
Abstract
Cystinosis is an autosomal recessive inherited lysosomal storage disease. It is characterized by generalized proximal tubular dysfunction known as renal Fanconi syndrome and causes end-stage renal disease by the age of about 10 years if left untreated. Extrarenal organs are also affected, including the thyroid gland, gonads, pancreas, liver, muscle, and brain. Treatment consists of administration of cysteamine, resulting in depletion of cystine that is trapped inside the lysosomes. Since cysteamine has a short half-life, it should be administered every 6 hours. Recently, a new delayed-release formulation was marketed, that should be administered every 12 hours. The first studies comparing both cysteamine formulations show comparable results regarding white blood cell cystine depletion (which serves as a measure for cystine accumulation in the body), while a slightly lower daily dose of cysteamine can be used.
Introduction
Cystinosis is a lysosomal storage disease. It is inherited in an autosomal recessive manner and is caused by mutations in the CTNS gene, which is located on chromosome 17p13.Citation1 These mutations cause intralysosomal cystine accumulation in cells throughout the body. Today, over 100 pathogenic mutations are known to cause cystinosis. The most frequent mutation found in western Europe is a large deletion of 57,257 base pairs.Citation2
CTNS encodes the lysosomal cystine transporter cystinosin, which is a 367 amino acid lysosomal membrane protein, with seven transmembrane domains. Cystinosin is a proton-cystine cotransporter, coupling the excretion of both cystine and protons out of the lysosome. Protons are carried back into the lysosome by H+-ATPase.Citation3 Besides the cystinosin that is localized on the lysosomal membrane, a second isoform exists due to alternative splicing of CTNS messenger RNA (mRNA). This isoform lacks one of the two lysosomal targeting motifs, consists of the amino acid sequence GYDQL, and was named cystinosin-LKG (the letters LKG representing its last three amino acids). Cystinosin-LKG is localized at the plasma membrane in lysosomes and several other organelles, including the Golgi apparatus and the endoplasmic reticulum. Its exact function remains unclear; however, it is known to facilitate the transport of cystine in an acidic environment, just like its counterpart cystinosin.Citation4
Cystinosis has an incidence of one per 100,000–200,000 live births, and in suspected cases, the diagnosis can be confirmed by measuring white blood cell (WBC) cystine levels and/or by CTNS gene analysis. Three clinical forms of cystinosis can be distinguished, the most frequent being infantile nephropathic cystinosis (OMIM 219800). Patients generally present before the age of 12 months with polyuria, polydipsia, and failure to thrive caused by generalized proximal tubular damage, also known as renal Fanconi syndrome. It is characterized by increased urinary excretion of several small molecules, including glucose, salts, amino acids, and low molecular weight proteins. Photophobia is caused by corneal cystine crystals, which can be recognized by an experienced ophthalmologist from the age of 12 months.Citation5 Further, several other organs can be involved during progression of the disease. These include the thyroid gland (hypothyroidism),Citation6 gonads (hypogonadism, azoospermia),Citation7,Citation8 pancreas (exocrine and endocrine insufficiency),Citation9,Citation10 muscles (hypotrophy, swallowing difficulties),Citation11 and the peripheral and central nervous systems.Citation11 Late-onset or juvenile nephropathic cystinosis (OMIM 219900) is characterized by the same symptoms as infantile cystinosis, but with a disease onset mostly after the first decade of life and a slower rate of disease progression. In contrast, patients with the ocular form of cystinosis (OMIM 219750) only have photophobia due to corneal cystine crystals while other organs remain spared. The latter two forms of cystinosis are very rare; over 95% of all cystinosis patients are diagnosed with the infantile form.Citation5
Treatment with cysteamine
Cysteamine (β-mercaptoethylamine) was first introduced as a possible therapeutic agent to treat cystinosis in 1976,Citation12 and until now has been the only available treatment for the disease. After its introduction as a treatment for patients with cystinosis, cysteamine was first prescribed in the form of cysteamine HCl. Because of the low bioavailability of the hydrochloride salt, it was replaced by phosphocysteamine in 1980.Citation13 In 1994, cysteamine bitartrate (Cystagon®, Mylan Pharma, Morgantown, WV, USA) was introducedCitation14 and this is currently the most widely used preparation of cysteamine. Recently, an enteric-coated preparation of cysteamine bitartrate (Procysbi™, Raptor Pharmaceuticals Inc., Novato, CA, USA) was approved by the US Food and Drug Administration and European Medicines Agency for the treatment of cystinosis.
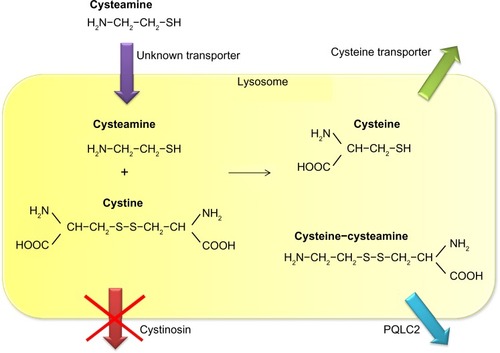
Cysteamine uses an unknown transporter to enter the lysosome and subsequently breaks the disulfide bond in cystine. This leads to formation of cysteine, which leaves the lysosome using the cystine transporter, and cysteine-cysteamine disulfide, which leaves the lysosome using the PQLC2 transporter ().Citation5,Citation15 Once in the cytosol, cysteine can be further metabolized to form glutathione, one of the cell’s most potent redox buffers. Indeed, in vitro experiments showed that the altered redox status demonstrated in cystinotic cells was corrected by administration of cysteamine, thereby increasing intracellular glutathione levels.Citation16
Figure 1 Mechanism of lysosomal cystine depletion by cysteamine. Cysteamine enters the lysosome through an unknown transporter and breaks the disulfide bond in cystine. This results in formation of cysteine and a new cysteine-cysteamine mixed disulfide, each of which can exit the lysosome through its own transporter.
One hour after ingestion of cysteamine, the drug’s plasma levels reach their maximum while WBC cystine levels are at their minimum, followed by a gradual decline in plasma cysteamine levels and a gradual increase of WBC cystine levels. Both reach their original values around 6 hours after ingestion.Citation17 This underscores the importance of a dosing regimen in which cysteamine is administered every 6 hours; ideally, it should also be administered during the night.Citation18 Understandably, such a dosing scheme causes a great burden in terms of compliance with cysteamine therapy.
Treatment of cystinosis patients with cysteamine is further hampered by the side effects of the drug. When cysteamine was first prescribed to patients with cystinosis, several of them developed hyperthermia, lethargy, and rash. However, all of these side effects were reversible after the treatment was discontinued, and none of these adverse events are noted today since it has become common practice to introduce cysteamine at a low dose with a subsequent gradual increase over several weeks.Citation19 Gastrointestinal complaints are due to increased gastric acid secretion after ingestion of cysteamine,Citation20 and thus concomitant use of proton pump inhibitors is advised.Citation21 After administration of cysteamine, a small amount is metabolized into the volatile sulfur compounds methanethiol and dimethylsulfide, causing halitosis.Citation22 Recently, eight cystinosis patients were found to have new adverse events associated with cysteamine, consisting of bruise-like lesions on the elbows, skin striae, and severe bone and muscular pain. Skin biopsies taken from the bruise-like lesions showed reactive angioendotheliomatosis and alterations in collagen fiber diameter.Citation23 The reactive angioendotheliomatosis could be attributed to increase survival of dermal microvascular endothelial cells after exposure to cysteamine,Citation24 while the collagen fiber abnormalities could be the result of a synergistic effect of cysteamine and copper deficiency (which was found in all patients identified as having these side effects) on inhibition of collagen cross-linking.Citation25
The newly reported side effects of cysteamine resulted in new recommendations regarding dosing. Instead of using body weight to calculate the dose of cysteamine (50 mg/kg/day, with a maximum of 90 mg/kg/day) it is now advised to use body surface area (1.30 g/m2/day, with a maximum of 1.95 g/m2/day). This difference in calculation method is particularly important in growing children, in whom body weight increases faster than body surface area. This may lead to higher (potentially toxic) doses when using body weight to calculate the daily cysteamine dose, since 90 mg/kg may exceed 1.95 g/m2.Citation23
Despite its side effects and four times daily dosing regimen, cysteamine remains the cornerstone of treatment for cystinosis and adequate administration of cysteamine significantly reduces the rate of progression towards end-stage renal disease, and postpones or even prevents the occurrence of extrarenal complications.Citation26–Citation29 Hypothyroidism, myopathy, pulmonary dysfunction, and diabetes mellitus (caused by pancreas insufficiency) occur less frequently in cystinosis patients who have been treated with cysteamine for a prolonged period of time.Citation27,Citation30 Further, while children with cystinosis show more pronounced growth retardation than children with other renal diseases, adequate treatment with cysteamine significantly improves growth parameters.Citation29,Citation31,Citation32 Nonetheless, most children with cystinosis also require treatment with recombinant human growth hormone to achieve a final height higher than two standard deviations below the mean.Citation29,Citation32
Unfortunately, not all complications of cystinosis are delayed by administration of cysteamine. Cysteamine does not influence the incidence of primary hypogonadism in male patients with cystinosis, nor does it improve delayed puberty. Also, male infertility due to azoospermia is not ameliorated by adequate treatment with cysteamine.Citation7,Citation8 Further, systemic administration of cysteamine does not prevent formation of corneal cystine crystals,Citation33 indicating a need for topical cysteamine eye drops which should be applied several times daily. Recently, a new gel formulation of cysteamine (Cystadrops®, Orphan Europe, Montluçon, France) was tested for its ability to diminish the formation of corneal cystine crystals, with better effects and less applications needed per day compared with the usual cysteamine hydrochloride formulation.Citation34
Other novel treatment strategies
The most promising results with regard to treatment of cystinosis without use of cysteamine come from the field of stem cell transplantation. In a CTNS−/− mouse model treated with wild-type allogeneic bone marrow and hematopoietic stem cells, the mice that received a bone marrow graft showed better renal function after 4 and 15 months, as well as decreased cystine content in a number of tissues, including the brain, eyes, heart, kidneys, liver, skeletal muscle, and spleen.Citation35,Citation36 However, despite these promising results, tissue cystine levels remained higher than those in asymptomatic heterozygous carriers, so administration of cysteamine may still be warranted.
In addition to bone marrow transplantation, gene therapy was also studied in the CTNS−/− mouse model in order to minimize the use of immunosuppressive agents, along with the risks of rejection and development of graft versus host disease. Hematopoietic stem cells from CTNS−/− mice were transduced with the human CTNS complementary DNA (cDNA) sequence followed by transplantation into CTNS−/− mice. Again, tissue cystine levels were significantly decreased after 4 and 8 months, including in the brain, eyes, kidneys, liver, muscle, and spleen, but were still higher than those after wild-type bone marrow transplantation.Citation37
Prodrugs are under development in order to diminish the bad taste and smell of cysteamine and to increase its half-life. Several prodrugs of cysteamine have been tested in vitro;Citation38,Citation39 however, none are as effective as cysteamine in decreasing intracellular cystine levels, and none have been tested in vivo.
Delayed-release cysteamine bitartrate
The observation that administration of cysteamine directly in the small intestine resulted in higher plasma cysteamine levels with a higher area under the curve when compared with gastric administration of the drugCitation40 led to development of the enteric-coated formulation of cysteamine bitartrate (Procysbi). In contrast with the immediate-release formulation of cysteamine bitartrate, this new delayed-release formulation can be administered every 12 hours instead of every 6 hours.Citation41 The delayed-release formulation consists of capsules containing small granules than can be administered through a feeding tube. This is important, given that most small children with cystinosis require a lot of fluids and electrolyte supplements, and thus have feeding tubes that are also used for administration of medication.
In a Phase III trial testing the noninferiority of the delayed-release formulation, 41 nontransplanted patients with cystinosis were randomized to treatment with either immediate-release or delayed-release cysteamine bitartrate for 3 weeks and then switched to the other formulation for a further 3 weeks. The mean difference in WBC cystine levels after treatment with the delayed-release formulation compared with the immediate-release formulation was 0.08 nmol ½ cystine/mg protein (95.8% confidence interval 0.00–0.16), meaning that the primary endpoint of noninferiority was met.Citation42
Interestingly, the average total daily steady-state dose of the delayed-release formulation was 82% of the previously established incoming dose of the immediate-release formulation,Citation42 indicating that similar results for depletion of WBC cystine can be achieved with a slightly lower daily amount of cysteamine. In theory, this could result in fewer side effects. An early pilot study was conducted comparing the excretion of volatile sulfur compounds in four patients after ingestion of immediate-release or delayed-release cysteamine bitartrate (as part of the crossover Phase III clinical trial). This study showed a trend towards a lower area under the curve for dimethylsulfide excretion in all four tested patients after ingestion of the delayed-release formulation. However, this trend did not reach statistical significance, patient numbers were small, and the measured levels of dimethylsulfide remained above those that cause halitosis at all time points.Citation43
A long-term study is presently underway using delayed-release cysteamine bitartrate in cystinosis patients with renal Fanconi syndrome and in those who have undergone renal transplantation. Again, as part of this study, excretion of volatile sulfur compounds in breath will be measured in a subgroup of patients, comparing the levels at study onset when patients are treated with the immediate-release formulation and again a few months after treatment with the delayed-release formulation. The final results of this study are pending, but preliminary data show that dimethylsulfide breath levels again remained persistently above the threshold that causes halitosis; however, further analysis should determine whether the area under the curve is influenced by the delayed-release formulation. Previous reports regarding the long-term treatment of two cystinosis patients with the delayed-release formulation show a good effect on the mean trough WBC cystine level, with no deterioration of renal, thyroid, or liver function observed during a treatment period of 6 years.Citation44
Prognosis
Overall, the quality of life for patients with cystinosis has improved significantly since the early 1980s. Since the use of cysteamine to treat cystinosis and the introduction of renal replacement therapy in pediatric populations (either through dialysis or by renal transplantation), patients with cystinosis are increasingly surviving into adulthood, whereas before the introduction of cysteamine as a possible treatment for cystinosis they generally died from end-stage renal disease by around the age of 10 years.Citation27 This increased life expectancy has caused an increase in the number of longer-term complications of the disease, that were not noticed in younger patients. This increased interest and recognition of longer-term complications of cystinosis prompted a search for a possible treatment for these complications. For instance, male infertility due to azoospermia was first described only a few years ago, awareness of which has enabled patients to seek further treatment options, such as testicular sperm extraction, which could open the possibility for male patients to produce their own offspring.Citation8 Another example is the severe growth retardation seen in patients with cystinosis. Since this is now treated more effectively (eg, by improving nutritional and electrolyte status, prescribing cysteamine, and administration of recombinant human growth hormone), today most cystinosis patients can reach a final height within the normal range,Citation29,Citation32 which further improves their quality of life.
Apart from better recognition and treatment of (late-term) complications of cystinosis, better treatment of the disease also improves the long-term outcome for the patient. Cystinosis is now more frequently recognized at a young age, resulting in more patients being adequately treated with cysteamine at a young age. This is very important, since early initiation of cysteamine therapy can significantly postpone or even prevent complications of the disease, including deterioration of renal function, hypothyroidism, and myopathy.Citation26–Citation29 However, one of the most important burdens regarding cysteamine therapy is the need to administer the drug every 6 hours in order to ensure adequate WBC cystine depletion.Citation18 Development of the new delayed-release formulation can overcome this problem, which will likely improve patient compliance with therapy, resulting in fewer long-term complications of cystinosis and improving patient life expectancy even further.
It remains uncertain to what degree potential new therapies for cystinosis (including bone marrow transplantation and/or gene therapy) will further improve the long-term outcomes. Although impressive results have been documented in the CTNS−/− mouse model, there are as yet no results of such treatment in humans.
Conclusion
From 1976Citation12 until now, cysteamine has been the only treatment available for cystinosis. The advent of cysteamine has significantly improved the outcome for patients with cystinosis, as have the better recognition and treatment of the complications of the disease. New treatment options, such as bone marrow transplantation are promising, but are not currently performed routinely in humans. Further, although intracellular cystine levels in a number of tissues in CTNS−/− mice decrease significantly after bone marrow transplantation derived from their wild-type litter mates, cystine levels remain above those of asymptomatic heterozygous carriers.Citation35,Citation36
Thus, even after bone marrow transplantation, there may still be a need for low doses of cysteamine to decrease the remaining accumulation of cystine. This indicates a need to further improve the treatment of cystinosis with cysteamine. In this regard, the new delayed-release formulation of cysteamine bitartrate is of importance to improve compliance with cysteamine therapy.
Disclosure
The authors report no conflicts of interest in this work
References
- TownMJeanGCherquiSA novel gene encoding an integral membrane protein is mutated in nephropathic cystinosisNat Genet1998183193249537412
- LevtchenkoEVan den HeuvelLEmmaFAntignacCClinical utility gene card for: cystinosisEur J Hum Genet2014225
- KalatzisVCherquiSAntignacCGasnierBCystinosin, the protein defective in cystinosis, is a H+-driven lysosomal cystine transporterEMBO J2001205940594911689434
- TarantaAPetriniSPalmaAIdentification and subcellular localization of a new cystinosin isoformAm J Physiol Renal Physiol2008294F1101F110818337546
- GahlWAThoeneJGSchneiderJACystinosisN Engl J Med200234711112112110740
- ChanAMLynchMJBaileyJDHypothyroidism in cystinosis. A clinical, endocrinologic and histologic study involving sixteen patients with cystinosisAm J Med1970486786925420554
- ChikCLFriedmanAMerriamGRPituitary-testicular function in nephropathic cystinosisAnn Intern Med19931195685758363167
- BesouwMTKremerJAJanssenMCLevtchenkoENFertility status in male cystinosis patients treated with cysteamineFertil Steril2010931880188319217094
- FivushBFlickJAGahlWAPancreatic exocrine insufficiency in a patient with nephropathic cystinosisJ Pediatr198811249513335962
- FivushBGreenOCPorterCCBalfeJWO’ReganSGahlWAPancreatic endocrine insufficiency in posttransplant cystinosisAm J Dis Child1987141108710893307383
- BroyerMTeteMJGuestGBerthéléméJPLabrousseFPoissonMClinical polymorphism of cystinosis encephalopathy. Results of treatment with cysteamineJ Inherit Metab Dis19961965758830179
- ThoeneJGOshimaRGCrawhallJCOlsonDLSchneiderJAIntracellular cystine depletion by aminothiols in vitro and in vivoJ Clin Invest197658180189932205
- ThoeneJGLemonsRCystine depletion of cystinotic tissues by phosphocysteamine (WR638)J Pediatr198096104310447373465
- SchneiderJAClarkKFGreeneAARecent advances in the treatment of cystinosisJ Inherit Metab Dis1995183873977494398
- JézégouALlinaresEAnneCHeptahelical protein PQLC2 is a lysosomal cationic amino acid exporter underlying the action of cysteamine in cystinosis therapyProc Natl Acad Sci U S A2012109E3434E344323169667
- WilmerMJKluijtmansLAvan der VeldenTJCysteamine restores glutathione redox status in cultured cystinotic proximal tubular epithelial cellsBiochim Biophys Acta2011181264365121371554
- SmolinLAClarkKFThoeneJGGahlWASchneiderJAA comparison of the effectiveness of cysteamine and phosphocysteamine in elevating plasma cysteamine concentration and decreasing leukocyte free cystine in nephropathic cystinosisPediatr Res1988236166203393396
- LevtchenkoENvan DaelCMde Graaf-HessACStrict cysteamine dose regimen is required to prevent nocturnal cystine accumulation in cystinosisPediatr Nephrol20062111011316252107
- CordenBJSchulmanJDSchneiderJAThoeneJGAdverse reactions to oral cysteamine use in nephropathic cystinosisDev Pharmacol Ther1981325307307866
- DohilRNewburyROSellersZMDeutschRSchneiderJAThe evaluation and treatment of gastrointestinal disease in children with cystinosis receiving cysteamineJ Pediatr200314322423012970638
- DohilRFidlerMBarshopBEsomeprazole therapy for gastric acid hypersecretion in children with cystinosisPediatr Nephrol2005201786179316133039
- BesouwMBlomHTangermanAde Graaf-HessALevtchenkoEThe origin of halitosis in cystinotic patients due to cysteamine treatmentMol Genet Metab20079122823317513151
- BesouwMTBowkerRDutertreJPCysteamine toxicity in patients with cystinosisJ Pediatr20111591004101121784456
- BesouwMvan den HeuvelLvan EijsdenRIncreased human dermal microvascular endothelial cell survival induced by cysteamineJ Inherit Metab Dis2013361073107723371451
- BesouwMTSchneiderJJanssenMCCopper deficiency in patients with cystinosis with cysteamine toxicityJ Pediatr201316375476023651769
- MarkelloTCBernardiniIMGahlWAImproved renal function in children with cystinosis treated with cysteamineN Engl J Med1993328115711628455682
- GahlWABalogJZKletaRNephropathic cystinosis in adults: natural history and effects of oral cysteamine therapyAnn Intern Med200714724225017709758
- Brodin-SartoriusATêteMJNiaudetPCysteamine therapy delays the progression of nephropathic cystinosis in late adolescents and adultsKidney Int20118117918921900880
- Van StralenKJEmmaFJagerKJImprovement in the renal prognosis in nephropathic cystinosisClin J Am Soc Nephrol201162485249121868618
- KimonisVETroendleJRoseSREffects of early cysteamine therapy on thyroid function and growth in nephropathic cystinosisJ Clin Endocrinol Metab199580325732617593434
- GahlWAReedGFThoeneJGCysteamine therapy for children with nephropathic cystinosisN Engl J Med19873169719773550461
- BesouwMLevtchenkoEGrowth retardation in children with cystinosisMinerva Pediatr20106230731320467383
- CantaniAGiardiniOCiarnellaCANephropathic cystinosis: ineffectiveness of cysteamine therapy for ocular changesAm J Ophthalmol1983957137146846466
- LabbéABaudouinCDeschênesGA new gel formulation of topical cysteamine for the treatment of corneal cystine crystals in cystinosis: the Cystadrops OCT-1 studyMol Genet Metab201411131432024440466
- SyresKHarissonFTadlockMSuccessful treatment of the murine model of cystinosis using bone marrow cell transplantationBlood20091142542255219506297
- YeagyBAHarrisonFGublerMCKoziolJASalomonDRCherquiSKidney preservation by bone marrow cell transplantation in hereditary nephropathyKidney Int2011791198120621248718
- HarrisonFYeagyBARoccaCJKohnDBSalomonDRCherquiSHematopoietic stem cell gene therapy for the multisystemic lysosomal storage disorder cystinosisMol Ther20132143344423089735
- OmranZKayGDi SalvoAKnottRMCairnsDPEGylated derivatives of cystamine as enhanced treatments for nephropathic cystinosisBioorg Med Chem Lett201121454721147534
- OmranZMoloneyKABenyllesAKayGKnottRMCairnsDSynthesis and in vitro evaluation of novel pro-drugs for the treatment of nephropathic cystinosisBioorg Med Chem2011193492349621536447
- FidlerMCBarshopBAGangoitiJAPharmacokinetics of cysteamine bitartrate following gastrointestinal infusionBr J Clin Pharmacol200763364017229040
- DohilRFidlerMGangoitiJATwice-daily cysteamine bitartrate therapy for children with cystinosisJ Pediatr2010156717519775699
- LangmanCBGreenbaumLASarwalMEffectiveness on white blood cell cystine levels and comparison of safetyClin J Am Soc Nephrol201271112112022554716
- BesouwMTangermanACornelissenERiouxPLevtchenkoEHalitosis in cystinosis patients after administration of immediate-release cysteamine bitartrate compared to delayed-release cysteamine bitartrateMol Genet Metab201210723423622832073
- DohilRCabreraBLTreatment of cystinosis with delayed-release cysteamine: 6-year follow-upPediatr Nephrol20132850751023001048