
Abstract
The thick ascending limb (TAL) of Henle’s loop is a crucial segment for many tasks of the nephron. Indeed, the TAL is not only a mainstay for reabsorption of sodium (Na+), potassium (K+), and divalent cations such as calcium (Ca2+) and magnesium (Mg2+) from the luminal fluid, but also has an important role in urine concentration, overall acid–base homeostasis, and ammonia cycle. Transcellular Na+ transport along the TAL is a prerequisite for Na+, K+, Ca2+, Mg2+ homeostasis, and water reabsorption, the latter through its contribution in the generation of the cortico-medullar osmotic gradient. The role of this nephron site in acid–base balance, via bicarbonate reabsorption and acid secretion, is sometimes misunderstood by clinicians. This review describes in detail these functions, reporting in addition to the well-known molecular mechanisms, some novel findings from the current literature; moreover, the pathophysiology and the clinical relevance of primary or acquired conditions caused by TAL dysfunction are discussed. Knowing the physiology of the TAL is fundamental for clinicians, for a better understanding and management of rare and common conditions, such as tubulopathies, hypertension, and loop diuretics abuse.
Physiology of the thick ascending limb (TAL)
Morphological features of TAL cells
The loop of Henle is a highly specialized nephron site, with two peculiar properties, its extreme heterogeneity and its anatomic configuration. It is composed of the pars recta of the proximal tubule (PT; thick descending limb), the thin descending and ascending limbs, the TAL, and the macula densa.Citation1 The descending limb penetrates the medulla, where an increasing interstitial osmotic gradient is guaranteed by the noticeable addition of NaCl through the water impermeable TAL, amplified by the hairpin-shaped loop of Henle’s, with the so-called countercurrent multiplier system.Citation2,Citation3 A similar arrangement of the surrounding vessels prevents osmotic gradient dissipiation.Citation4 Electron microscopy studies demonstrated the presence of two distinct cell types in the TAL, differing in the configuration of the apical membrane: rough-surface cells (R cells), characterized by abundant apical microvilli, and smooth-surface cells (S cells) with no microprojections, but with several subapical vesicles.Citation5 The former is abundant in the cortex, and the latter in the medulla. Functional studies have demonstrated that R cells have high apical and low basolateral K+ conductance, while S cells have low apical and high basolateral K+ conductance, in hamster.Citation6 The main differences in salt absorption between the two cell types are unclear. Both cell types show similar mechanisms of transport; however, Nielsen et al demonstrated that S cells showed an intense labeling of the Na-K-2Cl cotransporter (NKCC2) on subapical vesicles, that may serve as a reservoir that can be recruited based on homeostatic needs.Citation7
Transcellular and paracellular salt absorption along the TAL
The loop of Henle is responsible for the reabsorption of ~40% of filtered Na+, mostly in the TAL.Citation3 Here, Na+ entry from the lumen into the cells occurs mainly via the luminal electroneutral Na+,K+,2Cl− cotransporter, NKCC2, encoded by the SLC12A1 gene.Citation8 This cotransporter mediates secondary active Na+, K+, and 2Cl− uptake across the apical membrane.Citation9
The protein belongs to the family of solute carrier family 12, that includes two isoforms in humans encoded by two genes, SLC12A1 (NKCC2) and SLC12A2 (NKCC1).Citation3 While NKCC1 is widely expressed in several organs and tissues, NKCC2 is exclusively localized along the TAL.Citation10 Once reabsorbed from the lumen, Na+ exits the cell via the sodium pump; ClC-Ka and ClC-Kb channels, with their Barttin subunit, mediate Cl− exit from the basolateral side, participating in the electronegative blood side.Citation11 The importance of ClC channels in human physiopathology is highlighted by the evidence that mutations in every channel lead to human disease.Citation12 Finally, K+ ions are delivered back to the lumen through the apical renal outer medulla K+ channels (ROMK). These channels perform a dual crucial role in the TAL: first, they ensure K+ recycling to the lumen, essential for salt reabsorption; second, they set a positive transepithelial voltage, that drives paracellular reabsorption of cations.Citation13,Citation14
Additional Na+ reabsorption is driven by the generated electrical field through the paracellular way; moreover, other cations (Ca2+ and Mg2+) are reabsorbed by this route.
There is no doubt that the bulk of Ca2+ reabsorption along the TAL occurs through the paracellular pathway, following the electrochemical gradient. NKCC2 and in particular ROMK generate the “driving force” for paracellular cation transport. Interestingly, the evidence that paracellular transport is regulated by the calcium sensing receptor (CaSR) signaling in the absence of changes in transepithelial voltage suggested that paracellular permeability may vary, with consequent changes in divalent cations reabsorption.Citation15 The CaSR, expressed on the basolateral membrane of TAL cells, has been shown to modulate Ca2+ absorption through two mechanisms: 1) by inhibiting ROMK activity, thus leading to a decreased lumen-positive voltage;Citation16 and 2) by modulating tight junction (TJ) structure.Citation17,Citation18 The latter is the main determinant of paracellular permeability. Claudins are some of the main components of TJ. Along the TAL, several claudins are expressed, including claudin-16, 19, 10, 3, and 18.Citation19 Their role in divalent cations absorption has been recently highlighted by the discovery of salt losing phenotype associated with their dysfunction due to genetic mutations.Citation20 However, several evidences suggest that a significant component of Ca2+ reabsorption occurs also via transcellular pathway.Citation21 The presence on the basolateral membrane of the Na+/Ca2+ exchanger (NCX) and the Ca2+ATPase (PMCA) confirms the presence of a basolateral exit, and suggests the possible presence of a luminal route for Ca2+entry.Citation22
Micropuncture experiments have demonstrated that nearly 60% of the filtered Mg2+ is reabsorbed in the TAL.Citation23 Passive paracellular transit is the main route, and it is driven by the lumen-positive voltage. The importance of claudin-16 (paracellin 1) and claudin-19 in Mg2+ homeostasis has been pointed out in the recent years, given the association of their mutation with familial hypomagnesemia.Citation24
The synergic activity of the main transporters and channels involved in salt absorption (NKCC2, ROMK, the chloride channel Kb [also known as ClC-Kb], with the Barttin subunit) and the integrity of TJs are the prerequisite to prevent electrolytes imbalance (, ). The loss of function of any of those proteins leads to a salt losing phenotype. As salt absorption in the TAL is under the control of hormones, local mediators, and several intracellular signaling pathways, it is not uncommon that impaired salt absorption is secondary to these dysfunctions. As an example, activating mutations of CASR have been described to lead to salt losing nephropathy;Citation25 similarly, dysfunction of uromodulin, Ste20-related proline–alanine-rich kinase (SPAK), and oxidative stress response kinase (OSR1) have been shown to influence NKCC2 activity and to determine fluid and electrolytes imbalance.Citation26–Citation28
Figure 1 Schematic representation of TAL cells, illustrating the major transcellular proteins involved in salt reabsorption.
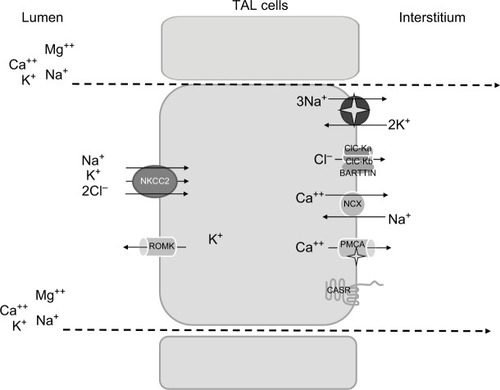
Table 1 Main channels, transporters, and other proteins relevant in salt absorption along the TAL
Generation of the cortico-medullar osmotic gradient
One of the major functions of TAL is the creation of the cortico-medullar osmotic gradient. In humans, interstitial osmolality progressively increases from the cortex, where it iŝ290 mOsm/kg, to the tip of the medulla, where it reaches 1200 mOsm/kg.Citation2 The U-shaped arrangement of the two branches of the loop of Henle, the different permeabilities of the descending and ascending limbs to Na+ and water, and the active Na+ reabsorption in TAL are fundamental factors in the creation of the medullary osmotic gradient.Citation4
Fluid entering the descending limb from the PT is isotonic with the plasma and the surrounding interstitium. The renal epithelium along the descending limb is highly permeable to water but not to solutes.Citation29 Here, the increasing interstitial osmolality from the cortex to the medulla provides the driving force for water reabsorption ().Citation30–Citation32 Thus, water diffuses from the lumen to the interstitium along the descending limb and luminal osmolality rises progressively up to the tip of Henle’s loop. Active Na+ and Cl− reabsorption along the TAL serves two roles: 1) first, it dilutes the luminal fluid, as it is impermeable to water, so the solutes that accumulate into the interstitium are not followed by the osmotic flow of water; 2) second, by adding Na+ and Cl− to the interstitium, it provides a strong contribution in the generation of the interstitial osmotic gradient in the outer medulla.Citation33
Figure 2 Mechanism of urine concentration.
Abbreviations: PT, proximal tubule; DT, distal tubule; UTA2, urea transporter; NKCC2, Na-K-2Cl cotransporter; TAL, thick ascending limb of the loop of Henle; CCD, cortical collecting duct; OMCD, outer medullar collecting duct; IMCD, inner medulla collecting duct.
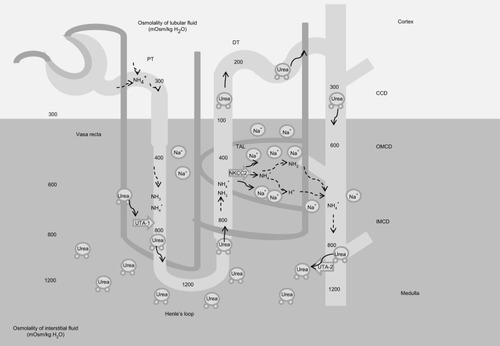
While Na+ is fundamental in the determination of the osmotic gradient in the outer medulla, in the inner medulla urea provides a strong contribution, thanks to its reabsorption from the lumen along the collecting duct (CD), and the recycling along the descending limb.Citation2 Similarly to the two branches of Henle’s loop, the descending and ascending vasa recta are arranged in a specific anatomic relationship. The countercurrent flow configuration establishes an axial osmolality gradient that is distributed along the corticomedullar axis.Citation34 Osmotic equilibration is achieved through a combination of water absorption and solute secretion, as vasa recta are permeable to water, urea, and sodium. However, the descending vasa recta, penetrating in the medulla, lose water and gain solutes, while the ascending vasa recta show the opposite behavior, preventing dissipation of the osmotic gradient.Citation35 Conditions that decrease medullary flow, such as dehydration, favor urine concentration as blood transit into the vasa recta has more time to achieve osmotic equilibration with the hypertonic interstitium. Conversely, an increased medullary flow decreases the urine concentrating ability.Citation34,Citation36
Acid–base homeostasis
The role of PT in bicarbonate reabsorption is well established.Citation37–Citation39 However, other downstream segments contribute to bicarbonate reabsorption. The loop of Henle reabsorbs a significant fraction (nearly 15%) of the filtered bicarbonate.Citation40 In vivo studies have demonstrated that the descending limb of Henle’s loop has low bicarbonate permeability in rats. In contrast, the final segment of the PT (S3 portion) has been shown to be able to reabsorb bicarbonate. Under physiologic conditions, its contribution in bicarbonate reabsorption is modest, as it is actively reabsorbed along the early PT segments and its luminal concentration is low.
Micropuncture studies have shown that bicarbonate concentration increases significantly at the tip of Henle’s loop, most probably as an indirect effect resulting from luminal fluid concentration due to water reabsorption along the descending limb.Citation41 In the TAL, bicarbonate is reabsorbed via the transcellular pathway; the main mechanism resembles bicarbonate reabsorption in the PT through Na+/H+ exchanger (NHE) activity.Citation42 Both NHE2 and NHE3 isoforms have been localized to the luminal membrane.Citation43 The contribution of NHE2 activity is considered low, as the addition of NHE2 knockdown in mice did not result in an overt acid–base disturbance compared with NHE3 knockout (KO) mice.Citation44 Perfusions studies, in vivo and ex vivo, showed that bicarbonate reabsorption requires carbonic anhydrase, and is stimulated by bumetanide.Citation41 Interestingly, de Bruijn et al have demonstrated that furosemide promotes luminal fluid acidification in the TAL by lowering the intracellular Na+ concentration, which in turn increases the driving force for NHE3 activity.Citation45 Bicarbonate exit from the cells is mediated by the Cl−/HCO−3 exchanger 2 (AE2) ().Citation46 Interestingly, at the luminal site, the presence of the Cl−/HCO−3 exchanger 1 (AE1) has been shown in rats, that has been proposed to participate in Na+ reabsorption by coupling the Na+/H+ activity.Citation47
Figure 3 Relevant proteins involved in acid–base homeostasis in the TAL.
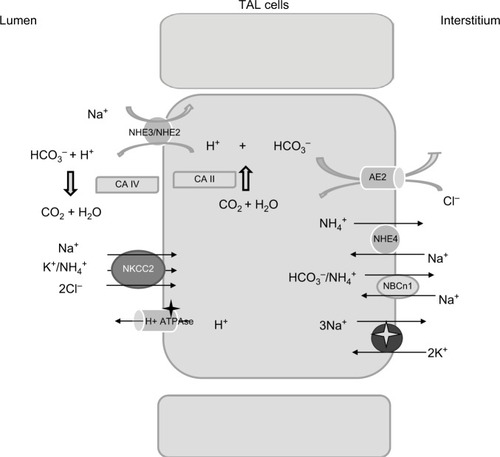
Besides NHE3, additional molecules contribute to bicarbonate reabsorption in the TAL. Functional studies have suggested the presence of an active H+-ATPase, and immunohistochemical analysis has reported the presence of proton ATPase along the TAL.Citation48 Its role in bicarbonate reabsorption is not considered as consistent as a Na+/H+ exchanger under physiological conditions.
Changes in acid–base balance modulate the rate of bicarbonate reabsorption along the TAL: both acute and chronic metabolic acidosis increase while metabolic alkalosis depresses bicarbonate reabsorption.Citation49 Functional studies have demonstrated that both H+-ATPase and Na+/H+ exchanger are able to adapt to changes in the acid–base status, probably via hormonal stimulation by glucocorticoids and aldosterone.Citation49
Ammonia handling is not of minor importance. Urine ammonia excretion derives mainly from renal ammoniagenesis, rather than glomerular filtration.Citation50 It is produced from glutamine in the PT as ammonium ion (NH4+) and is released to the luminal fluid. TAL has a crucial role in ammonia reabsorption; this process occurs via NKCC2, at the K+ binding site.Citation51 K+/NH4+ exchange and conductive K+ transport have also been described in the TAL, but their contribution is less significant compared with NKCC2.Citation36 Basolateral exit is mediated by the Na+/NH4+ exchanged via NHE4, the Na+-bicarbonate NBCn1 cotransporter, and by a Cl−-dependent pathway.Citation51–Citation53 Interstitial ammonia is at least in part passively recycled into the thin descending limb of Henle’s loop, predominantly as ammonia (NH3). This recycling increases the axial ammonia gradient, paralleling the increasing tonicity of the interstitial fluid.Citation54 Ammonia is then secreted into the luminal fluid along the CD, by the ammonia-specific transporters, Rhesus glycoproteins Rhbg and Rhcg.Citation55
Pathophysiology of TAL dysfunction
Inherited and acquired conditions leading to an imbalance in salt reabsorption along the TAL have a significant impact on human pathophysiology.
Several water and electrolytes disturbances are attributed to TAL dysfunction. TAL hypofunction is responsible for water and electrolytes wasting, while increased salt absorption is considered a contributing factor in the pathogenesis of hypertension.
Bartter syndrome (BS)
BS is a rare disorder caused by genetic mutations leading to impaired salt reabsorption along the TAL. Five genetic BS subtypes have been described; BS type I is caused by mutations in SLCA12A1 (NKCC2) gene, while BS type II is associated to KCNJ1 (ROMK) mutations; type III and IV are caused by CLCNKB (ClC-Kb) and BSND (Barttin subunit) genetic aberrations; and BS type V results from activating mutation of the CASR (CaSR).Citation56 Animal models of BS have provided a significant contribution in understanding the mechanism of the disease. Both NKCC2 and ROMK null mice are characterized by early lethality.Citation57,Citation58 The recessive mutant mouse line Slc12a1(I299F) resembled the features of patients suffering from type I BS, including polyuria, metabolic alkalosis, and hypercalciuria.Citation59 Interestingly, ROMK null mice develop transient hyperkalemia after birth, consistent with ROMK hypofunction in the CD, where the channel mediates K+ secretion.Citation60 Later, infants develop classic features of BS, including hypokalemia, sustained by K+ secretion along the CD mediated by the flow-dependent maxi-K+ channels, despite ROMK deficiency. All BS patients share similar clinical features and biochemical abnormalities, including fluid loss leading to low-normal blood pressure, hypokalemia, and metabolic alkalosis. Often, hypercalciuria occurs, while Mg2+ loss is generally balanced by increased reabsorption through downstream nephron sites, mainly the distal tubule (DT).Citation61,Citation62 Fluid wasting causes the activation of the renin–angiotensin–aldosterone system (RAAS) axis, while Ca2+ loss may lead to secondary hyperparathyroidism.Citation63 Two major clinical forms of BS have been described, the antenatal (also known as hyperprostaglandin E syndrome) and the classic variant. The antenatal form is characterized by polyhydramnios and growth retardation in utero, with severe hypokalemia and fluid loss requiring intensive treatment at birth. This form is generally characteristic of type I, II, and IV. The latter is the only one that is associated with hearing loss, due to the presence of the Barttin subunit in the inner ear. The classic form tends to be less severe, with a late onset, generally in childhood.Citation63 As a result of chronic volume depletion, increased PT urate reabsorption and consequent hyperuricemia may occur.Citation64,Citation65 Type III BS has a variable phenotype. It is linked to the CLCNKB gene, expressed on the basolateral site of both TAL and DT. The disease may have features of BS (either antenatal or classic) and/or Gitelman syndrome.Citation66 ClC-K2 deficient mice exhibit salt losing phenotype, with compensatory increased RAAS axis, hypotension, and increased prostaglandin E2 generation; interestingly, the mice show a blunted response to both furosemide and thiazides, indicating a combined defective salt absorption along both TAL and DT.Citation67 Activating mutations of the CaSR have been associated with hypocalcemia and hypercalciuria, and sometime hypomagnesemia.Citation68 The pathogenesis of Ca2+ and Mg2+ wasting is multifactorial in this condition. CaSR is a well-known modulator of parathyroid hormone (PTH). In the presence of high plasma Ca2+ levels, it inhibits PTH secretion. Gain of function mutation in the CASR has been shown to determine autosomal dominant hypoparathyroidism, with consequent effects on serum levels of Ca2+, phosphate, and Mg2+.Citation69 However, the activation of CaSR in the kidney is believed to contribute to Ca2+ wasting, as suggested by the evidence that patients carrying CASR mutations have more severe urine Ca2+ loss compared with patients with primary hypoparathyroidism.Citation70
Recently, cases of some male infants showing features of transient antenatal BS, associated with mutations in the MAGE-D2 gene, have been reported.Citation71 The gene product is expressed in the DT and in the TAL in both adult and fetal human kidney. Immunostaining studies revealed reduced NKCC2 and Na+-Cl− cotransporter (NCC) apical abundance in one of these patients, with increased NKCC2 cytoplasmic retention. In vitro, MAGE-D2 promotes trafficking to the apical membrane of both NCC and NKCC2.
Familial hypomagnesemia
Claudins are transmembrane proteins involved in intercellular adhesion and paracellular barrier formation.Citation20 In mammals, over 24 claudin genes have been described; some genes exhibit peculiar tissue-specificity expression, others are expressed in several organs and tissues. In the kidney, several claudins have been mapped along the nephron.Citation72 In 1999, Simon et al discovered a mutation in a gene encoding a protein belonging to this family, CLDN16 (encoding claudin-16, also named paracellin), causing familial hypercalciuric hypomagnesemia (FHHNC).Citation73 Recessive mutations of CLDN16 and CLDN19 (encoding claudin-19) are the most common causes of FHHNC.Citation20 The disease is believed to be the result of a deficient reabsorption of divalent cations in the TAL due to a defective paracellular barrier. This defect results in increased fractional excretion of Ca2+ and Mg2+, leading to hypocalcemia and secondary hyperparathyroidism and sometimes to hypomagnesemia.Citation74 Nephrocalcinosis is the leading cause of renal failure. Patients with CLDN19 mutations will also exhibit severe ocular involvement.Citation75 Interestingly, the role of claudin-10 in salt absorption along the TAL has been recently highlighted. Breiderhoff et al showed that claudin-10 deficiency affected paracellular permeability to monovalent and divalent cations in the TAL, causing nephrocalcinosis and hypermagnesemia.Citation76 Its role in TAL salt absorption has been confirmed also in humans by the study of Bongers et al.Citation77 They described two patients carrying CLDN10 mutations with a hypokalemic salt losing nephropathy, with hypermagnesemia. Accordingly, it has been recently shown that patients suffering from the so-called HELIX syndrome (characterized by hypohidrosis, salt losing nephropathy, lacrimal gland dysfunction, ichthyosis, and xerostomia) carried biallelic mutations of CLDN10 gene. Functional studies demonstrated that salt wasting depended on impaired salt absorption along the TAL.Citation78 The role of claudins in salt homeostasis is further supported by a genome-wide association study demonstrating that claudin-14 variants are a risk factor for hypercalciuric nephrolithiasis.Citation79 In addition, Corre et al have recently described a genome-wide association study demonstrating that claudin-14 variants influence differential urine excretion of Mg2+ and Ca2+, suggesting that there is still a lot to be discovered in paracellular electrolyte handling along the distal nephron.Citation80
CNNM2 (hypomagnesemia with seizures and mental retardation)
The CNNM2 gene is mainly expressed in the kidney, brain, and lung in mammalian tissues and performs the function of a divalent cation transporter, including Mg2+.Citation81 The protein has been localized on the basolateral membrane of both TAL and DT cells in human kidney sections. The presence along the DT is not surprising, as transcellular Mg2+ reabsorption from the lumen has been largely proven. The presence in the TAL may either indicate a role in the control of paracellular Mg2+ absorption, or a possible presence of apical Mg2+ uptake, as suggested by some authors.Citation82
Mutations in this gene cause a rare disease characterized by cognitive impairment, seizure, and hypomagnesemia. The latter is believed to result from impaired luminal Mg2+ reabsorption. A genome-wide association study demonstrated a correlation between CNNM2 variants and plasma Mg2+ levels, supporting a significant role of CNNM2 in human Mg2+ homeostasis.Citation83
The EAST syndrome
This is a rare recessive genetic disorder associated with mutations in the KCNJ10 gene, encoding the K+ channel Kir 4.1.Citation84 The acronym EAST indicates the most important clinical features, including epilepsy, ataxia, senso-neural deafness, and tubulopathy. The Kir 4.1 channel is in fact expressed in several organs, including the central nervous system, the inner ear, and the nephron.Citation85 In the latter, it has been shown on the basolateral membrane of the distal nephron, from the macula densa to the early CD and in cortical TAL.Citation72 The salt losing phenotype resembles Gitelman syndrome, with hypocalciuria and hypomagnesemia, besides metabolic alkalosis and hypokalemia. Fan et al demonstrated that the vasopressin-dependent stimulation of the 80–150 pS basolateral K+ channel in Kcnj10 (Kir 4.1) KO mice counteracts the loss of function of Kir 4.1 in the TAL, preventing defective salt absorption at this nephron site.Citation86
Mitochondrial diseases
Inherited mitochondrial diseases are a heterogeneous class of clinical conditions characterized by impaired function of the mitochondrial respiratory chain, due to genetic mutation of either genomic or mitochondrial DNA.Citation87 The spectrum of clinical features is highly variable and the most common morbidities are neurologic and myopathic diseases. Some studies have described renal involvement, with a wide spectrum of tubular disorders, including Fanconi and Bartter-like syndrome; in addition, glomerular defects (focal and segmental glomerulosclerosis) and cystic diseases have been reported.Citation88 The Kearns–Sayre syndrome is a rare mitochondrial disease caused by a large deletion of mitochondrial DNA.Citation89 The clinical findings are Bartter-like syndrome, with hypokalemic metabolic alkalosis and nephrocalcinosis. Some reports described Fanconi-like phenotypes. Extrarenal features include ophthalmoplegia, cerebellar, heart, and several endocrine and metabolic dysfunctions.Citation88
Increased salt absorption along the TAL: a potential role in salt-sensitive hypertension
Hypertension is a major public health problem, and it is considered to be of multifactorial origin. High dietary salt intake is considered one of the major environmental factors favoring its development. However, the effect of salt intake on blood pressure varies among individuals, a phenomenon called salt sensitivity.Citation90 The molecular mechanism underlying salt sensitivity is only partially understood. A significant contribution is mainly attributable to an impaired ability to excrete Na+. Evidences demonstrate that increased Na+ reabsorption along the distal nephron plays a crucial role in the pathogenesis of hypertension.Citation91,Citation92 Monogenic forms of hypertension include clinical conditions characterized by increased salt absorption along the DT (Gordon syndrome) and the CD. The latter may be caused by gain of function mutations of the principal apical channel mediating Na+ entry, epithelial sodium channel (Liddle syndrome), or by the activation of mineralocorticoid receptors (autosomal dominant pseudoaldosteronism type I).Citation93 Indirect data suggest that the TAL also has a crucial role in fluid balance and the pathogenesis of hypertension.Citation94 TAL hypofunction, as in BS, clearly leads to fluid loss and low-normal blood pressure. Animal models of salt-sensitive hypertension show increased avidity in salt absorption along the TAL. Milan hypertensive strain rats displayed increased NKCC2 activity during the induction phase of hypertension.Citation95 In Dahl salt-sensitive rats, increased TAL Cl− uptake has been demonstrated by in vivo micropuncture studies.Citation96 In accordance with animal studies, several data suggest that also in humans TAL contributes to salt sensitivity and hypertension. African-American individuals show a much higher incidence of salt-sensitive hypertension than white people. In the first group of subjects, a lower plasma renin activity (PRA) has been shown, suggesting a primary increase in renal salt reabsorption and consequent compensatory PRA suppression.Citation97 Results of Luft et al’s studies, showing a higher kaliuretic response after furosemide administration compared with white people, considered TAL as the major nephron site responsible for increased salt reabsorption in black individuals.Citation98
In the Framingham heart study population, heterozygote genetic variants of NKCC2, ROMK, and NCC correlated with low blood pressure.Citation99 In addition, a recent genome-wide association study indicated UMOD gene variants as risk factors for both salt-sensitive hypertension and kidney damage,Citation27 further supporting the hypothesis of TAL contribution in salt-sensitive hypertension.Citation100,Citation101 In fact, besides other known functions, uromodulin (UMOD) has been proposed to modulate salt absorption in the TAL. Graham et al showed that UMOD−/− mice were resistant to salt-induced changes of blood pressure,Citation102 while an independent study showed that UMOD overexpression increased blood pressure.Citation102 In vitro studies suggest that UMOD increases either NKCC2 or ROMK activities.Citation27 Moreover, the evidence that cyclosporine-induced hypertension is associated with NKCC2 upregulation further supports the hypothesis that increased avidity in salt absorption along the TAL contributes to the pathogenesis of hypertension.Citation103,Citation104
Acquired conditions resulting in TAL hypofunction: electrolytes disorders, loop diuretics, bilateral ureteral obstruction
Chronic electrolytes imbalance
Hypokalemia is known to inhibit urine concentrating ability and Na+ and Cl− absorption along the TAL.Citation13 In vivo microperfusion studies have demonstrated that chronic hypokalemia reduced Na+ and fluid absorption in the TAL.Citation105,Citation106 In a study by Gutsche et al, impaired salt absorption along the TAL in rats under a low K+ diet was restored by acute K+ infusion; the evidence of impaired Na+ efflux across the peritubular membrane suggested hypokalemia-dependent inhibition of the basolateral Na+-K+-ATPase.Citation107 The resulting increased Na+ and fluid loading to the distal K+-secretory segments of the nephrons, that is known to enhance K+ secretion, causes further K+ urinary excretion.Citation105 Recent studies have addressed the role of the intracellular proteins SPAK and OSR1 in electrolytes homeostasis and salt absorption along the distal nephron. Both proteins have been shown to phosphorylate and activate NKCC2 and NCC in vitro.Citation28 However, their function in vivo is controversial. In fact, SPAK null mice showed a Gitelman-like phenotype, suggesting a pivotal role of the protein in NCC regulation, rather than NKCC2, while kidney-specific OSR1 null mice had a Bartter-like phenotype, confirming its role in modulating NKCC2 function.Citation108,Citation109
Interestingly, total double KO mice is embryonic lethal, while constitutive SPAK KO mice with inducible OSR1 knockdown at steady state showed mild volume contraction and hypokalemia, a less severe phenotype than expected by combined NCC and NKCC2 hypofunction. Accordingly, phospho-NKCC2 abundance was unaffected despite the absence of SPAK and OSR1; conversely, phospho-NCC abundance in response to low K+ diet was reduced, suggesting a significant role of SPAK and OSR1 in NCC regulation by K+ plasma levels.Citation28
Extracellular Ca2+ has a potent inhibitory effect on salt absorption. Chronic hypercalcemia leads to urinary salt loss and fluid wasting. By binding the CaSR on the basolateral membrane of the TAL, extracellular Ca2+ has been shown to inhibit ROMK activity via phospholipase A2, reducing the lumen-positive voltage and consequently paracellular cation reabsorption.Citation16
As stated earlier, recent studies suggest that CaSR regulates paracellular calcium reabsorption along the TAL without affecting the transepithelial electrical gradient. CaSR activation is supposed to enhance CLD14 expression, leading to the downregulation of claudin-16/19, with consequent hypercalciuria. The requirement of calcineurin in the pathway regulating claudin-14 expression may provide an explanation for the cyclosporine-induced hypercalciuria.Citation110
Loop diuretics
Diuretics are widely used in several clinical fields. Furosemide, torasemide, and bumetanide bind NKCC2 in a reversible fashion. The resulting NKCC2 inhibition leads to the reduction in Na+, K+, and Cl− absorption, an effect that overrules that the cortico-medullar osmotic gradient increases urine output and impairs paracellular cations reabsorption. This property has predictable beneficial effects in several conditions, and loop diuretics are the main therapy in fluid retentive states and hypercalcemic conditions.Citation111 After 1 hour from the oral administration of furosemide, normal subjects experienced increased urine output and increased excretional fraction of Na+, K+, Cl−, Ca2+, and Mg2+ in a time-dependent manner, an effect that decreases after 4–6 hours. The effect is dose-dependent, and larger doses are required when the glomerular filtration rate (GFR) decreases.Citation112 Low-dose loop diuretics rarely cause important side effects; conversely, high dosage or long-term treatments, as usually is needed in chronic conditions, result in electrolytes and acid–base imbalance. Hypokalemic metabolic alkalosis is a common side effect of loop diuretics treatment. The underlying mechanism is based on increased salt delivery to the distal nephron, that enhances K+ and H+ secretion along the CD.Citation113 Extracellular volume contraction and the activation of the RAAS axis further contribute to metabolic alkalosis maintenance. Conversely, hyponatremia is uncommon in patients taking loop diuretics compared with patients under thiazides treatment, but still this complication can occur in the presence of concomitant pathologic conditions.Citation114 By increased free water clearance, loop diuretics are expected to increase rather than decrease plasma Na+ concentration. However, high dose of loop diuretics, as often can be necessary in chronic heart failure or in advanced chronic kidney disease, can cause over-diuresis, intravascular volume depletion, and increased vasopressin incretion, worsening hyponatremia.Citation115
Post-obstructive syndrome
Post-obstructive diuresis is a polyuric condition resulting after the relief of bilateral urinary tract obstruction. Diuresis is considered a physiological response to counteract water and salt retention during the obstruction. However, in some patients, diuresis does not cease when the excess of volume and salt is resolved, resulting in pathologic diuresis.Citation116 Several factors have been implicated in the pathophysiology of this condition. Earlier studies from the 1950s suggested that increased urine and salt excretion was the result of reduced Na+ reabsorption, with some studies placing the site of reduced reabsorption in the PT, and others in the distal nephron.Citation117
Micropuncture studies showed that post-obstructive diuresis resulted from impaired Na+ reabsorption at nephron sites beyond the PT in rats.Citation118 McDougal and Wright showed that Na+ absorption was decreased either in PT, paralleling the reduction of the GFR, or in the TAL, leading to increased Na+ loading to the CD in rats.Citation117 Subsequent studies have shown that polyuria following bilateral obstruction relief was associated with reduced expression of the major Na+ transporters, including NHE3, NKCC2, type II Na-Pi cotransporter, and NCC.Citation101 Some authors have demonstrated the role of reduced responsivity of the CD to vasopressin, leading to a diabetes insipidus-like syndrome.Citation119 Recent studies suggest that hydrogen sulphide (H2S), a gasotransmitter modulating a wide range of physiologic functions, may represent a novel therapeutic approach in this condition, given the evidence that increasing H2S levels may reduce renal fibrosis.Citation120,Citation121 It would be of interest to address its effect on salt absorption in this setting, considering that it has been shown to modulate urinary Na+ and K+ excretion, probably by affecting the function of both NKCC2 and Na-K ATPase.Citation122
Conclusion
The TAL has a central role in water and electrolyte homeostasis and acid–base balance, and its dysfunction has a crucial role in human pathophysiology.
Increased awareness and understanding of its specific functions will enable physicians to better manage acquired and inherited conditions caused by TAL-defective salt absorption, and to reduce the incidence and the complications of potentially life-threatening conditions.
Disclosure
The authors report no conflicts of interest in this work.
References
- MountDBThick ascending limb of the loop of HenleClin J Am Soc Nephrol20149111974198625318757
- SandsJMLaytonHEThe physiology of urinary concentration: an updateSemin Nephrol200929317819519523568
- GregerRIon transport mechanisms in thick ascending limb of Henle’s loop of mammalian nephronPhysiol Rev19856537607952409564
- DantzlerWHPannabeckerTLLaytonATLaytonHEUrine concentrating mechanism in the inner medulla of the mammalian kidney: role of three-dimensional architectureActa Physiol (Oxf)2011202336137821054810
- ImaiMTsuruokaSYoshitomiKJunichiTSuzukiMMutoSMorphological and functional heterogeneity of the thick ascending limb of Henle’s loopClin Exp Nephrol199931917
- TsuruokaSKosekiCMutoSTabeiKImaiMAxial heterogeneity of potassium transport across hamster thick ascending limb of Henle’s loopAm J Physiol19942671 Pt 2F121F1298048551
- NielsenSMaunsbachABEcelbargerCAKnepperMAUltra-structural localization of Na-K-2Cl cotransporter in thick ascending limb and macula densa of rat kidneyAm J Physiol19982756 Pt 2F885F8939843905
- MutigKTrafficking and regulation of the NKCC2 cotransporter in the thick ascending limbCurr Opin Nephrol Hypertens201726539239728614115
- MarkadieuNDelpireEPhysiology and pathophysiology of SLC12A1/2 transportersPflugers Arch201446619110524097229
- AresGRCaceresPSOrtizPAMolecular regulation of NKCC2 in the thick ascending limbAm J Physiol Renal Physiol20113016F1143F115921900458
- HenningsJCAndriniOPicardNThe ClC-K2 chloride channel is critical for salt handling in the distal nephronJ Am Soc Nephrol201728120921727335120
- StöltingGFischerMFahlkeCCLC channel function and dysfunction in health and diseaseFront Physiol2014537825339907
- TrepiccioneFZacchiaMCapassoGPhysiopathology of potassium deficiencyAlpernRJCaplanMMoeOWSeldin and Giebisch’s The Kidney, Physiology and Pathophysiology5th edSan DiegoElsevier201317131738
- ZacchiaMAbategiovanniMLStratigisSCapassoGPotassium: from physiology to clinical implicationsKidney Dis (Basel)201622727927536695
- LoupyARamakrishnanSKWootlaBPTH-independent regulation of blood calcium concentration by the calcium-sensing receptorJ Clin Invest201212293355336722886306
- WangWLuMBalazyMHebertSCPhospholipase A2 is involved in mediating the effect of extracellular Ca2+ on apical K+ channels in rat TALAm J Physiol19972733 Pt 2F421F4299321915
- GongYReniguntaVHimmerkusNClaudin-14 regulates renal Ca++ transport in response to CaSR signalling via a novel microRNA pathwayEMBO J20123181999201222373575
- RiccardiDValentiGLocalization and function of the renal calcium-sensing receptorNat Rev Nephrol201612741442527157444
- Kiuchi-SaishinYGotohSFuruseMTakasugaATanoYTsukitaSDifferential expression patterns of claudins, tight junction membrane proteins, in mouse nephron segmentsJ Am Soc Nephrol200213487588611912246
- GongYHouJClaudins in barrier and transport function-the kidneyPflugers Arch2017469110511327878608
- BlaineJChoncholMLeviMRenal control of calcium, phosphate, and magnesium homeostasisClin J Am Soc Nephrol20151071257127225287933
- HoenderopJGHartogAStuiverMDoucetAWillemsPHBindelsRJLocalization of the epithelial Ca2+ channel in rabbit kidney and intestineJ Am Soc Nephrol20001171171117810864572
- EllisonDHDivalent cation transport by the distal nephron: insights from Bartter’s and Gitelman’s syndromesAm J Physiol Renal Physiol20002794F616F62510997911
- GodronAHarambatJBoccioVFamilial hypomagnesemia with hypercalciuria and nephrocalcinosis: phenotype–genotype correlation and outcome in 32 patients with CLDN16 or CLDN19 mutationsClin J Am Soc Nephrol20127580180922422540
- WatanabeSFukumotoSChangHAssociation between activating mutations of calcium-sensing receptor and Bartter’s syndromeLancet2002360933469269412241879
- CarmosinoMProcinoGSveltoMNa+-K+-2Cl- cotransporter type 2 trafficking and activity: the role of interacting proteinsBiol Cell2012104420121222211456
- TruduMJanasSLanzaniCCommon noncoding UMOD gene variants induce salt-sensitive hypertension and kidney damage by increasing uromodulin expressionNat Med201319121655166024185693
- FerdausMZBarberKWLópez-CayuqueoKISPAK and OSR1 play essential roles in potassium homeostasis through actions on the distal convoluted tubuleJ Physiol2016594174945496627068441
- KimWYLeeHWHanKHDescending thin limb of the intermediate loop expresses both aquaporin 1 and urea transporter A2 in the mouse kidneyHistochem Cell Biol2016146111227091563
- YangBBankirLUrea and urine concentrating ability: new insights from studies in miceAm J Physiol Renal Physiol20052885F881F89615821253
- ZacchiaMDi IorioVTrepiccioneFCaterinoMCapassoGThe kidney in Bardet–Biedl syndrome: possible pathogenesis of urine concentrating defectKidney Dis (Basel)201732576528868293
- ZonaEZacchiaMDi IorioVCapolongoGRinaldiLCapassoGIl coinvolgimento renale nella sindrome di Bardet-Biedl [Pathophysiology of renal dysfunction in Bardet–Biedl syndrome]G Ital Nefrol20173456272 Italian28963828
- FentonRAKnepperMAUrea and renal function in the 21st century: insights from knockout miceJ Am Soc Nephrol200718367968817251384
- ZimmerhacklBLRobertsonCRJamisonRLThe medullary microcirculationKidney Int19873126416473550235
- FentonRAUrea transporters and renal function: lessons from knockout miceCurr Opin Nephrol Hypertens200817551351818695393
- ZacchiaMCapassoGDehydration: a new modulator of klotho expressionAm J Physiol Renal Physiol20113014F743F74421835769
- AmbühlPMAmemiyaMDanczkayMChronic metabolic acidosis increases NHE3 protein abundance in rat kidneyAm J Physiol19962714 Pt 2F917F9258898023
- PreisigPAThe acid-activated signaling pathway: starting with Pyk2 and ending with increased NHE3 activityKidney Int200772111324132917882150
- LiuLZacchiaMTianXAcid regulation of NaDC-1 requires a functional endothelin B receptorKidney Int201078989590420703215
- CapassoGUnwinRGiebischGRole of the loop of Henle in urinary acidificationKidney Int Suppl199133S33S351890799
- CapassoGUnwinRAgulianSGiebischGBicarbonate transport along the loop of Henle. I. Microperfusion studies of load and inhibitor sensitivityJ Clin Invest19918824304371830889
- GoodDWWattsBA3rdFunctional roles of apical membrane Na+/H+ exchange in rat medullary thick ascending limbAm J Physiol19962704 Pt 2F691F6998967348
- PaillardMH+ and HCO3− transporters in the medullary thick ascending limb of the kidney: molecular mechanisms, function and regulationKidney Int Suppl199865S36S419551430
- LedoussalCLorenzJNNiemanMLSoleimaniMSchultheisPJShullGERenal salt wasting in mice lacking NHE3 Na+/H+ exchanger but not in mice lacking NHE2Am J Physiol Renal Physiol20012814F718F72711553519
- de BruijnPILarsenCKFrischeSFurosemide-induced urinary acidification is caused by pronounced H+ secretion in the thick ascending limbAm J Physiol Renal Physiol20153092F146F15325995110
- HouillierPBourgeoisSMore actors in ammonia absorption by the thick ascending limbAm J Physiol Renal Physiol20123023F293F29722088435
- EladariDBlanchardALevielFFunctional and molecular characterization of luminal and basolateral Cl-/HCO-3 exchangers of rat thick limbsAm J Physiol19982753 Pt 2F334F3429729505
- CapassoGUnwinRRizzoMPicaAGiebischGBicarbonate transport along the loop of Henle: molecular mechanisms and regulationJ Nephrol200215Suppl 5S88S9612027225
- CapassoGUnwinRCianiFBicarbonate transport along the loop of Henle. II. Effects of acid-base, dietary, and neurohumoral determinantsJ Clin Invest19949428308388040339
- WeinerIDVerlanderJWRenal ammonia metabolism and transportCompr Physiol20133120122023720285
- AmlalHPaillardMBicharaMNH4+ transport pathways in cells of medullary thick ascending limb of rat kidney. NH4+ conductance and K+/NH4+(H+) antiportJ Biol Chem19942693521962219718071316
- BlanchardAEladariDLevielFTsimaratosMPaillardMPodevinRANH4+ as a substrate for apical and basolateral Na+-H+ exchangers of thick ascending limbs of rat kidney: evidence from isolated membranesJ Physiol1998506Pt 36896989503331
- LeeSLeeHJYangHSThornellIMBevenseeMOChoiISodium-bicarbonate cotransporter NBCn1 in the kidney medullary thick ascending limb cell line is upregulated under acidic conditions and enhances ammonium transportExp Physiol201095992693720591978
- WeinerIDMitchWESandsJMUrea and ammonia metabolism and the control of renal nitrogen excretionClin J Am Soc Nephrol20151081444145825078422
- BiverSBelgeHBourgeoisA role for Rhesus factor Rhcg in renal ammonium excretion and male fertilityNature2008456722033934319020613
- JainGOngSWarnockDGGenetic disorders of potassium homeostasisSemin Nephrol201333330030923953807
- TakahashiNChernavvskyDRGomezRAIgarashiPGitelmanHJSmithiesOUncompensated polyuria in a mouse model of Bartter’s syndromeProc Natl Acad Sci U S A200097105434543910779555
- LorenzJNBairdNRJuddLMImpaired renal NaCl absorption in mice lacking the ROMK potassium channel, a model for type II Bartter’s syndromeJ Biol Chem200227740378713788012122007
- KemterERathkolbBBankirLMutation of the Na(+)-K(+)-2Cl(−) cotransporter NKCC2 in mice is associated with severe polyuria and a urea-selective concentrating defect without hyperreninemiaAm J Physiol Renal Physiol20102986F1405F141520219826
- BaileyMACantoneAYanQMaxi-K channels contribute to urinary potassium excretion in the ROMK-deficient mouse model of type II Bartter’s syndrome and in adaptation to a high-K dietKidney Int2006701515916710355
- McCormickJAEllisonDHDistal convoluted tubuleCompr Physiol201551459825589264
- ZacchiaMCapassoGParvalbumin: a key protein in early distal tubule NaCl reabsorptionNephrol Dial Transplant20082341109111118083762
- KoulouridisEKoulouridisIMolecular pathophysiology of Bartter’s and Gitelman’s syndromesWorld J Pediatr201511211312525754753
- PresslerCAHeinzingerJJeckNLate-onset manifestation of antenatal Bartter syndrome as a result of residual function of the mutated renal Na+-K+-2Cl− co-transporterJ Am Soc Nephrol20061782136214216807401
- ZacchiaMCapolongoGRinaldiLCapassoGFisiopatologia dell’handling renale dell’acido urico [Renal handling of uric acid]G Ital Nefrol201532Suppl 62 Italian
- SeysEAndriniOKeckMClinical and genetic spectrum of Bartter syndrome type 3J Am Soc Nephrol20172882540255228381550
- GrillASchießlIMGessBFremterKHammerACastropHSalt-losing nephropathy in mice with a null mutation of the Clcnk2 geneActa Physiol (Oxf)2016218319821127421685
- HannanFMThakkerRVCalcium-sensing receptor (CaSR) mutations and disorders of calcium, electrolyte and water metabolismBest Pract Res Clin Endocrinol Metab201327335937123856265
- RiccardiDBrownEMPhysiology and pathophysiology of the calcium sensing receptor in the kidneyAm J Physiol20102983F485F499
- Vargas-PoussouRHuangCHulinPFunctional characterization of a calcium-sensing receptor mutation in severe autosomal dominant hypocalcemia with a Bartter-like syndromeJ Am Soc Nephrol20021392259226612191970
- LaghmaniKBeckBBYangSSPolyhydramnios, transient antenatal Bartter’s syndrome, and MAGED2 mutationsN Engl J Med2016374191853186327120771
- HouJRajagopaMYuASClaudins and the kidneyAnnu Rev Physiol20137547950123140368
- SimonDBLuYChoateKAParacellin-1, a renal tight junction protein required for paracellular Mg2+ resorptionScience1999285542410310610390358
- SikoraPZaniewMHaischLRetrospective cohort study of familial hypomagnesaemia with hypercalciuria and nephrocalcinosis due to CLDN16 mutationsNephrol Dial Transplant201530463664425477417
- KonradMSchallerASeelowDMutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvementAm J Hum Genet200679594995717033971
- BreiderhoffTHimmerkusNStuiverMDeletion of claudin-10 (Cldn10) in the thick ascending limb impairs paracellular sodium permeability and leads to hypermagnesemia and nephrocalcinosisProc Natl Acad Sci U S A201210935142411424622891322
- BongersEMHFSheltonLMMilatzSA novel hypokalemic-alkalotic salt-losing tubulopathy in patients with CLDN10 mutationsJ Am Soc Nephrol201728103118312828674042
- Hadj-RabiaSBrideauGAl-SarrajYMultiplex epithelium dysfunction due to CLDN10 mutation: the HELIX syndromeGenet Med Epub201783
- ThorleifssonGHolmHEdvardssonVSequence variants in the CLDN14 gene associate with kidney stones and bone mineral densityNat Genet200941892693019561606
- CorreTOlingerEHarrisSECommon variants in CLDN14 are associated with differential excretion of magnesium over calcium in urinePflugers Arch201746919110327915449
- StuiverMLainezSWillCCNNM2, encoding a basolateral protein required for renal Mg2+ handling, is mutated in dominant hypomagnesemiaAm J Hum Genet201188333334321397062
- FunatoYYamazakiDMikiHRenal function of cyclin M2 Mg2+ transporter maintains blood pressureJ Hypertens201735358559228033128
- MeyerTEVerwoertGCHwangSJGenome-wide association studies of serum magnesium, potassium, and sodium concentrations identify six loci influencing serum magnesium levelsPLoS Genet201068e100104520700443
- AbdelhadiOIancuDStanescuHKletaRBockenhauerDEAST syndrome: clinical, pathophysiological, and genetic aspects of mutations in KCNJ10Rare Dis201641e119504327500072
- ReicholdMZdebikAALiebererECNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel functionProc Natl Acad Sci U S A201010732144901449520651251
- FanLWangXZhangDVasopressin-induced stimulation of the Na(+)-activated K(+) channels is responsible for maintaining the baso-lateral K(+) conductance of the thick ascending limb (TAL) in EAST/SeSAME syndromeBiochim Biophys Acta20151852112554256226319417
- SuomalainenABattersbyBJMitochondrial diseases: the contribution of organelle stress responses to pathologyNat Rev Mol Cell Biol Epub201789
- EmmaFSalviatiLMitochondrial cytopathies and the kidneyNephrol Ther201713Suppl 1S23S2828577739
- EmmaFMontiniGParikhSMSalviatiLMitochondrial dysfunction in inherited renal disease and acute kidney injuryNat Rev Nephrol201612526728026804019
- TrepiccioneFZacchiaMCapassoGThe role of the kidney in salt-sensitive hypertensionClin Exp Nephrol2012161687222038257
- FrameAAWainfordRDRenal sodium handling and sodium sensitivityKidney Res Clin Pract201736211713128680820
- PetrazzuoloOTrepiccioneFZacchiaMCapassoGHypertension and renal calcium transportJ Nephrol201023Suppl 16S112S11721170867
- WarnockDGGenetic forms of human hypertensionCurr Opin Nephrol Hypertens200110449349911458030
- GrahamLADominiczakAFFerreriNRRole of renal transporters and novel regulatory interactions in the TAL that control blood pressurePhysiol Genomics201749526127628389525
- CapassoGRizzoMGaravagliaMUpregulation of apical sodium-chloride cotransporter and basolateral chloride channels is responsible for the maintenance of salt-sensitive hypertensionAm J Physiol Renal Physiol20082952F556F56718480177
- KirchnerKAIncreased loop chloride uptake precedes hypertension in Dahl salt-sensitive ratsAm J Physiol19922622 Pt 2R263R2681539735
- AvivAHollenbergNKWederAUrinary potassium excretion and sodium sensitivity in blacksHypertension200443470771314967834
- LuftFCGrimCEFinebergNWeinbergerMCEffects of volume expansion and contraction in normotensive whites, blacks, and subjects of different agesCirculation1979594643650421305
- MonetteMYRinehartJLiftonRPForbushBRare mutations in the human Na-K-Cl cotransporter (NKCC2) associated with lower blood pressure exhibit impaired processing and transport functionAm J Physiol Renal Physiol20113004F840F84721209010
- ZacchiaMCapassoGThe importance of uromodulin as regulator of salt reabsorption along the thick ascending limbNephrol Dial Transplant201530215816025422312
- ZacchiaMZacchiaEZonaERenal phenotype in Bardet–Biedl syndrome: a combined defect of urinary concentration and dilution is associated with defective urinary AQP2 and UMOD excretionAm J Physiol Renal Physiol20163114F686F69427488999
- GrahamLAPadmanabhanSFraserNJValidation of uromodulin as a candidate gene for human essential hypertensionHypertension201463355155824324041
- BlankensteinKIBorschewskiALabesRCalcineurin inhibitor cyclosporine A activates renal Na-K-Cl cotransporters via local and systemic mechanismsAm J Physiol Renal Physiol20173123F489F50128003191
- Esteva-FontCGuillén-GómezEDiazJMRenal sodium transporters are increased in urinary exosomes of cyclosporine-treated kidney transplant patientsAm J Nephrol201439652853524942911
- UnwinRCapassoGGiebischGPotassium and sodium transport along the loop of Henle: effects of altered dietary potassium intakeKidney Int1994464109210997861703
- McKayAJPetersonLNK infusion corrects thick ascending limb Cl reabsorption in K-depleted rats by an aldosterone-independent mechanismAm J Physiol19932645 Pt 2F792F7998498531
- GutscheHUPetersonLNLevineDZIn vivo evidence of impaired solute transport by the thick ascending limb in potassium-depleted ratsJ Clin Invest19847349089166707211
- YangSSLoYFWuCCSPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstrictionJ Am Soc Nephrol201021111868187720813865
- LinSHYuISJiangSTImpaired phosphorylation of Na+-K+-2Cl− cotransporter by oxidative stress-responsive kinase-1 deficiency manifests hypotension and Bartter-like syndromeProc Natl Acad Sci U S A201110842175381754321972418
- ChangCTHungCCTianYCYangCWWuMSCiclosporin reduces paracellin-1 expression and magnesium transport in thick ascending limb cellsNephrol Dial Transplant20072241033104017299004
- HoornEJEllisonDHDiuretic resistanceAm J Kidney Dis201769113614227814935
- BartoliERossiLSolaDCastelloLSainaghiPPSmirneCUse, misuse and abuse of diureticsEur J Intern Med20173991728233622
- KokotFHyla-KlekotLDrug-induced abnormalities of potassium metabolismPol Arch Med Wewn20081187–843143418714739
- SicaDACarterBCushmanWHammLThiazide and loop diureticsJ Clin Hypertens (Greenwich)201113963964421896142
- LapsiaVKazoryALoop diuretics for heart failure-associated hyponatremiaAm J Med20101238e5e6
- HalbgewachsCDomesTPostobstructive diuresis: pay close attention to urinary retentionCan Fam Physician201561213714225821871
- McDougalWSWrightFSDefect in proximal and distal sodium transport in post-obstructive diuresisKidney Int1972263043174670908
- YargerWEAynedjianHSBankNA micropuncture study of postobstructive diuresis in the ratJ Clin Invest19725136256375011104
- LiCWangWKwonTHKnepperMANielsenSFrøkiaerJAltered expression of major renal Na transporters in rats with bilateral ureteral obstruction and release of obstructionAm J Physiol Renal Physiol20032855F889F90112865255
- HanSJNohMRJungJMHydrogen sulfide-producing cystathionine γ-lyase is critical in the progression of kidney fibrosisFree Radic Biol Med201711242343228842346
- PernaAFDi NunzioAAmoresanoADivergent behavior of hydrogen sulfide pools and of the sulfur metabolite lanthionine, a novel uremic toxin, in dialysis patientsBiochimie20161269710727129884
- XiaMChenLMuhRWLiPLLiNProduction and actions of hydrogen sulfide, a novel gaseous bioactive substance, in the kidneysJ Pharmacol Exp Ther200932931056106219246614