
Abstract
Introduction
X-linked Alport syndrome (XLAS) is caused by pathogenic variants in COL4A5 which lead to abnormalities of the glomerular basement membrane (GBM) structural and is characterized by progressive kidney disease, hearing loss, and ocular abnormalities. The aim of this study was to identify gene mutations in a Chinese family with XLAS by whole-exome sequencing (WES) and verified the pathogenicity of the mutation in vitro experiments.
Case Presentation
A five-generation pedigree with a total of 49 family members originating from Hainan province of China was investigated in this study. The proband was a 23-year-old male who developed microscopic hematuria, proteinuria and end-stage kidney disease (ESKD) at age 17. WES identified a novel splicing mutation c.321+5G>A of COL4A5, which cause exon skip. Further co-segregation analysis confirmed that this mutation exists in relatives who had renal abnormalities using Sanger sequencing. According to American College of Medical Genetics and Genomics guidelines (ACMG), the mutation was determined to be of uncertain significance (VUS). In vitro splicing experiments have shown that the COL4A5 variant induces aberrant mRNA splicing and transcript deletion.
Conclusion
We identified a novel intronic COL4A5 pathogenic mutation (c.321+5G>A) in a Chinese XLAS family and described the phenotypes of affected relatives. This study expands the mutation spectrum of COL4A5 gene in XLAS and demonstrates the importance of gene screening for AS.
Introduction
Alport syndrome (AS), also known as hereditary progressive nephritis, is an inherited glomerulopathy characterized by hematuria, proteinuria, and progressive kidney failure, with some patients having extra-renal manifestations such as sensorineural deafness and ocular abnormalities.Citation1 The abnormalities of the glomerular basement membrane (GBM) structural caused by mutations in COL4A5 on chromosome X, which encodes type IV collagen α5 chain and causes X-linked AS (XLAS).Citation2 There are three inheritance patterns of AS, the X-linked genetic phenotype resulting from mutations in COL4A5 gene is the most common, accounting for approximately 85% of cases,Citation3 followed by autosomal recessiveCitation4 and autosomal dominant.Citation5
Over 90% of maleCitation6 and 12% of female XLAS patients are reported to develop end-stage kidney disease (ESKD) by age 40, respectively. Genotypic phenotypes exhibit a high correlation in males XLAS.Citation6 Males exhibit proteinuria and hematuria in early childhood compared to female patients with XLAS, and the disease develops to ESKD by 25 to 3.Citation7 The severity of male patients with early onset to ESKD correlated with the type of variants, patients with nonsense variants have the most severe phenotypes, whereas patients with splicing variants have moderate phenotypes and patients with missense variants have mild phenotypes,Citation8 in contrast to female patients with XLAS, in whom no genotype–phenotype correlation was presented.Citation9,Citation10 Genetic testing is more sensitive and specific than renal biopsy for the diagnosis of AS and can provide predictive information about disease severity and prognosis. It has been shown that 10% of the pathogenic variants in XLAS are typically aberrant splicing mutations,Citation11 which could exhibit a different prognosis depending on whether the variants result in truncated transcript.Citation12 In this study, we used whole-exome sequencing (WES) to identify one splicing variant in a Chinese family with XLAS and verified the pathogenicity of the mutation in vitro experiments.
Materials and Methods
A five-generation pedigree with a total of 49 family members originating from the remote rural areas in Hainan province of China was investigated in this study (). For all participants, we collected clinical data including family relationship, gender, age at onset of disease, renal function (hematuria, proteinuria, stage of chronic kidney disease (CKD)), sensorineural hearing loss, ocular lesions and medical history. Patients’ CKD was staged according to the K/DOQI clinical practice guideline.Citation13 There are no consanguineous marriages in the family. WES and Sanger sequencing were performed for proband (IV-7) and their parents. In vitro splicing analysis was performed to verify the effect of mutation on mRNA splicing. Peripheral blood samples were collected from 17 family members and verified by Sanger sequencing. Renal pathology of the proband’s brother (IV-6) was assessed using hematoxylin-eosin (HE) staining, periodic acid-Schiff (PAS) staining, periodic acid silver methenamine (PASM) staining, Masson examination, immunofluorescence staining and electron microscopy (EM) analysis. Consent was obtained from all patients to collect and publish the data.
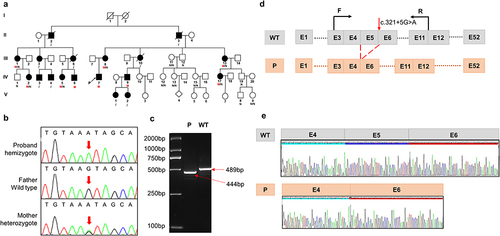
Figure 1 Sequence analysis of the pedigree and in vitro splicing experiments (a) pedigree of the Chinese family with XLAS. Black filled symbols represent affected individuals; The symbols with a slash correspond to deceased individuals; Arrow indicates the proband; M represent mutation (c.321+5G>A), N represent normal, / indicate not detect. (b) Sanger sequencing of the proband and his parents. (c) Electrophoresis for amplification of COL4A5 cDNA from the skin tissue of healthy individual and proband. (d) Schematic overview of the region harboring c.321+5G>A mutation in COL4A5 and representation of the abnormal splicing. Representation of the alternative splicing of exon 5 in COL4A5. (e) Sequencing chromatogram of COL4A5 wild-type cDNA and splice variants.
Case Presentation
As shown in , the progression and severity of AS varied considerably between affected individuals in the family. There were no ocular lesions or hearing loss in the proband, his mother, or other family members. WES and Sanger sequencing were performed for proband (IV-7) and their parents. Peripheral blood samples were collected from 17 family members and verified by Sanger sequencing. Co-segregation analysis revealed that several members of the family had c.321+5G>A mutation in COL4A5; however, the severity of their clinical phenotypes was inconsistent ( and ).
Table 1 Clinical Data of Patients with the COL4A5 Gene Variant
Iv-7
The proband was a 23-year-old male who developed foamy urine, microscopic hematuria (BC250/UL), and moderate-to-severe proteinuria at the age of 11 years, and soon progressed to ESKD, with serum creatinine more than 700 μmol/L, which was treated with peritoneal dialysis. Ultrasound showed both kidneys to be normal size, with enhanced parenchymal echogenicity and unclear demarcation of the cortex from the medulla.
Comprehensive whole-exome sequencing revealed a hemizygous splice variant in COL4A5 (NM_000495: c.321+5G>A) in the proband, which inherited from his mother. The Sanger sequencing in the proband’s parents confirmed that the mutation was indeed inherited from the mother (). Prediction tools such as Varseak (https://varseak.bio), Splice AI (https://spliceailookup.broadinstitute.org/), and Mutation Taster (http://www.mutationtaster.org/) suggested that the mutation may affect the function of 5 ‘donor splice site. According to the American College of Medical Genetics and Genomics Guidelines (ACMG), the variant was initially determined to be of uncertain significance (VUS), with no or very low incidence in healthy populations (PM2), and multiple calculations predicted that the mutation could potentially have deleterious effects on genes or gene products (PP3). Thus, it is necessary to confirm the pathogenicity by further functional studies.
To investigate whether the COL4A5 variant could lead to mRNA splicing defects, we isolated total RNA from the skin tissue of the proband and a wild-type individual. The RNA was then reverse transcribed into cDNA. To confirm the splicing predictions, cDNA sequencing was performed. Alongside normal splicing, aberrant splicing products were observed. The presence of the splicing mutation c.321+5G>A in the proband, which was inherited from his mother, resulted in the generation of an abnormal transcript lacking the entire exon 5, in addition to the normal-length transcript. Electrophoresis of the amplified COL4A5 cDNA from the proband and wild-type individuals revealed a large-sized fragment (489 bp) corresponding to the correctly spliced mRNA, and a small-sized fragment (444 bp) corresponding to the skipping of exon 5. This skipping caused a 45 bp deletion in the coding sequence of the mRNA ( and ). The sequencing chromatogram of the normal COL4A5 cDNA and the splicing variant are shown in .
III-8
The proband’s mother presented with microhematuria and proteinuria at the age of 34 with normal renal function and developed ESKD and started hemodialysis by the age of 43. Ultrasound showed increased parenchymal echogenicity and decreased blood flow in both kidneys. WES showed a heterozygous variant c.321+5G>A in COL4A5 gene.
IV-2
The female cousin of proband, 23 years old, presented with leg edema, hypertension (230/140mmHg), microscopic hematuria and proteinuria, blood creatinine of 256μmol/l, and moderate to severe renal function loss (CKD stage 3). Abdominal ultrasound revealed echogenic enhancement of both renal parenchyma with indistinct corticomedullary demarcation and echogenic enhancement of the left renal calyx margin. After 11 months of blood pressure control, proteinuria decreased, and renal function partially recovered with a creatinine of 179μmol/l.
Iv-5
Another female cousin of proband, 23-year-old, presented with microscopic hematuria (RBC84/ul) at the age of 17 years, no proteinuria, normal renal function, and ultrasound showed punctate strong echogenicity at the dermo-medullary junction with calcification, which could be easily confused with medullary sponge kidneys. Blood pressure was normal (110/70 mmHg).
Iv-6
Proband’ male cousin, 15 years old, presented with microscopic hematuria, a small amount of proteinuria (516 mg/L), RBCs with full field of view. About one year after the onset of the disease, it progressed to moderate proteinuria (1–2g/L). Uric acid 518 umol/l, blood pressure 128/85mmHg. Ultrasound showed enhanced echo at the margins of the renal calyces in both kidneys, suggesting calcification. In addition to this, the patient had strabismus.
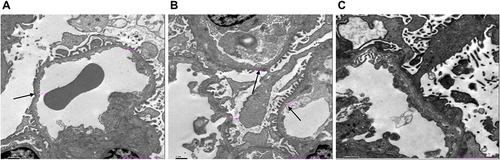
Renal pathology of IV-6 was assessed using hematoxylin-eosin (HE) staining, periodic acid-Schiff (PAS) staining, periodic acid silver methenamine (PASM) staining, Masson examination, immunofluorescence staining and electron microscopy images (EM) analysis. The renal histopathology of IV-6 showed focal segmental glomerulosclerosis with mild enlargement of the kidney volume. The renal tubular epithelial cells exhibited granular degeneration, and a few tubular lumens were dilated. (SFigure 1A). EM images showed glomerular capillary endothelial cells, renal tubular epithelial cells and visceral epithelial cells with vacuolar degeneration. It also revealed that segmental fusion of foot processes is irregular with an uneven density of GBM thickness (). No deposition of immune complexes was observed via Immunofluorescence staining (SFigure 1B).
Figure 2 Electron microscopy images of his kidney of IV-6 (A-C) show two glomeruli under electron microscopy. Electron microscopy of renal showed glomerular capillary endothelial cells, renal tubular epithelial cells, and visceral epithelial cells with vacuolar degeneration; segmental fusion of foot processes; irregular with an uneven density of glomerular basement membrane thickness. The black arrow indicates an abnormally thick basement membrane.
IV-9
The other male cousin of the proband is 34 years old. At the early stage of illness at the age of 21, there was microscopic hematuria, a small amount or trace proteinuria, 24-hour urine protein of 0.26 with normal renal function. B-ultrasound showed punctate hyperechoicity, with numerous patchy dense shadows clustered around the renal calyx in the left renal medulla. The diagnosis is medullary sponge kidney, and no abnormalities were found in the right kidney. After 7 years, the disease progressed to ESKD and began hemodialysis.
IV-17
The other cousin of the proband is 28 years old, who developed microscopic hematuria and proteinuria (162 mg/l) at the age of 11, with RBC250/UL, blood creatinine as high as 700μmol/l or above. As the disease progressed, peritoneal dialysis begins. Blood pressure was normal at 118/81mmHg, and ultrasound showed no renal abnormalities.
III-1
The proband’s aunt, 43 years old, presented with intermittent microscopic hematuria at the age of 29, with normal urine protein and creatinine levels. At that time, her renal function and blood pressure were normal. Afterwards, she reported an increase in nocturia. At the age of 43, a physical examination revealed abnormal kidney function, with hematuria, urine trace protein 1726 mg/l, urine albumin 1203 mg/l, creatinine 147 umol/l, uric acid 402 umol/L, Hb113 g/L, and blood pressure 159/99mmHg. B-ultrasound showed normal size and morphology of both kidneys, with diffuse changes. As renal function deteriorated, she was in CKD-III at age of 43.
III-13
The older aunt (III-13) had a later onset and presented with microscopic hematuria without proteinuria and normal renal function at the age of 54. At the age of 51, she developed stomach cancer and underwent surgery.
Other Cases
The proband’s grandfather, II-3, as well as his younger brother, II-2, developed ESRD at the age of 70, and 30, respectively, and both of them have passed away. The proband’s aunts III-6 developed ESRD at the age of 31 and began hemodialysis. She passed away at the age of 36. Another aunt III-10 died of ESRD at the age of 28. The two nieces of the proband, V-1 and V-2, presented with gross hematuria, and Sanger sequencing was not performed for them. The other aunt of the proband III-3 and two male cousins IV-3 and IV-4 did not undergo Sanger sequencing, so their genotypes could not be obtained. According to family recollections, they all experienced hematuria, but no further clinical phenotypes were provided.
Discussion
In this study, a novel intronic mutation in COL4A5 was identified by WES and Sanger sequencing in an affected Chinese family with a history of kidney disease, which demonstrated the benefit of genetic sequencing in determining the etiology of hereditary kidney disease.
In the 19th century, Fliter and Gregory et al proposed the AS clinical diagnostic criteria that most cases could be clearly diagnosed by clinical manifestations, family history, renal histopathological examination and type IV collagen α-chain staining.Citation14 However, in actual clinical work, AS patients often show atypical renal histopathological changes and lack of unique symptoms, which usually exhibit low awareness rates of chronic kidney diseases. Unfortunately, due to the lack of symptoms, patients often do not seek medical help until renal failure has already occurred. Approximately 10% of affected individuals, however, develop kidney failure by the age of 60, making it difficult to provide effective treatments.Citation15 Male XLAS patients with hemizygous pathogenic variants in COL4A5 have a 100% risk of progression to ESRD, and female patients with heterozygous pathogenic variants have an approximately 25% risk of developing ESRD,Citation1 which is consistent with our findings. Therefore, genetic diagnosis became an effective method for diagnosing AS,Citation16 which promoted a demand for a comprehensive understanding of the genetic causes of AS. Genetic analysis, such as WES, may be effective in increasing awareness and enabling early intervention in the treatment of AS, which may delay the course of AS and reduce the severity of the disease.
Studies have shown that almost all females with XLAS and kidney failure had a history of proteinuria.Citation17 This is consistent with our findings that IV-5 and III-3 did not present a history of proteinuria and had stage I of ESKD compared with those females in families with a history of proteinuria. Furthermore, girls with proteinuria detected before the age of 15 years developed to ESKD more rapidly than those without proteinuria, suggesting that proteinuria is associated with a risk of serious disease. Hemizygous male patients with AS are typically more severely affected than females with heterozygotes variants,Citation18,Citation19 who have recurrent hematuria, early-onset proteinuria and ESKD, often requiring dialysis or kidney transplantation before thirty years old.Citation20 Female carriers of X-linked AS exhibit a lack of genotype–phenotype correlation, with significant intrafamilial variability in disease severity. The same observation is evident in our family. Despite being of the same age as IV-5, IV-2 progressed to CKD stage 3 with massive proteinuria, while IV-5 manifested a milder phenotype. III-8 developed ESKD at 48 years, while III-13, at 55 years, presented with microscopic hematuria with a small amount of proteinuria. This intrafamilial heterogeneity might be attributed to random X-chromosome inactivation. Research has shown that 90% of female patients with severe AS phenotype have X-chromosome inactivation of the normal COL4A5 allele in their kidneys.Citation21 In peripheral leukocytes, the relative activity of the mutated X chromosome compared to the normal allele varies among AS heterozygotes, and this ratio is not associated with renal function.Citation22 Furthermore, proteinuria onset and progression, as well as hearing loss, are risk factors for renal failure. In this family, female patients with significant proteinuria have relatively more severe disease severity,Citation9 and there are no patients with hearing abnormalities.
Literatures have reported that some individuals with AS are associated with a phenotype of hypertension.Citation23 We aimed to investigate whether a genotype–phenotype correlation exists within this large family. However, we only observed hypertension in patient IV-2 (230/140mmHg), while no such phenotype was present in the other family members. This discrepancy might be attributed to clinical heterogeneity.
It is widely acknowledged that there is a strong correlation between mutation type and phenotype in patients with XLAS. The type of variants and the location of the variant affecting the collagen α chain are the predominant factors of the age of onset of ESKD, and the likelihood of sensorineural hearing loss and ocular abnormalities.Citation24 It was found that the male with splicing variants leading to in-frame transcription had a milder phenotype than those with out-of-frame transcription.Citation7,Citation12 Nonsense mutations have a 90% chance of developing ESRD at age 30, and splicing mutations and missense mutations have a 70% and 50% risk of developing ESRD, respectively.Citation6 A study analyzing 14 XLAS pedigrees with atypical splicing mutations showed that the median age of ESKD onset was 20 years and 29 years for families with truncating variants and non-truncating variants, respectively.Citation12 In our study, we identified a novel mutation, c.321+5G>A, in COL4A5 gene. This mutation resulted in the production of an abnormal transcript lacking exon 5, in addition to the normal-length transcript. The expression level of COL4A5 RNA in peripheral blood is extremely low, while the expression level in skin and kidney tissues is relatively high. The most direct and effective way to determine whether an intronic mutation affects splicing is to extract RNA from the patient’s kidney or skin tissue for analysis, which is an invasive experiment. There are also studies that use in vitro Minigene technology to analyze potential splicing mutations.Citation25
Splice variants account for a significant proportion of the total variants in XLAS, and this proportion is likely to increase with future advances in genetic analysis. It is essential to clarify the pathogenicity and transcript patterns by in vitro functional splicing assays.
Conclusion
Our study identified an intronic variant and demonstrated its pathogenicity in vitro experiment. This finding contributes to the enrichment of database resources for AS. AS should be suspected when there is a family history of persistent glomerular hematuria and renal impairment. Furthermore, we also highlight the critical role that genetic analysis can play in personalized AS diagnosis.
Statement of Ethics
Written informed consent was obtained from all participants in accordance with principles of the Declaration of Helsinki. The protocol of the study was approved by the Medical Ethics Committee of Central South University Xiangya School of Medicine Affiliated Haikou Hospital in Haikou, Hainan, China (ZY-IRB-FOM-063).
Disclosure
Jingmin Yang and Daru Lu should be regarded as co-corresponding authors for this study. All authors declare no competing interests in this work.
Additional information
Funding
References
- Kashtan CE, Ding J, Garosi G, et al. Alport syndrome: a unified classification of genetic disorders of collagen IV α345: a position paper of the Alport syndrome classification working group. Kidney Int. 2018;93(5):1045–1051. doi:10.1016/j.kint.2017.12.018
- Nozu K, Nakanishi K, Abe Y, et al. A review of clinical characteristics and genetic backgrounds in Alport syndrome. Clin Exp Nephrol. 2019;23(2):158–168. doi:10.1007/s10157-018-1629-4
- Savige J, Gregory M, Gross O, Kashtan C, Ding J, Flinter F. Expert guidelines for the management of Alport syndrome and thin basement membrane nephropathy. J Am Soc Nephrol. 2013;24(3):364–375. doi:10.1681/asn.2012020148
- Heidet L, Arrondel C, Forestier L, et al. Structure of the human type IV collagen gene COL4A3 and mutations in autosomal Alport syndrome. J Am Soc Nephrol. 2001;12(1):97–106. doi:10.1681/asn.V12197
- Feingold J, Bois E, Chompret A, Broyer M, Gubler MC, Grünfeld JP. Genetic heterogeneity of Alport syndrome. Kidney Int. 1985;27(4):672–677. doi:10.1038/ki.1985.63
- Jais JP, Knebelmann B, Giatras I, et al. X-linked Alport syndrome: natural history in 195 families and genotype- phenotype correlations in males. J Am Soc Nephrol. 2000;11(4):649–657. doi:10.1681/asn.V114649
- Yamamura T, Horinouchi T, Nagano C, et al. Genotype-phenotype correlations influence the response to angiotensin-targeting drugs in Japanese patients with male X-linked Alport syndrome. Kidney Int. 2020;98(6):1605–1614. doi:10.1016/j.kint.2020.06.038
- Bekheirnia MR, Reed B, Gregory MC, et al. Genotype-phenotype correlation in X-linked Alport syndrome. J Am Soc Nephrol. 2010;21(5):876–883. doi:10.1681/asn.2009070784
- Yamamura T, Nozu K, Fu XJ, et al. natural history and genotype-phenotype correlation in female X-Linked Alport Syndrome. Kidney Int Rep. 2017;2(5):850–855. doi:10.1016/j.ekir.2017.04.011
- Jais JP, Knebelmann B, Giatras I, et al. X-linked Alport syndrome: natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a “European Community Alport Syndrome Concerted Action” study. J Am Soc Nephrol. 2003;14(10):2603–2610. doi:10.1097/01.asn.0000090034.71205.74
- Savige J, Storey H, Watson E, et al. Consensus statement on standards and guidelines for the molecular diagnostics of Alport syndrome: refining the ACMG criteria. Eur J Hum Genet. 2021;29(8):1186–1197. doi:10.1038/s41431-021-00858-1
- Horinouchi T, Nozu K, Yamamura T, et al. Detection of splicing abnormalities and genotype-phenotype Correlation in X-linked Alport Syndrome. J Am Soc Nephrol. 2018;29(8):2244–2254. doi:10.1681/asn.2018030228
- K/DOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis. 2003;42(4 Suppl 3).
- Churg J, Sherman RL. Pathologic characteristics of hereditary nephritis. Arch Pathol. 1973;95(6):374–379.
- Pierides A, Voskarides K, Athanasiou Y, et al. Clinico-pathological correlations in 127 patients in 11 large pedigrees, segregating one of three heterozygous mutations in the COL4A3/ COL4A4 genes associated with familial haematuria and significant late progression to proteinuria and chronic kidney disease from focal segmental glomerulosclerosis. Nephrol Dial Transp. 2009;24(9):2721–2729. doi:10.1093/ndt/gfp158
- Kashtan CE. Alport Syndrome: Achieving early diagnosis and treatment. Am J Kidney Dis. 2021;77(2):272–279. doi:10.1053/j.ajkd.2020.03.026
- Gibson JT, de Gooyer M, Huang M, Savige J. A systematic review of pathogenic COL4A5 variants and proteinuria in women and girls with X-linked Alport Syndrome. Kidney Int Rep. 2022;7(11):2454–2461. doi:10.1016/j.ekir.2022.08.021
- Migeon BR. X inactivation, female mosaicism, and sex differences in renal diseases. J Am Soc Nephrol. 2008;19(11):2052–2059. doi:10.1681/asn.2008020198
- Temme J, Peters F, Lange K, et al. Incidence of renal failure and nephroprotection by RAAS inhibition in heterozygous carriers of X-chromosomal and autosomal recessive Alport mutations. Kidney Int. 2012;81(8):779–783. doi:10.1038/ki.2011.452
- Gross O, Netzer KO, Lambrecht R, Seibold S, Weber M. Meta-analysis of genotype-phenotype correlation in X-linked Alport syndrome: impact on clinical counselling. Nephrol Dial Transp. 2002;17(7):1218–1227. doi:10.1093/ndt/17.7.1218
- Guo C, Van Damme B, Vanrenterghem Y, Devriendt K, Cassiman JJ, Marynen P. Severe Alport phenotype in a woman with two missense mutations in the same COL4A5 gene and preponderant inactivation of the X chromosome carrying the normal allele. J Clin Invest. 1995;95(4):1832–1837. doi:10.1172/jci117862
- Vetrie D, Flinter F, Bobrow M, Harris A. X inactivation patterns in females with Alport’s syndrome: a means of selecting against a deleterious gene? J Med Genet. 1992;29(9):663–666. doi:10.1136/jmg.29.9.663
- Kalmár T, Turkevi-Nagy S, Bitó L, et al. Phenotype-genotype correlations in three different cases of adult-onset genetic focal segmental glomerulosclerosis. Int J Mol Sci. 24(24). doi:10.3390/ijms242417489
- Savige J, Huang M, Croos Dabrera MS, Shukla K, Gibson J. Genotype-Phenotype Correlations for Pathogenic COL4A3-COL4A5 Variants in X-Linked, Autosomal Recessive, and Autosomal Dominant Alport Syndrome. Front Med Lausanne. 2022;9:865034. doi:10.3389/fmed.2022.865034
- Malone AF, Funk SD, Alhamad T, Miner JH. Functional assessment of a novel COL4A5 splice region variant and immunostaining of plucked hair follicles as an alternative method of diagnosis in X-linked Alport syndrome. Pediatr Nephrol. 2017;32(6):997–1003. doi:10.1007/s00467-016-3565-4