric fluid (SGF, pH 1.2), and simulated intestinal fluid (SIF, pH 6.8) using the Higuchi and Connor method.Citation21 In brief, each IRB formulation equivalent to 20 mg IRB was added to different vials containing 20 mL solvent and vortexed for 2 minutes to obtain a homogenous mixture. All the mixtures were incubated in an HZQ-A shaking water bath (Weier, People’s Republic of China) operated at 50 rpm for 48 hours at 37°C. Samples were withdrawn at appropriate intervals and centrifuged at 6,000× g and 37°C for 10 minutes. The supernatant was passed through a 0.2 μm polyvinylidene fluoride membrane filter, and then diluted in the corresponding solvent prior to measuring the drug concentration by HPLC.
Dissolution study
The dissolution of each IRB formulation was tested according to the USP32-NF27 type II paddle method using a D-800 LS dissolution apparatus (Tianjin University Radio Factory, People’s Republic of China). The stirring speed was 75 rpm, and the temperature was maintained at 37°C±0.1°C. Enzyme-free SGF (pH 1.2) and SIF (pH 6.8) were used as different dissolution media. A fixed weight of crude IRB (75 mg) or each prepared sample containing an equivalent mass of drug was added to 900 mL dissolution medium. Subsequently, 5 mL samples were withdrawn from the dissolution vessel at defined time intervals (5, 10, 15, 20, 30, and 45 minutes) and passed through a 0.2 μm polyvinylidene fluoride membrane filter. The drug concentration of each withdrawn sample was quantified by HPLC.
Cell viability assays
HT-29 human colon carcinoma cells were obtained from the American Type Culture Collection. For the PrestoBlue assay, HT-29 cells were cultured in 96-well microplates (4,000 cells/well) with DMEM containing 10% fetal bovine serum, 100 U/mL penicillin, and 0.1 mg/mL streptomycin at 37°C in a 5% CO2/95% air atmosphere. After confluence took place, the cells were exposed to various concentrations of HP-β-CD/CC or SBA-16 (25, 50, 75, 100, 150, and 200 μg/mL prepared in DMEM medium without fetal bovine serum) for 48 hours. Control cells were cultured with DMEM alone. After exposure, the culture medium was replaced with 10% PrestoBlue solution and the cells were incubated for an additional 30 minutes. Finally, the absorbance of each well was measured at 570 nm with a reference wavelength of 600 nm using an enzyme-linked immunosorbent assay reader. The cell viability was calculated as: absorbance of samples ×100/absorbance of control.
Bioavailability study
Beagle dogs weighing between 10 and 14 kg were used to determine drug absorption following oral administration of the IRB-loaded HP-β-CD/CC formulation and the control formulation (Pulining®; Harbin Pharmaceutical Group Sixth Pharmaceutical, Harbin, People’s Republic of China). All animal experiments were approved by the animal ethics committee of Xuzhou Medical College and performed in accordance with the National Institute of Health and Nutrition Guidelines for the Care and Use of Laboratory Animals. Six dogs were divided into two groups, and a single-dose, randomized, crossover study was performed. A washout period of 1 week was allowed between two consecutive treatments. The dogs were randomly selected to receive a single dose of the preparations containing 150 mg IRB after a 12 hours fast. At designated time intervals, about 2 mL blood sample was obtained from each animal by venipuncture and then transferred to anticoagulant-coated tubes. Plasma was separated by centrifugation (1,600× g, 15 minutes) and then stored at −20°C prior to analysis.
IRB was extracted using a protein precipitation method. Briefly, 0.1 mL plasma was mixed with 0.5 mL methanol containing an internal standard (telmisartan, 0.1 μg/mL) in a 4 mL centrifuge tube. After vortex mixing (4 minutes) and centrifugation (1,600× g, 10 minutes) at 20°C, the supernatant was transferred to another tube and a 10 μL aliquot was injected into the HPLC system. The blood concentrations of IRB were determined using a HPLC method that was validated to ensure acceptable accuracy and precision (accuracy more than 94% with a relative standard deviation less than 9.0%). The chromatographic system (Shimadzu, Japan) consisted of a LC-10ATvp binary pump and a RF 535 fluorescence detector. Chromatographic separation was achieved on a 200×4.6 mm Shim-pack VP-ODS C18 column with a security guard column. The isocratic mobile phase, acetonitrile: 0.02 M phosphate buffer (50:50, v/v), was pumped at a flow rate of 1.0 mL/min. The column eluent was monitored using a fluorescence detector set at excitation and emission wavelengths of 250 nm and 370 nm, respectively. Under these chromatographic conditions, the retention time was 5.9 minutes for IRB and 7.5 minutes for telmisartan.
The pharmacokinetic parameters were calculated using standard non-compartmental methods with DAS software (version 2.1.1, Mathematical Pharmacology Professional Committee, People’s Republic of China). Standard pharmacokinetic parameters, including the area under the plasma concentration–time curve from time zero to time t hours (AUC0→t), the time to reach the maximum plasma concentration (Tmax), and the peak plasma concentration (Cmax), were determined. The relative bioavailability (Fr) was calculated as: AUC0→t (test) ×100/AUC0→t (reference).
Physical stability test
To assess the physical stability of the IRB-loaded HP-β-CD/CC formulation, an accelerated stability test was carried out. Each sample was put into a screw-capped plastic bottle and then maintained at 40°C and 75% relative humidity in a stability oven. Small aliquots of the formulation were obtained after designated incubation times (1, 2, 3, and 6 months), and the changes in the solid state and dissolution of the IRB formulation were monitored by DSC, PXRD, and dissolution test.
Results and discussion
The morphology and microstructures of the prepared HP-β-CD/CC microparticles and drug-loaded HP-β-CD/CC samples were observed by SEM. It can be seen from that the prepared HP-β-CD/CC powder consisted of many well-dispersed microspheres with a particle diameter of 3–6 μm. The surface of the HP-β-CD/CC microparticles appears porous and looks similar to that of the reported β-CD/CC microparticles.Citation17 Remarkably, a high-magnification image in inset shows that some of the nanometer-sized primary particles with different orientations overlap each other, confirming the formation of the 3D interconnected network-like structure during the co-precipitation process. In addition, TEM image () also demonstrated that the HP-β-CD/CC matrix is composed of numerous nanoparticles (about 50 nm). Based on SEM observations, the morphology of raw IRB was plate-shaped with a length ranging from 10 to 50 μm (). In addition, it can be seen that the SBA-16 sample was made up of nearly spherical microparticles with a uniform particle size of 1–2 μm (). The corresponding TEM image, as shown in , clearly showed their ordered mesostructure. Interestingly, the morphology of the drug-loaded HP-β-CD/CC particles was the same as that of the pure HP-β-CD/CC matrix, while the outer surface of the particles (likely coated with a layer of drug) became even compared with the pure matrix (). The SEM micrographs of drug-loaded HP-β-CD/CC indicate that most of IRB had been highly dispersed and absorbed in the nanopores of HP-β-CD/CC or adsorbed uniformly on the outer surface of HP-β-CD/CC, as confirmed by nitrogen physisorption analysis. The nitrogen adsorption/desorption isotherms of the prepared HP-β-CD/CC and SBA-16 materials are shown in Supporting information . According to the International Union of Pure and Applied Chemistry (IUPAC), the HP-β-CD/CC samples are identified as type IV isotherms with a H3 hysteresis loop deriving from nanoparticle aggregates with slit-shaped pores. In contrast, SBA-16 exhibit type IV isotherms and a broad H2 hysteresis loop, which are typical for 3D ordered mesoporous materials with cage-type pores.Citation22 The textural properties of both samples before and after drug loading, including the BET specific surface area (SBET), total pore volume (Vt), pore diameter (WBJH), and degree of drug loading, are listed in . It can be seen that the HP-β-CD/CC matrix has a high SBET and Vt, but lower than those of SBA-16 probably due to the lack of small mesopores in the HP-β-CD/CC matrix. It is well known that both micropores and mesopores with sizes of less than 10 nm can help to increase the SBET of the porous matrices.Citation9,Citation22 The study of the change in the SBET, WBJH, and Vt of the prepared porous matrices before and after loading provided important information about the space occupied by the drug molecules. After drug loading, a decrease in SBET, WBJH, and Vt was observed and these changes were attributed to the successful incorporation of IRB into the cavities of each matrix. On the other hand, it is evident that IRB-HP-β-CD/CC has a larger WBJH compared with IRB-SBA-16. As is well-known, a large WBJH might be contributed to the fast release of loading drugs owing to faster diffusional transport, better accessibility, and reduced restriction of drug molecules out of the pore channels.Citation23,Citation24
Table 1 Specific surface area, pore volume, pore diameter, particle size, and drug loading of the samples (n=3)
Figure 1 SEM photographs of (A) and (B) HP-β-CD/CC, (C) crude IRB, and (D) IRB-HP-β-CD/CC.
Notes: (B inset) primary particles of HP-β-CD/CC, and (D inset) high-magnification of IRB-HP-β-CD/CC.
Abbreviations: HP-β-CD/CC, hydroxypropyl-β-cyclodextrin functionalized calcium carbonate; IRB-HP-β-CD/CC, irbesartan loaded-HP-β-CD/CC; SEM, scan ning electron microscopy.

Figure 2 TEM photographs of (A) HP-β-CD/CC and (C) SBA-16; SEM photographs of (B) SBA-16 and (D) IRB-SBA-16.
Note: (A inset) primary particles of HP-β-CD/CC.
Abbreviations: HP-β-CD/CC, hydroxypropyl-β-cyclodextrin functionalized calcium carbonate; SBA-16, Santa Barbara amorphous materials-16; IRB-SBA-16, irbesartan loaded-SBA-16; TEM, transmission electron microscopy.

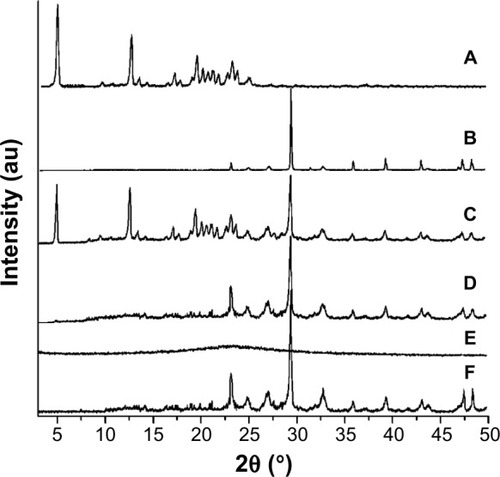
In order to verify the final solid state of the drug, the diffraction pattern of crude IRB, the physical mixture (PM, mass ratio of IRB: HP-β-CD/CC was 1:4), and IRB-loaded samples are compared in . It can be seen that crude IRB exhibits characteristic peaks at 4.7°, 12.6°, 19.5°, and 23.2° (), whereas HP-β-CD/CC shows a prominent peak at 29.4° (). In the case of the PM (), the characteristic peaks of IRB were retained, suggesting that IRB remained in a crystalline state. Unlike the PM, no crystalline IRB was detected in the IRB-HP-β-CD/CC and IRB-SBA-16 samples (). From the PXRD measurements, it can be concluded that the encapsulated IRB was molecularly well dispersed in both porous matrices, and amorphous IRB was formed in the solid dispersions.Citation13,Citation23,Citation25 This is related to finite-size effects, preventing the drug molecules from rearranging themselves in a crystal lattice.Citation26 In addition, it was found that no characteristic peaks assigned to the crystalline form of IRB could be detected in the IRB-HP-β-CD/CC samples after the storage tests for 6 months (). The PXRD results indicated that IRB-HP-β-CD/CC exhibited excellent stability against re-crystallization.
Figure 3 PXRD patterns of (A) crude IRB, (B) HP-β-CD/CC, (C) PM, (D) IRB-HP-β-CD/CC, (E) IRB-SBA-16, and (F) IRB-HP-β-CD/CC after 6 months of storage.
Abbreviations: HP-β-CD/CC, hydroxypropyl-β-cyclodextrin functionalized calcium carbonate; IRB-HP-β-CD/CC, irbesartan loaded-HP-β-CD/CC; SBA-16, Santa Barbara amorphous materials-16; IRB-SBA-16, irbesartan loaded-SBA-16; PXRD, powder X-ray diffraction; PM, physical mixture.
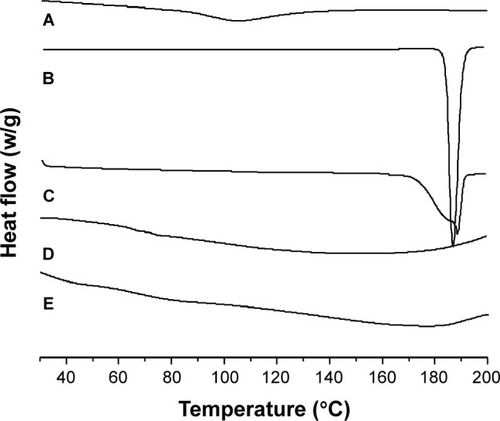
shows the DSC thermograms of HP-β-CD/CC, crude IRB, the PM, and IRB-loaded samples. HP-β-CD/CC showed a broad endothermic peak over a temperature range of 90°C–120°C, which corresponded to the loss of water molecules at around 100°C (). The crude IRB exhibited a sharp endothermic peak at 187.5°C () which was the melting point of IRB. A similar endothermic peak was also detected in the DSC curve of the PM (), suggesting that the drug was present in an unchanged crystalline state in the PM. In contrast, no such endothermic peak was observed for the IRB-HP-β-CD/CC samples before and after storage (). For the IRB-SBA-16 samples, the endothermic peak also disappeared (not shown here). The absence of an endothermic peak in the DSC thermograms of both IRB-loaded samples clearly indicates the amorphous state of the IRB entrapped in both carriers.
Figure 4 DSC thermograms of (A) HP-β-CD/CC, (B) crude IRB, (C) PM, (D) IRB-HP-β-CD/CC and (E) HP-β-CD/CC after 6 months of storage.
Abbreviations: HP-β-CD/CC, hydroxypropyl-β-cyclodextrin functionalized calcium carbonate; IRB-HP-β-CD/CC, irbesartan loaded-HP-β-CD/CC; PM, physical mixture; DSC, differential scanning calorimetry.
As listed in , the experimental data indicated that the crude IRB possesses a very low solubility in purified water (9.2±1.6 μg/mL). However, both IRB-loaded samples showed an increase in solubility. The saturated solubility of IRB for IRB-HP-β-CD/CC and IRB-SBA-16 was 56.4±3.3 and 72.1±3.8 μg/mL, respectively. An increase in the solubility of the model drug may be largely attributed to the changes in the solid state and particle size of IRB. The nano-scale pore channels of the porous matrices reduced the particle size of IRB from the micron range to the low nanometer range (as confirmed by the SEM study). The difference in saturated solubility between IRB-HP-β-CD/CC and IRB-SBA-16 could be attributed to the wetting and solubilization properties of HP-β-CD. As is well-known, HP-β-CD is a cyclic oligosaccharide with a hydrophilic outer surface and a lipophilic inner cavity. This allows small molecule lipophilic drugs to be entrapped within the inner cavity of HP-β-CD while its outer surface permits aqueous miscibility. Numerous studies have shown that HP-β-CD can interact with BCS class II drugs to form inclusion complexes with enhanced aqueous solubility.Citation27,Citation28
Table 2 Aqueous solubility of different IRB formulations (n=3)
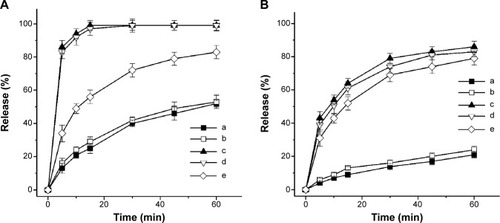
In , dissolution tests were performed in enzyme-free SGF and SIF in order to investigate the drug release behavior in different gastrointestinal (GI) tract environments. In both dissolution media, the dissolution of crude IRB was limited due to its very low solubility, hydrophobic surface, and large crystal size. As expected, the dissolution profiles of the PM was similar to that of crude IRB, showing that the mechanical physical mixing of crude IRB and HP-β-CD/CC had little effect on the dissolution of IRB. Interestingly, the dissolution rate of IRB from the drug-loaded microparticles was significantly increased and it was seen that differences in the dissolution profile were more evident in SGF (). This outstanding increase in drug dissolution may be due to many factors such as the increase in drug wettability and specific surface area, in addition to the decrease in particle size and crystallinity of IRB in the particles. Due to their hydrophilic nature, both nanoporous HP-β-CD/CC and mesoporous silica could be rapidly wetted by water, and the incorporated drug molecules were surrounded by aqueous medium thus allowing fast dissolution.Citation29,Citation30 Furthermore, a high specific surface area of IRB-HP-β-CD/CC (36 m2/g versus 0.72 m2/g for crude IRB powder) was also beneficial for drug release. According to the modification of the Noyes–Whitney equation, the dissolution rate of the drugs is proportional to the effective surface area available for dissolution.Citation31 In gastric acid medium, although the release from both nanoporous HP-β-CD/CC and mesoporous silica can be considered as rapid, there is a substantial difference between both formulations in terms of the initial release; IRB-HP-β-CD/CC releases more than 90% within the first 10 minutes, whereas the corresponding amount was only 47% for IRB-SBA-16. The difference between the release rates of IRB-HP-β-CD/CC and IRB-SBA-16 could be attributed to the solubility of the carrier. Since both HP-β-CD and calcium carbonate are readily soluble in acidic conditions, IRB molecules loaded into pore systems of HP-β-CD/CC had a greater chance of diffusing into the dissolution medium. As shown in , at pH 6.8, the cumulative release of IRB from IRB-HP-β-CD/CC and IRB-SBA-16 was 79% and 70% respectively, after 30 minutes. For the IRB-HP-β-CD/CC samples, the dissolution improvement in SIF was not as significant as in SGF due to calcium carbonate hardly soluble at neutral pH. Compared with IRB-SBA-16, a relatively fast release rate of IRB-HP-β-CD/CC may be attributed to its relatively larger pore size (13.6±3.8 nm). For IRB-SBA-16, the small pore size (3.2±0.4 nm) may hinder the rapid diffusion of IRB molecules into the dissolution media.Citation9,Citation23 In addition, it can be seen clearly from that the release profiles of IRB from IRB-HP-β-CD/CC were similar to that of the freshly prepared ones after 6 months of storage.
Figure 5 Dissolution of IRB from different formulations in (A) SGF and (B) SIF.
Notes: (a) crude IRB, (b) PM, (c) IRB-HP-β-CD/CC, (d) IRB-HP-β-CD/CC after 6 months of storage, and (e) IRB-SBA-16. Each data point represents the mean ± SD (n=3).
Abbreviations: IRB, irbesartan; SGF, simulated gastric fluid; SIF, simulated intestinal fluid; SBA-16, Santa Barbara amorphous materials-16; IRB-SBA-16, irbesartan loaded-SBA-16; HP-β-CD/CC, hydroxypropyl-β-cyclodextrin functionalized calcium carbonate; IRB-HP-β-CD/CC, irbesartan loaded-HP-β-CC; PM, physical mixture; SD, standard deviation.
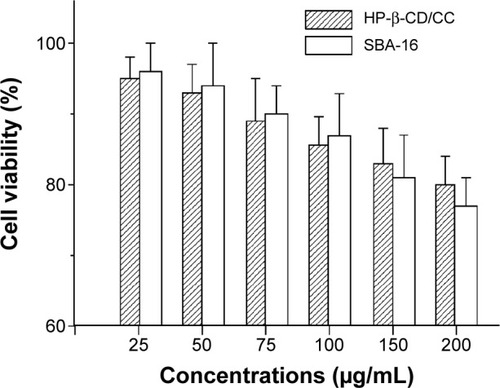
In , HT-29 human colon carcinoma cells were treated with different porous matrices (HP-β-CD/CC and SBA-16) at different concentrations (25–200 μg/mL) for 48 hours, and the cell viability was evaluated using Presto-Blue reagent. The cell viability results, as shown in , revealed that HP-β-CD/CC (or SBA-16) is almost completely noncytotoxic at low concentrations (<50 μg/mL) and exhibit only very weak cytotoxic effects at a high concentration (200 μg/mL). Thus, HP-β-CD/CC can be safely used for oral drug delivery.
Figure 6 Effect of HP-β-CD/CC and SBA-16 on HT-29 cell viability at various concentrations (n=6).
Abbreviations: HP-β-CD/CC, hydroxypropyl-β-cyclodextrin functionalized calcium carbonate; SBA-16, Santa Barbara amorphous materials-16.
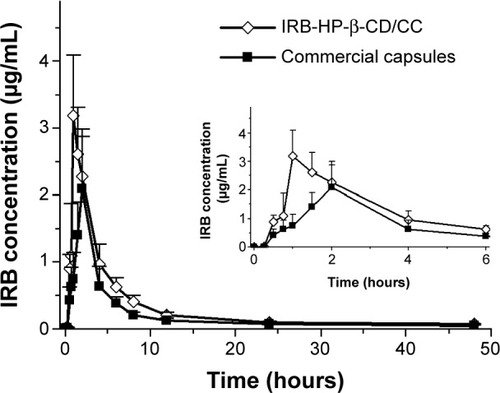
Generally, the bioavailability of a BCS class II drug is rate-limited by its dissolution, so that even a small increase in the dissolution rate sometimes results in a large increase in bioavailability. Therefore, an increase in the dissolution rate of the drug is thought to be a key factor for improving the bioavailability of BCS class II drugs.Citation4 A pharmacokinetic study was performed in beagle dogs to investigate whether IRB-HP-β-CD/CC could increase the oral bioavailability of IRB. The mean plasma IRB concentration after the oral administration of each IRB formulation is presented in and the corresponding pharmacokinetic parameters of IRB are listed in . Orally administered IRB-HP-β-CD/CC demonstrated a higher Cmax of 3.19±1.20 μg/mL and a shorter Tmax of 1.25±0.27 hours compared with oral commercial capsules (Cmax 2.04±0.73 μg/mL and Tmax 1.92±0.66 hours). These results are in agreement with the dissolution studies showing that IRB was released from IRB-HP-β-CD/CC much faster than that from crude IRB powder. The AUC0→48h value was also found to be significantly higher for the IRB-HP-β-CD/CC formulation than for the commercial capsules. The high AUC0→48h value of HP-β-CD/CC based formulation suggested that marked adsorption occurred after the quick release of IRB from HP-β-CD/CC in the GI tract. This underlines the important role of HP-β-CD/CC based formulation in producing a high concentration gradient between the drug and the GI epithelium produced by more drug release. Furthermore, the high surface area makes microparticles more likely to adhere to the epithelial gut wall, prolonging the absorption and leading to increased bioavailability. Compared with the commercial formulation, the relative bioavailability judged from the AUC0→48h of IRB was found to be 151.6%±36.2%. From the above result, it can conclude that using HP-β-CD/CC based porous amorphous solid dispersion (ASD) technique was beneficial to the absorption of IRB.
Table 3 Pharmacokinetic parameters of the IRB formulations tested (n=6)
Figure 7 Plasma concentration–time profiles of the IRB formulations tested.
Note: Each data point represents the mean ± SD (n=6).
Abbreviations: IRB, Irbesartan; HP-β-CD/CC, hydroxypropyl-β-cyclodextrin fun-ctionalized calcium carbonate; IRB-HP-β-CD/CC, irbesartan loaded-HP-β-CC; SD, standard deviation.
Conclusion
In this study, a novel nanoporous HP-β-CD/CC based IRB formulation was designed, and the particle size, morphology, specific surface area, physical state, solubility, dissolution, and pharmacokinetics were studied. The IRB-loaded HP-β-CD/CC microparticles showed a significantly higher drug release rate than IRB powder or IRB-loaded SBA-16 in vitro. The pharmacokinetic data clearly showed that the IRB-loaded HP-β-CD/CC formulation produced a significant improvement in the bioavailability compared with the commercial capsules. More importantly, the stability test confirmed that the IRB-loaded HP-β-CD/CC formulation exhibited no clear change in crystallinity and dissolution velocity during a 6-month storage period. These results demonstrated that HP-β-CD/CC based porous ASD is a promising formulation approach to improve the solubility and the in vivo absorption performance of a water-insoluble compound such as IRB.
Acknowledgments
This work was financially supported by the National Science Foundation of China (No 81402879), the Natural Science Foundation of Jiangsu Province of China (No BK20140221), and Project for Excellent Talents of Xuzhou Medical College (No 53631309). We would like to thank Dr David Jack for ensuring that the document is free of language errors.
Supplementary materials
Figure S1 Nitrogen adsorption/desorption isotherms of (A) HP-β-CD/CC and (B) SBA-16.
Note: Nitrogen adsorption isotherm (♦); nitrogen desorption isotherm (⋄).
Abbreviations: HP-β-CD/CC, hydroxypropyl-β-cyclodextrin functionalized calcium carbonate; SBA-16, Santa Barbara amorphous materials-16; STP, standard temperature and pressure.
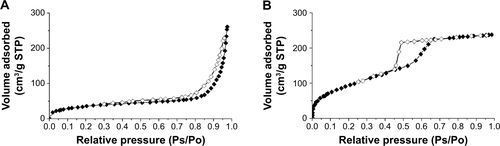
Table S1 The materials used in the study
Disclosure
The authors report no conflicts of interest in this work.
References
- LipinskiCALombardoFDominyBWFeeneyPJExperimental and computational approaches to estimate solubility and permeability in drug discovery and development settingsAdv Drug Deliv Rev20014632611259830
- SinghSParikhTSandhuHKSupersolubilization and amorphization of a model basic drug, haloperidol, by interaction with weak acidsPharm Res2013301561157323430485
- BroughCWilliamsROAmorphous solid dispersions and nano-crystal technologies for poorly water-soluble drug deliveryInt J Pharm201345315716623751341
- KawabataYWadaKNakataniMYamadaSOnoueSFormulation design for poorly water-soluble drugs based on biopharmaceutics classification system: basic approaches and practical applicationsInt J Pharm201142011021884771
- MüllerRHJacobsCKayserONanosuspensions as particulate drug formulations in therapy: rationale for development and what we can expect for the futureAdv Drug Deliv Rev20114731911251242
- Merisko-LiversidgeELiversidgeGGNanosizing for oral and parenteral drug delivery: a perspective on formulating poorly-water soluble compounds using wet media milling technologyAdv Drug Deliv Rev20116342744021223990
- VasconcelosTSarmentoBCostaPSolid dispersions as strategy to improve oral bioavailability of poor water soluble drugsDrug Discov Today2007121068107518061887
- ThomasMJSlipperIWalunjAInclusion of poorly soluble drugs in highly ordered mesoporous silica nanoparticlesInt J Pharm201038727227720025947
- WangSOrdered mesoporous materials for drug deliveryMicroporous Mesoporous Mater200911719
- KuangDBrezesinskiTSmarslyBHierarchical porous silica materials with a trimodal pore system using surfactant templatesJ Am Chem Soc2004126105341053515327299
- WaniAMuthuswamyESavithraGHMaoGBrockSOupickýDSurface functionalization of mesoporous silica nanoparticles controls loading and release behavior of mitoxantronePharm Res2012292407241822555380
- ZhangYWangJBaiXJiangTZhangQWangSMesoporous silica nanoparticles for increasing the oral bioavailability and permeation of poorly water soluble drugsMol Pharm2012950551322217205
- KinnariPMäkiläEHeikkiläTSalonenJHirvonenJSantosHAComparison of mesoporous silicon and non-ordered mesoporous silica materials as drug carriers for itraconazoleInt J Pharm201141414815621601623
- ParakhonskiyBVHaaseAAntoliniRSub-micrometer vaterite containers: synthesis, substance loading, and releaseAngew Chem Int Ed Engl2012511195119722375283
- PreisigDHaidDVarumFJDrug loading into porous calcium carbonate microparticles by solvent evaporationEur J Pharm Biopharm20148754855824568926
- SvenskayaYParakhonskiyBHaaseAAnticancer drug delivery system based on calcium carbonate particles loaded with a photosensitizerBiophys Chem2013182111523932207
- KurapatiaRRaichurAMComposite cyclodextrin–calcium carbonate porous microparticles and modified multilayer capsules: novel carriers for encapsulation of hydrophobic drugsJ Mater Chem B2013131753184
- Kassler-TaubKLittlejohnTElliottWRuddyTAdlerEComparative efficacy of two angiotensin II receptor antagonists, irbesartan and losartan in mild-to-moderate hypertension. Irbesartan/Losartan Study InvestigatorsAm J Hypertens1998114454539607383
- FaresMMSalemMSKhanfarMInulin and poly(acrylic acid) grafted inulin for dissolution enhancement and preliminary controlled release of poorly water-soluble Irbesartan drugInt J Pharm201141020621121421037
- StevensWJLebeauKMertensMVan TendelooGCoolPVansantEFInvestigation of the morphology of the mesoporous SBA-16 and SBA-15 materialsJ Phys Chem B20061109183918716671732
- HiguchiTConnorsKAPhase-solubility techniquesAdv Anal Chem Instrum19654117212
- KrukMAccess to ultralarge-pore ordered mesoporous materials through selection of surfactant/swelling-agent micellar templatesAcc Chem Res2012451678168722931347
- ShenSCNgWKChiaLHuJTanRBPhysical state and dissolution of ibuprofen formulated by co-spray drying with mesoporous silica: effect of pore and particle sizeInt J Pharm201141018819521419202
- GuoZLiuXMMaLEffects of particle morphology, pore size and surface coating of mesoporous silica on naproxen dissolution rate enhancementColloids Surf B Biointerfaces201310122823523010024
- ZhangPForsgrenJStrømmeMStabilisation of amorphous ibuprofen in Upsalite, a mesoporous magnesium carbonate, as an approach to increasing the aqueous solubility of poorly soluble drugsInt J Pharm201447218519124950364
- JacksonCLMcKennaGBVitrification and crystallization of organic liquids confined to nanoscale poresChem Mat1996821282137
- LiuMCaoWSunYHeZPreparation, characterization and in vivo evaluation of formulation of repaglinide with hydroxypropyl-b-cyclodextrinInt J Pharm201447715916625455768
- ZoellerTDressmanJBKleinSApplication of a ternary HP-β-CD-complex approach to improve the dissolution performance of a poorly soluble weak acid under biorelevant conditionsInt J Pharm201243017618322531855
- Vallet-RegiMBalasFArcosDMesoporous materials for drug deliveryAngew Chem Int Ed Engl2007467548755817854012
- Vallet-RegiMNanostructured mesoporous silica matrices in nanomedicineJ Intern Med2010267224320059642
- DokoumetzidisAMacherasPA century of dissolution research: from Noyes and Whitney to the biopharmaceutics classification systemInt J Pharm200632111116920290