Abstract
Secondary (AA) amyloidosis is a multisystem disorder complicating chronic infections or inflammatory diseases. It is characterized by extracellular deposit of fibrils composed of fragments of serum amyloid A (SAA), an acute phase reactant protein. The kidney is the most frequent organ involved, manifesting as progressive proteinuria and renal impairment. Attenuation of the level of circulating SAA protein by treating the underlying inflammatory condition remains the primary strategy in treating AA amyloidosis. However, at times, achieving adequate control of protein production can prove difficult. In addition, relapse of renal function often occurs rapidly following any subsequent inflammatory stimulus in patients with existing amyloidosis. Recently there has been an interest in finding other potential strategies targeting amyloid deposits themselves. Eprodisate is a sulfonated molecule with a structure similar to heparan sulfate. It competitively binds to the glycosaminoglycan-binding sites on SAA and inhibits fibril polymerization and amyloid deposition. Recent randomized clinical trial showed that it may slow down progressive renal failure in patients with AA amyloidosis. However confirmatory studies are needed and results of a second Phase III study are eagerly awaited to clarify whether or not eprodisate has a place in treating renal amyloid disease.
Keywords:
Introduction
The amyloidoses are a group of heterogeneous, life-threatening diseases, characterized by the deposition of highly organized, insoluble protein aggregates within organs and normal tissue. The abnormal deposition of β-sheet fibrillar proteins results in either localized or systemic disease and can be hereditary or acquired in nature. In the systemic form of the disease, it is possible for deposits to occur in all tissues of the human body with the exception of the brain (this is seen in locally occurring amyloid deposits in Alzheimer’s disease). The binding of Congo red stain to the β-pleated sheet emits a characteristic apple-green birefringence under polarized light, allowing a diagnosis of amyloidosis to be made.
To date, 28 structurally unrelated precursor proteins have been described that have the capacity to adopt a β-sheet structure.Citation1 All of these proteins are naturally soluble, and through processes of cleavage, aggregation, and misfolding, form fibrils with an abnormal β-sheet morphology.Citation2 The two most common precursor proteins implicated in the formation of amyloid proteins include monoclonal light chains leading to primary or AL amyloidosis, and serum amyloid A (SAA), which in turn causes secondary (reactive) or AA amyloidosis.
AA amyloidosis mainly occurs as a result of chronic inflammatory states, such as rheumatoid arthritis, ankylosing spondylitis, or psoriatic arthritis but can also result from chronic infections, Familial Mediterranean Fever (FMF), or Crohn’s disease (). Less commonly, Castleman’s disease, vasculitis, and cancers have also been implicated.Citation3 A recent single-center study detailing 20 years’ Florentine experience of AA amyloidosis showed that rheumatoid arthritis contributed to 45% of cases and 67% of patients had some form of renal involvement.Citation4 Other recent studies have shown that 5%–17% of patients with rheumatoid arthritis have amyloidosis reported as the cause of death.Citation5 In a study of 374 patients with AA amyloidosis, Lachmann et al reported that patients can experience symptoms of inflammatory disease for a median duration of approximately 17 years before a diagnosis of amyloidosis is made.Citation3
Table 1 List of causes of AA amyloid in order of incidence
The kidney is the organ chiefly affected by AA amyloidosis, manifesting as progressive proteinuria, nephrotic syndrome, and renal dysfunction.Citation3,Citation6 Up to 40% of patients with AA amyloidosis develop end stage renal failure.Citation3 The complications arising as a result of renal failure are the cause of death in 40%–60% of cases, with a median survival after diagnosis of 4–8 years.Citation7–Citation9 Old age, end stage renal failure at presentation, reduced serum albumin level, and persistent higher levels of SAA concentration are associated with poor prognosis.Citation3
Hepatic amyloid deposits are present in nearly a quarter of the patients affected with secondary amyloidosis.Citation3 Gastrointestinal involvement, which manifests as diarrhea and malabsorption syndrome, is present in approximately 20% of patients. The incidence of gastrointestinal disease is increasing as the availability of renal replacement therapy has led to the extension of patients’ lives and their subsequent ability to accumulate increasing amounts of amyloid deposits.Citation3,Citation10
In contrast with primary (AL) amyloidosis, other clinical features such as thyroid, splenic, cardiac, and autonomic nerve involvement are less frequent.Citation11 The survival rates have been shown to improve upon treatment of the underlying inflammatory condition.Citation12–Citation14
Pathophysiology of AA amyloidosis
The precursor SAA is a 104-amino-acid protein that is produced in response to inflammatory stimuli but its exact function remains unclear. This is an acute phase reactant and is produced in the liver in response to pro-inflammatory cytokines, and can be up regulated by up to 1000 times during episodes of acute inflammation.Citation15,Citation16 The principal cytokines involved in SAA induction are tumor necrosis factor (TNF), interleukin 1 (IL-1), interleukin 6 (IL-6), transcription factors such as SAA activating factor (SAF-1), and lipopolysaccharide (LPS).Citation17–Citation19 In addition, it has also been demonstrated that hepatic clearance of SAA during episodes of inflammation (both acute and chronic) is reduced, hence the elevated levels witnessed.Citation20
SAA is found in the serum as an apolipoprotein.Citation21 Under normal circumstances it is degraded without the development of amyloid fibrils.Citation22 In patients with amyloidosis however, the intermediate SAA products combine to form fibrils. These fibrils are deposited in the extracellular space of tissues and organs. Glycosaminoglycans such as heparan or dermatan sulfate, and serum amyloid P (SAP) then bind to these fibrils, which are then rendered invulnerable to degradation ().Citation23–Citation25 However, not all patients with chronic inflammation develop this complication and it remains largely unclear why some patients are prone to develop secondary (AA) amyloidosis while the others are protected. Certain genetic factors have been shown to increase the propensity for developing amyloidosis, such as polymorphisms in the gene coding for SAA1. Patients who are homozygous for the SAA1 isotype have a three- to sevenfold increased risk of developing amyloidosis.Citation26–Citation28 Other factors such as matrix metalloproteinase proteins (MMP), isoprenoid metabolism, heat shock proteins, and cathepsin D have also been implicated in the pathogenesis of AA amyloidosis.Citation29
Figure 1 The pathogenesis of amyloid fibrils.
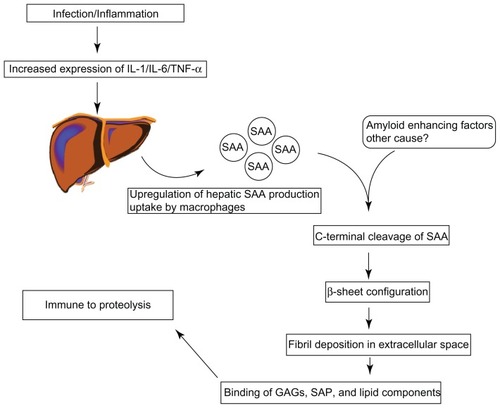
The exact mechanism by which the deposits affect organ function is not well known. Clearly large deposits can distort the integrity and function of a tissue on a structural level. Whilst positive correlations exist between the size of amyloid deposits and disease severity, there is a variable relationship between changes in renal function due to changes in size of amyloid deposits.Citation30
It has been suggested that amyloidogenic precursor proteins and intermediate filaments have direct toxic effects on the tissues, which are independent of the amyloid deposits. These toxicities might directly contribute to the disease manifestations. This view is supported by observations that proteinuria decreases rapidly after chemotherapy for primary AL amyloidosis and treatment of the underlying inflammatory condition in secondary AA amyloidosis, leading to a marked reduction in the production of the precursor amyloidogenic proteins.Citation31,Citation32 However, in a small published series, there was a lack of improvement in the size of amyloid deposits on repeat kidney biopsy in patients with primary AL amyloidosis following treatment, despite significant improvement in proteinuria.Citation33,Citation34 The measured level of serum SAA protein has been shown to correlate with disease activity and is a useful prognostic marker, and the outcome is thought to be favorable if the plasma SAA concentration is kept below 10 mg/L.Citation35
Current treatment options and challenges
Attenuation of the level of circulating SAA protein by way of treating the underlying inflammatory condition remains the primary strategy in treating AA amyloidosis. The median plasma concentration of SAA in healthy individuals is 3 mg/L, but the concentration can be 650-fold higher during an acute-phase response.Citation36 The relative risk of death amongst patients with a plasma SAA concentration of >155 mg/L is 18 times greater than those with a plasma SAA concentration of <4 mg/L, and four times greater than in patients with an SAA concentration of 4–9 mg/L.Citation3 Lachmann et al reported an improvement in renal functions in patients with a median SAA concentration of 6 mg/L and deterioration in patients with a median SAA concentration of 28 mg/L.Citation3 Proteinuria reduces significantly in patients with AA amyloidosis when the underlying inflammatory disease becomes quiescent.Citation31
Chronic pyogenic and granulomatous diseases (particularly tuberculosis) previously accounted for a far higher proportion of cases and were successfully treated with antimicrobial therapy and surgery for those with abscesses and osteomyelitis. Other therapeutic interventions include colchicine, which has been successfully used to treat FMF, thus preventing or treating the associated secondary AA amyloidosis.Citation3,Citation37 Conditions previously associated with AA amyloidosis such as Hodgkin’s disease have virtually been eliminated as a cause with increasingly effective chemotherapeutic agents. The majority of cases now seen in the developed world reflect the increasingly efficient treatment of infectious diseases, and are seen in those with inflammatory arthritides. Regression of amyloid deposits in these patients is dependent upon successful treatment with immunosuppressive strategies or biological agents such as anti-TNF therapies or interleukin-1 receptor antagonists.Citation38,Citation39 The increase in use of disease-modifying agents and biological treatments for rheumatic diseases has led to a subsequent decline in the incidence of patients requiring renal replacement therapy for amyloid.Citation40
Targeting the production of amyloidogenic proteins is a potentially effective strategy, but achieving adequate control of protein production can prove difficult. In addition, relapse of renal function often occurs rapidly following any subsequent inflammatory stimulus in patients with existing amyloidosis. Murine studies have suggested that the presence of an amyloid-enhancing factor (AEF) fast-tracks further surges of SAA into amyloid formation.Citation41 Other potential strategies that have been attempted include targeting of the amyloid deposits themselves. Serum amyloid P (SAP) is a plasma glycoprotein that is bound to the β-sheets and ubiquitous in all amyloid tissues, regardless of the sub-type. The presence of SAP has formed the basis for a key assessment tool allowing quantification of amyloid deposition in tissues, by the binding of radioisotope-labeled SAP to amyloid deposits.Citation42 Therapies targeting SAP have previously been developed that target the SAP–amyloid fibril interaction and allow the removal of SAP by the liver.Citation43 SAP monoclonal antibodies have also been developed in murine models and have been shown to reduce visceral amyloid deposits by triggering a complement-dependent macrophage-derived reaction.Citation44 Recently, antisense oligonucleotides have also been successfully used to downregulate expression of SAA in animal models.Citation45
Introduction to eprodisate
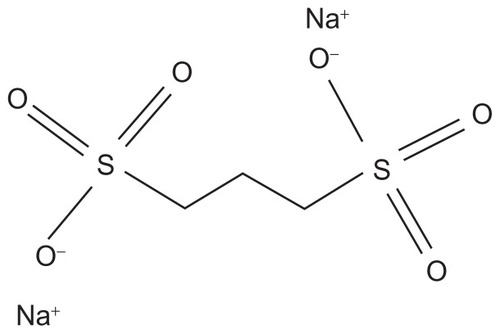
As mentioned above, sulfated glycosaminoglycans, such as dextran and heparan sulfate, are always associated anatomically with amyloid deposits, regardless of the nature of the protein deposited, and are necessary for the assembly of the fibrils as well as the stability of tissue amyloid deposits.Citation46–Citation49 Heparan sulfate is considered to be responsible for the stability of SAA2, which is the immediate precursor to AA amyloid in adopting the β-sheet structure that is characteristic of the amyloid protein-folding patterns.Citation50 Pre-clinical studies using small-molecule anionic sulfonates or sulfates significantly reduced murine splenic AA amyloid progression by disruption of the heparan sulfate-β-peptide fibril aggregate by competitively binding to the glycosaminoglycan-binding sites, thus inhibiting fibril polymerization.Citation51 Four compounds were shown to be significantly effective in a dose-dependent manner in animal models, and of these, 1–3 propanedisulfonic acid disodium salt or eprodisate (NC-503, Kiacta®, previously Fibrillex™, marketed by Neurochem, Laval, Canada), was taken forward to human studies and used in the Phase III eprodisate for AA amyloidosis (EFAAT) trial. Eprodisate is similar in structure to heparan sulfate and is a low molecular weight, negatively charged, sulfonated molecule ().Citation51,Citation52
Figure 2 The chemical structure of 1,3 propanedisulfonic acid disodium salt.
Pharmacokinetics safety and efficacy
Pre-clinical pharmacokinetic studies showed that eprodisate has good bioavailability when administered orally and is not protein-bound.Citation52 Pharmacokinetic analysis of Phase I studies involving healthy volunteers showed a high inter-individual variability in plasma concentration following oral doses ranging from 100–2400 mg. Maximum plasma concentrations were reached within 15–60 minutes post-dose. A plasma half-life of approximately 10–20 hours was estimated from a multiple rising oral dose study.Citation49 Eprodisate is primarily excreted by the kidneys and the plasma concentration is increased in patients with renal impairment requiring dose adjustment.Citation49,Citation52 Animal studies using eprodisate at a high daily dose of 2 gm/kg/day over a 10-month period showed that the drug was well tolerated with low toxicity potential and was found to be nonmutagenic.Citation53
Eprodisate was assessed in a multicenter, randomized, double-blind, placebo-controlled trial to evaluate its efficacy and safety in patients with AA amyloidosis and renal involvement.Citation54 Renal involvement was defined as proteinuria of greater than 1 g/day or a creatinine clearance of <60 mL/minute. Patients were excluded if their creatinine clearance was <20 mL/minute or they had renal disease attributable to other than AA amyloidosis. Significant liver dysfunction and diabetes were also amongst the exclusion criteria.
One hundred and eighty-three patients were assigned to the treatment drug, or placebo, and were well matched for age, gender and weight. Drug dosages were calculated according to baseline renal function. The study drug was administered orally twice daily at least 1 hour before or 2 hours after a meal. Patients with creatinine clearance rates of less than 30 mL per minute received a total of 800 mg of eprodisate per day in two divided doses, those with rates of 30–80 mL per minute received a total of 1600 mg per day in two divided doses, and those with rates of more than 80 mL per minute received a total of 2400 mg of eprodisate per day in two divided doses. Doses were decreased during the study if creatinine clearance decreased. Treatment of the underlying inflammatory disease was determined by the patient’s physician. Patients were followed up 4 monthly for a total of 2 years.
The primary endpoint of the study was a composite of decline in renal function or death. Worsening renal function was defined as doubling of serum creatinine, 50% reduction in creatinine clearance from the baseline, or progression to end-stage renal failure. Of the 183 patients, 124 completed the study. The adverse events were similar between the two groups. There were five deaths in each group during or within 15 days after completion of administration of the study drug. Two patients in the eprodisate group died of ischemic stroke, one patient had nephrotic syndrome as cause of death while one each had gastrointestinal hemorrhage and pneumonia. The deaths in the placebo group were due to ischemic stroke, amyloid cardiomyopathy, bowel perforation, sepsis, and pancytopenia in one patient each. None of the deaths were considered to be related to eprodisate itself.
There was a clear trend towards arresting the decline in renal function in the treatment group compared to the placebo group with a 42% reduction seen (0.37–0.93, P = 0.02), which was independent of baseline renal function or circulating SAA concentration. Urinary protein excretion, however, was not affected. Some discrepancies were evident; the placebo arm had significantly more patients with a higher diastolic blood pressure and had a higher median serum creatinine level. The placebo arm also tended towards higher median SAA and C-reactive protein (CRP) levels although the differences between the two arms were not statistically significant (P = 0.14 for both). The treatment arm had significantly more patients with chronic infections (21 versus 9, P = 0.01) raising the possibility that treatment with antimicrobial therapy may have partially accounted for some of the effect seen, given that median SAA and CRP levels were lower in this group. The acknowledged mechanism of action of eprodisate means that the glycosaminoglycan-amyloid fibril complex is disrupted, thereby preventing formation of new amyloid deposits. This is possibly why renal function in the treatment arm declined more slowly than in the placebo arm. There appeared to be no improvement in the levels of proteinuria in the treatment group suggesting that glomerular damage previously incurred by the kidney was not affected by the putative lack of development of additional amyloid deposits.
The future
In 2006, Neurochem Inc, announced that a letter had been received from the US Food and Drug Administration requesting further efficacy and safety information that would need to be addressed through additional clinical trials before approval would be given for its use in AA amyloidosis. One-year follow-up data from the original randomized trial revealed that eprodisate continued to be safe, and that comparisons looking at progression to dialysis/ESRD, or the composite endpoint of renal decline and death favored those patients in the treatment arm.Citation55 In 2008, it was announced that a second Phase III trial would be initiated as recommended by both European and US regulatory agencies. Recruitment commenced in November 2010 and the estimated date of completion of the study is May 2014. The primary outcome measures are a decrease in creatinine clearance of 40% from baseline, an increase in serum creatinine of 80%, or progression to end-stage renal disease.Citation56
Conclusion
Clinicians currently have a limited repertoire of therapeutic options when confronted with a patient with AA amyloidosis, although treating the underlying inflammatory condition remains the primary strategy. The preliminary results from Dember et al’s studyCitation54 are certainly encouraging and the results from the second Phase III study will clarify whether or not eprodisate has a place in treating renal amyloid disease. At this stage, it is not known whether using eprodisate as an adjunctive therapy earlier on in the course of disease may affect outcomes, perhaps when SAA levels start to rise consistently above 4 mg/L. Strategies such as eprodisate that are designed to disturb the formation of amyloid fibrils, and other molecular techniques that target the stability of amyloid fibrils offer a new way of treating a potentially devastating condition.
Disclosure
The authors report no conflicts of interest in this work.
References
- SipeJDBensonMDBuxbaumJNAmyloid fibril protein nomenclature: 2010 recommendations from the nomenclature committee of the International Society of AmyloidosisAmyloid2010173–410110421039326
- MerliniGBellottiVMolecular mechanisms of amyloidosisN Engl J Med2003349658359612904524
- LachmannHJGoodmanHJGilbertsonJANatural history and outcome in systemic AA amyloidosisN Engl J Med2007356232361237117554117
- CaniaABergesioFCurciarelloGThe Florence Register of amyloidosis: 20 years’ experience in the diagnosis and treatment of the disease in the Florence district areaAmyloid201118Suppl 1818321838443
- KoivuniemiRPaimelaLSuomalainenRLeirisalo-RepoMAmyloidosis as a cause of death in patients with rheumatoid arthritisClin Exp Rheumatol200826340841318578961
- GertzMAKyleRASecondary systemic amyloidosis: response and survival in 64 patientsMedicine (Baltimore)19917042462562067409
- SchnitzerTJAnsellBMAmyloidosis in juvenile chronic polyarthritisArthritis Rheum1977202 Suppl245252263902
- MoroniGBanfiGMontoliAChronic dialysis in patients with systemic amyloidosis: the experience in northern ItalyClin Nephrol199238281851516284
- AkpolatTAkpolatIKandemirBBehcet’s disease and AA-type amyloidosisAm J Nephrol2000201687010644872
- SattianayagamPTHawkinsPNGillmoreJDSystemic amyloidosis and the gastrointestinal tractNat Rev Gastroenterol Hepatol200961060861719724253
- TuglularSYalcinkayaFPaydasSA retrospective analysis for aetiology and clinical findings of 287 secondary amyloidosis cases in TurkeyNephrol Dial Transplant200217112003200512401861
- BerglundKThysellHKellerCResults, principles and pitfalls in the management of renal AA-amyloidosis; a 10–21 year followup of 16 patients with rheumatic disease treated with alkylating cytostaticsJ Rheumatol19932012205120578014932
- GoldsmithDJRobertsISShortCDMallickNPComplete clinical remission and subsequent relapse of bronchiectasis-related (AA) amyloid induced nephrotic syndromeNephron19967435725768938683
- HazenbergBPvan RijswijkMHClinical and therapeutic aspects of AA amyloidosisBaillieres Clin Rheumatol1994836616907954868
- MarhaugGThree assays for the characterization and quantitation of human serum amyloid AScand J Immunol19831843293386417768
- HoffmanJSBendittEPChanges in high density lipoprotein content following endotoxin administration in the mouse. Formation of serum amyloid protein-rich subfractionsJ Biol Chem19822571710510105177107614
- ThornCFLuZYWhiteheadASRegulation of the human acute phase serum amyloid A genes by tumour necrosis factor-alpha, interleukin-6 and glucocorticoids in hepatic and epithelial cell linesScand J Immunol200459215215814871291
- UtsunomiyaINagaiSOh-ishiSSequential appearance of IL-1 and IL-6 activities in rat carrageenin-induced pleurisyJ Immunol19911476180318091716281
- RayAShakyaAKumarDBensonMDRayBKInflammation- responsive transcription factor SAF-1 activity is linked to the development of amyloid A amyloidosisJ Immunol200617742601260916888022
- GollaherCJBaussermanLLHepatic catabolism of serum amyloid A during an acute phase response and chronic inflammationProc Soc Exp Biol Med199019432452502113283
- ParmeleeDCTitaniKEricssonLHEriksenNBendittEPWalshKAAmino acid sequence of amyloid-related apoprotein (apoSAA1) from human high-density lipoproteinBiochemistry19822114329833037115671
- HusebekkASkogenBHusbyGMarhaugGTransformation of amyloid precursor SAA to protein AA and incorporation in amyloid fibrils in vivoScand J Immunol19852132832873922050
- TennentGALovatLBPepysMBSerum amyloid P component prevents proteolysis of the amyloid fibrils of Alzheimer disease and systemic amyloidosisProc Natl Acad Sci U S A19959210429943037753801
- LiJPGalvisMLGongFIn vivo fragmentation of heparan sulfate by heparanase overexpression renders mice resistant to amyloid protein A amyloidosisProc Natl Acad Sci U S A2005102186473647715843464
- GellermannGPAppelTRTannertARaft lipids as common components of human extracellular amyloid fibrilsProc Natl Acad Sci U S A2005102186297630215851687
- Gershoni-BaruchRBrikRZacksNShinawiMLidarMLivnehAThe contribution of genotypes at the MEFV and SAA1 loci to amyloidosis and disease severity in patients with familial Mediterranean feverArthritis Rheum20034841149115512687559
- CazeneuveCAjrapetyanHPapinSIdentification of MEFV-independent modifying genetic factors for familial Mediterranean feverAm J Hum Genet20006751136114311017802
- AltiokOSeguretFTouitouIMEFV sequence variants and amyloidosis: still an enigmatic questionHum Mutat2003211969712497636
- van der HilstJCHRecent insights into the pathogenesis of type AA amyloidosisScientific World Journal20111164165021403980
- SnanoudjRDurrbachAGauthierEChanges in renal function in patients with familial amyloid polyneuropathy treated with orthotopic liver transplantationNephrol Dial Transplant20041971779178515150345
- ElkayamOHawkinsPNLachmannHYaronMCaspiDRapid and complete resolution of proteinuria due to renal amyloidosis in a patient with rheumatoid arthritis treated with infliximabArthritis Rheum200246102571257312384913
- LeungNDispenzieriAFervenzaFCRenal response after high-dose melphalan and stem cell transplantation is a favorable marker in patients with primary systemic amyloidosisAm J Kidney Dis200546227027716112045
- ZeierMPerzJLinkeRPNo regression of renal AL amyloid in monoclonal gammopathy after successful autologous blood stem cell transplantation and significant clinical improvementNephrol Dial Transplant200318122644264714605290
- KyleRAWagonerRDHolleyKEPrimary systemic amyloidosis: Resolution of the nephrotic syndrome with melphalan and prednisoneArch Intern Med19821428144514477103624
- GillmoreJDLovatLBPerseyMRPepysMBHawkinsPNAmyloid load and clinical outcome in AA amyloidosis in relation to circulating concentration of serum amyloid A proteinLancet20013589275242911454373
- LedueTBWeinerDLSipeJDPoulinSECollinsMFRifaiNAnalytical evaluation of particle-enhanced immunonephelometric assays for C-reactive protein, serum amyloid A and mannose-binding protein in human serumAnn Clin Biochem199835Pt 67457539838988
- GillmoreJDHawkinsPNPepysMBAmyloidosis: a review of recent diagnostic and therapeutic developmentsBr J Haematol19979922452569375734
- KeersmaekersTClaesKKuypersDRde VlamKVerschuerenPWesthovensRLong-term efficacy of infliximab treatment for AA-amyloidosis secondary to chronic inflammatory arthritisAnn Rheum Dis200968575976119366896
- Fernandez-NebroAOliveACastroMCVarelaAHRieraEIrigoyenMVLong-term TNF-alpha blockade in patients with amyloid A amyloidosis complicating rheumatic diseasesAm J Med2010123545446120399323
- von HuttenHMihatschMLobeckHRudolphBErikssonMRockenCPrevalence and origin of amyloid in kidney biopsiesAm J Surg Pathol20093381198120519561448
- KisilevskyRYoungIDPathogenesis of amyloidosisBaillieres Clin Rheumatol1994836136267954865
- HawkinsPNLavenderJPPepysMBEvaluation of systemic amyloidosis by scintigraphy with 123I-labeled serum amyloid P componentN Engl J Med199032385085132377176
- PepysMBHerbertJHutchinsonWLTargeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosisNature2002417688625425912015594
- BodinKEllmerichSKahanMCAntibodies to human serum amyloid P component eliminate visceral amyloid depositsNature20104687320939720962779
- Kluve-BeckermanBHardwickJDuLAntisense oligonucleotide suppression of serum amyloid A reduces amyloid deposition in mice with AA amyloidosisAmyloid201118313614621830877
- SnowADWillmerJKisilevskyRA close ultrastructural relationship between sulfated proteoglycans and AA amyloid fibrilsLab Invest19875766876982447384
- LyonAWNarindrasorasakSYoungIDCo-deposition of basement membrane components during the induction of murine splenic AA amyloidLab Invest19916467857902046330
- StenstadTMagnusJHHusbyGCharacterization of proteoglycans associated with mouse splenic AA amyloidosisBiochem J1994303Pt 26636707980430
- KisilevskyRThe relation of proteoglycans, serum amyloid P and apo E to amyloidosis current status, 2000Amyloid200071232510842701
- McCubbinWDKayCMNarindrasorasakSKisilevskyRCircular- dichroism studies on two murine serum amyloid A proteinsBiochem J198825637757833223951
- KisilevskyRLemieuxLJFraserPEKongXHultinPGSzarekWAArresting amyloidosis in vivo using small-molecule anionic sulfonates or sulfates: implications for Alzheimer’s diseaseNat Med1995121431487585011
- GandhiNSManceraRLHeparin/heparan sulfate-based drugsDrug Discov Today20101523–241058106920974281
- ManentiLTansindaPVaglioAEprodisate in amyloid A amyloidosis: a novel therapeutic approach?Expert Opin Pharmacother20089122175218018671471
- DemberLMHawkinsPNHazenbergBPEprodisate for the treatment of renal disease in AA amyloidosisN Engl J Med2007356232349236017554116
- NeurochemNeurochem submits a complete response to FDA approvable letter for Kiacta2006 [cited November 22, 2011]. Available from: http://drugs.com/nda/kiacta_061016.html
- CT Development America, IncEfficacy and safety study of KIACTA in preventing renal function decline in AA amyloidosisClinicalTrials gov [website on the Internet]Bethseda, MDUS National Library of Medicine2011 [updated December 6, 2011]. Available from: http://clinicaltrials.gov/ct2/show/NCT01215747?term=eprodisate&rank=1. NLM identifier:NCT01215747Accessed February 7, 2012