
Abstract
Primary ovarian insufficiency is a condition that represents impaired ovarian function on a continuum with intermittent ovulation. This condition commonly leads to premature menopause, defined as cessation of ovulation prior to the age of 40 years. Because there are potential immediate and long-term consequences of hypoestrogenism, a timely diagnosis is invaluable. This comprehensive review will discuss identifiable causes for primary ovarian insufficiency, including genetic disorders and metabolic abnormalities, as well as review current strategies for diagnosis, evaluation, and management of women with this condition.
Introduction
It is estimated that approximately 90% of women undergoing menopause will do so between the age of 45 and 55 years, with the average age of menopause occurring at 51 years.Citation1,Citation2 Unlike the age of menarche, which has decreased over the past decade, there has been no apparent drift in the age of natural menopause. These observations suggest that the age of menopause is probably an evolutionarily conserved trait. Premature menopause describes menopause that occurs prior to the age of 40 years. This condition affects approximately 1% of the population in the US.Citation3 An earlier onset of menopause can be spontaneous or the result of medical interventions such as surgical removal of the ovaries or be due to chemotherapy or radiation treatment with subsequent ovarian damage. When menopause is surgically induced, it is associated with a rapid decline in ovarian hormone levels and consequently more severe menopausal symptoms. These symptoms include hot flashes, sleep disturbances, mood liability, and decreased energy.
Primary ovarian insufficiency
Primary ovarian insufficiency (POI) is a term that is increasingly used and has been adopted to encompass diagnostically similar conditions, including hypergonadotropic hypogonadism, premature ovarian failure, and ovarian dysgenesis. This terminology is semantically more accurate because it is a term used to describe impaired ovarian function on a continuumCitation4 rather than a specific endpoint. This condition can be transient or progressive, and usually results in eventual premature menopause.Citation4 The condition affects one in 10,000 women by the age of 20 years and one in 100 by the age of 40 years.Citation5 Conceptually, POI is characterized by one of two processes, ie, ovarian follicular dysfunction or depletion of functional primordial follicles before the age of 40 years. The diagnosis should be confirmed by obtaining two follicle-stimulating hormone (FSH) levels in the menopausal range (>30 U/L) at least 1 month apart in the setting of 4–6 months of amenorrhea.
Patients typically present with oligomenorrhea or amenorrhea and may exhibit increasing symptoms of estrogen deficiency. Additionally, patients can experience long-term consequences of hypoestrogenism, including osteoporosis, accelerated cardiovascular aging, and neurocognitive disorders. Infertility is another consequence of POI. Although most patients will present with amenorrhea, about 50% will have varying degrees of residual ovarian function. It is estimated that approximately 5%–10% are able to conceive spontaneously.Citation1
Three potential mechanisms can be associated with POI, ie, a congenital decrease in primordial follicles, accelerated follicular atresia, and an inability to recruit primordial follicles.Citation6 Unfortunately, for most patients presenting with POI, the cause is largely unexplained. Potential etiologies for POI can be divided into genetic, autoimmune, metabolic dysfunction, infectious, and iatrogenic categories. Each are separately discussed belowCitation6 and are listed in .
Table 1 Causes of primary ovarian insufficiency
Genetic disorders associated with POI
X chromosome disorders
Disorders that involve the X chromosome and loci that regulate germ cell development and viability are linked to POI. Turner syndrome (45,X) is associated with streak gonads and other stigmata, including short stature, a broad and webbed neck, coarctation of the aorta, a shortened fourth metacarpal, pigmented nevi, an ogival palate, cognitive deficits, vertebral abnormalities, and renal anomalies. The prevalence is about one in 2,500 female births, with 80% of cases being maternal in origin.Citation7 Because animal models with monosomy X have normal ovarian development, it is assumed that crucial genes on the normal X chromosome are inactivated.Citation8
Levels of circulating adrenal androgens and their conversion to estrogen account in part for development of pubic/axillary hair. Müllerian derivatives and female external genitalia are well differentiated, but without significant estrogen exposure remain small and infantile in size. A small percentage (3%–5%) of adults with Turner syndrome will menstruate spontaneously and exhibit breast development. In these cases, 45,X/46,XX mosaicism should be suspected. If pregnancy is desired, hormone replacement therapy can be initiated to increase uterine size, followed by assisted reproductive technology, namely in vitro fertilization with an oocyte donor. The success rate of clinical pregnancies using this approach is approximately 50% per cycle without an increased risk of chromosomal abnormalities in the offspring.Citation9 However, coexisting cardiac abnormalities associated with Turner syndrome may increase the risk of pregnancy for the mother. Because the maternal risk in individuals with Turner syndrome is unpredictable and there is a risk of unanticipated maternal cardiac decompensation, the American Society for Reproductive Medicine discourages this type of approach to achieve pregnancy.
X isochromosome
Normal division of the centromere occurs in the longitudinal plane. When the centromere splits abnormally in the transverse plane, the resulting chromosome pair contains structurally identical arms with identical genes. The isochromosome for the long arm (q) is the most common X structural abnormality. These patients have streak gonads and tend to have the Turner stigmata.Citation7
Microdeletions on the X chromosome undetectable by conventional karyotyping are also found in women with POI. Although there are regions on the short (p) and long arms (q) of the X chromosome that are designated ovarian genes (POF1 and POF2, respectively), women with premature ovarian failure have been noted to have alterations outside these designated areas. A more detailed list of known gene mutations are, listed in and .
Table 2 Proposed gene mutations on Xp associated with primary ovarian insufficiency
Table 3 Proposed gene mutations on Xq associated with primary ovarian insufficiency
FMR1 gene
Fragile X syndrome is an inherited X-linked dominant disorder that is a leading cause of inherited cognitive disability. The degree of cognitive disability is typically more severe in males although both males and females can be affected.Citation10 The Fragile X mental retardation 1 (FMR1) gene is mapped on the X chromosome at position q27.3. The fragile site of the X chromosome contains a trinucleotide (CGG) repeat in the 5′ region of the gene. In normal variants, the trinucleotide repeat ranges from six to 55 repeats. When the repeat expands to 55–200, the individuals are considered premutation carriers, and those above 200 are considered affected.Citation10 It has been shown that females carrying a premutation have up to a 23% rate of POI and experience earlier menopause by approximately 5 years.Citation10 In female premutation carriers, the ovarian dysfunction depends on CGG repeat size, although the relationship is not linear. The underlying molecular mechanisms of how Fragile X causes POI are unknown, although there is a hypothesis that the FMR1 premutation may have a toxic RNA gain-of-function effect on ovarian follicle dynamics.Citation10
46,XX associated with POI
XX gonadal dysgenesis not associated with phenotypic anomalies are most commonly inherited in an autosomal recessive fashion. There is variance in phenotypic penetration noted among siblings.Citation11–Citation13 It has been challenging to identify the specific autosomal genes responsible for various forms of XX gonadal dysgenesis. There are sporadic cases associated with reciprocal autosomal translocations that have not been easily reproduced. Mouse models suggest alterations at the following gene regions may impact POI: 4p11–q12 (tyrosine kinase receptor), 12q22 (mast cell growth factor), 9q33 (nuclear receptor factor), 6p21.3 (DNA mismatch repair), and 18q21.3 (cell death repressor protein).Citation14
Syndromic ovarian failure
Studies of individuals with cerebellar ataxia with XX gonadal dysgenesis have shown variance in the manifestation of mental retardation, cataracts, and neurosensory deafness, suggesting a single mutant gene is unlikely to explain all causes.Citation15
Gonadal dysgenesis and multiple malformation syndromes
Other variants presenting with XX gonadal dysgenesis have associated somatic features, including microcephaly, arachnodactyly, epibulbar dermoids, short stature, metabolic acidosis, blepharophimosis-ptosis-epicanthus, Malouf syndrome (dilated cardiomyopathy, mental retardation, blepharoptosis), and limb-mammary syndrome (ectrodactyly, ectodermal dysplasia, cleft lip/palate).Citation16–Citation18
Metabolic abnormalities associated with POI
A number of inherited enzymatic pathway disorders have been associated with ovarian follicular dysfunction leading to POI. Galactose 1-phosphate uridylyltransferase deficiency (galactosemia) was one of the first to be characterized.Citation19 The gene encoding this enzyme is located on 9p and the pathogenesis is believed to involve toxic accumulation of galactose during infancy. Phenotypic features of this disorder involve defects in the ocular, renal, and hepatic systems.Citation19 Because galactosemia requires treatment in childhood to prevent mental retardation, it is unlikely to be diagnosed in an otherwise healthy adult presenting only with POI.
Carbohydrate-deficient glycoprotein deficiency
This condition is most commonly caused by a missense mutation located on chromosome 16p13, a phosphomannomutase gene mutation (PMM1). The enzyme defect results in accumulation of mannose-6-phosphate which cannot be converted to mannose-1-phospate, and is associated with a wide variety of neurologic abnormalities, including ataxia, hyperreflexia, hypotonia, joint contractions, and epilepsy.Citation20
17 α-hydroxylase/17,20 desmolase deficiency
Defects in the sex steroid biosynthetic pathway can lead to predictable consequences depending on the enzyme deficiency. When 17 α-hydroxylase/17,20-lyase is deficient, pregnenolone cannot be converted to 17 α-hydroxypregnenolone. This leads to a reduced ability to produce cortisol, androstenedione, testosterone, and estrogens. This condition in XX genetic females will be associated with normal appearing external genitalia, but affected individuals fail to undergo secondary sexual development at puberty. The ovaries do respond to exogenous gonadotropins. The disorder is a result of an autosomal recessive mutation isolated to a single gene, ie, CYP17 located on 10q24–25.Citation21
Aromatase mutations
Deficiency of aromatase activity prevents conversion of androgens to estrogens and is often associated with clitoral hypertrophy and primary amenorrhea in females.Citation22 Treatment with exogenous estrogen will induce the growth spurt, breast development, and menstrual cycles.
Other disorders leading to ovarian follicular dysfunction
Other disorders leading to ovarian follicular dysfunction include mutations in hormone receptors and downstream post-receptor signaling. Although extremely rare, mutations in the structure, metabolism, or action of gonadotropins can cause POI. Mutations in the FSH receptor, first described in a Finnish population, will cause elevation of FSH levels. These patients often present with primary or secondary amenorrhea and elevated FSH levels, but will have normal-appearing ovarian follicles visualized on transvaginal ultrasound.Citation23
Savage syndrome describes individuals with gonadotropin post-receptor defects. These women present with elevated luteinizing hormone and FSH levels; however, their ovaries contain multiple immature ovarian follicles. These women have normal karyotypes and are resistant to exogenous gonadotropin stimulation.Citation23
Autoimmune-associated causes of POI
POI can be associated with a variety of autoimmune disorders, most commonly thyroiditis.Citation23 There is also a strong association between POI and autoimmune polyendocrine syndrome. This condition may include hypothyroidism, adrenal insufficiency, hypoparathyroidism, and type 1 diabetes mellitus.Citation23 Other associated autoimmune disorders include dry eye syndrome, myasthenia gravis, rheumatoid arthritis, systemic lupus erythematosus, and congenital thymic aplasia. Currently, there are no standardized tests to identify the presence of ovarian autoantibodies but it is now possible to screen for antiadrenal antibodies.Citation23
Infectious causes of POI
Mumps oophoritis has been directly linked to POI, although data are scarce. It should be suspected in women presenting with mumps parotitis and lower abdominal pain. More recently, there has been suggestion that human immunodeficiency virus (HIV) infection (or antiviral therapy) can lead to POI. A prospective study in 2010 evaluated 78 HIV-positive women and found that these women tended to have abnormal levels of antral follicular count (63%), FSH (36%), inhibin B (57%), and anti-Müllerian hormone (AMH, 23%) relative to the normal population.Citation24
Iatrogenic causes of POI
Medical treatment for neoplastic conditions can be associated with POI. Chemotherapy and radiotherapy are well documented causes of POI. Both significantly reduce ovarian function. In general, the rate of follicular atresia is directly related to level of exposure. Chemotherapy induces apoptosis of mature ovarian follicles. Histologic examination of ovarian tissue after treatment will often show cortical fibrosis, vascular damage, and decreased follicular numbers.Citation25 Vinca alkaloids, anthracycline antibiotics, and antimetabolites appear to be low risk for inducing gonadotoxicity. Alkylating agents (ie, cyclophosphamide) carry a much higher risk and are cytotoxic even if the ovary is in a resting state. These agents will lead to POI in approximately 40% of women. Suppression of luteinizing hormone and FSH secretion with a gonadotropin-releasing hormone agonist prior to chemotherapy appears to reduce ovarian damage,Citation26 whereas administration of oral contraceptives appears to have little protective effect.
Direct radiation to the pelvis for treatment of Hodgkin lymphoma accelerates atresia in ovarian follicles. Even with pelvic shielding, it is difficult to protect ovarian function which is highly sensitive to radiation. One study concluded that 26% of women with total abdominal radiation for approximately 3.5 years developed POI by age 23 years.Citation27 Surgical transposition of the ovaries outside the pelvis, well away from the radiation field, is effective at minimizing radiation exposure. For women using this approach, assisted reproductive technology procedures are often required to achieve fertility.
Surgical causes of POI
Aside from surgical menopause due to bilateral oophorectomy, surgical removal of the uterus can result in earlier menopause in some women. This is likely explained by inadvertent damage to the ovarian blood supply during surgery. One study noted the average age of menopause in women who had undergone isolated hysterectomies was 45.0±4.0 years. This is significantly lower than the average age of menopause in women who have not undergone this surgery.Citation28,Citation29
Racial differences in ovarian reserve
Recent evidence supports the notion that ovarian reserve differs among varying racial/ethnic groups. Seifer et al provided evidence of racial differences in ovarian reserve as measured by AMH.Citation30 This study compared AMH levels at two time points in a racially diverse, multicenter cohort study of HIV-infected women (median age 37.5 years and 43.3 years). Black women demonstrated average AMH levels approximately 25.2% lower than those of whites after controlling for age, smoking, body mass index, and HIV status. Additionally, AMH levels in Hispanic women were 24.6% lower than those in white women, although this difference did not reach statistical significance.Citation30
Diagnosis and evaluation
Current studies have failed to determine specific biomarkers or signs/symptoms of POI that will accurately predict when menopause will occur. Patients may present with a shortening or increase in the intermenstrual cycle interval, menstrual irregularities including oligomenorrhea, dysfunctional uterine bleeding, or amenorrhea. Women may note symptoms of estrogen deficiency such as vasomotor symptoms, mood disturbances, and atrophic vaginitis, although these later symptoms are usually delayed. Patients can also experience long-term consequences of hypoestrogenism which include osteoporosis, accelerated cardiovascular aging, and neurocognitive disorders.Citation1
Unfortunately, standardized diagnostic criteria for POI have yet to be established. It is advisable to perform a complete history and physical examination to exclude secondary causes of amenorrhea. These conditions include pregnancy, polycystic ovarian syndrome, hypothalamic amenorrhea, chronic medical illness secondary to poorly controlled diabetes or celiac disease, lifestyle habits (extreme exercise, poor caloric intake), hypothalamic or pituitary lesions, hyperprolactinemia, hypothyroidism, and hyperthyroidism. The screening history should also focus on a family history of early menopause, previous ovarian/pelvic surgery, as well as chemotherapy or radiation therapy which may identify a cause. The clinician should probe for a personal or family history of autoimmune disorders (eg, thyroid disorders, diabetes, Addison’s disease, vitiligo, systemic lupus, rheumatoid arthritis, celiac disease), fragile X syndrome, or intellectual disability.Citation4
The physical examination should focus on body habitus, evidence of normal secondary sexual characteristics, as well as evidence of vaginal atrophy secondary to hypoestrogenism. For women with 3 consecutive months of amenorrhea, laboratory testing should include human chorionic gonadotropin, FSH, thyroid-stimulating hormone, prolactin, and estradiol levels. If serum estradiol is low and FSH is elevated, these values should be repeated in 1 month. If FSH remains elevated, additional testing, including a peripheral karyotype, fragile X screen, anti adrenal antibodies, serum calcium, and a morning cortisol, should be obtained (see and ). Should a Y chromosome be identified, the patient should be counseled regarding gonadal removal because these individuals have an increased potential for malignancy.
Figure 1 Laboratory testing to confirm diagnosis of primary ovarian insufficiency.
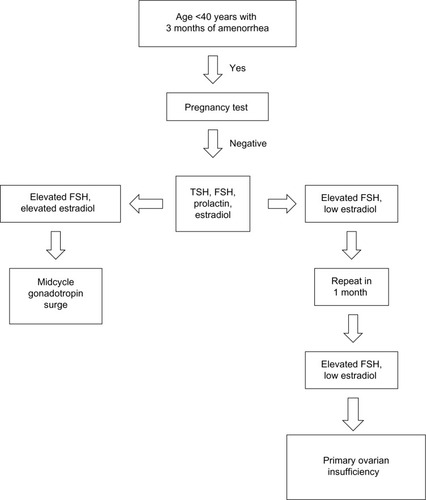
Table 4 Diagnostic testing for conditions associated with primary ovarian insufficiency
For those patients with an estradiol level >50 pg/mL and/or intermittent uterine bleeding, there may be some evidence for residual ovarian function. For those desiring fertility, low-dose hormone replacement (ie, estradiol 2 mg orally or estradiol 50 μg/day transdermally with cyclic progesterone 200 mg/day for 12 days each month) will allow ovulation to occur and should be effective in treating hypoestrogenic symptoms. For women on this type of regimen, failure to have a withdrawal bleed during progesterone treatment may indicate pregnancy.
Because it is possible to have periodic estrogen production in POI, administering a progesterone withdrawal test may not be particularly helpful. Other diagnostic tests include transvaginal ultrasound imaging of the ovaries. Findings of a normal ovarian size/volume and presence of a high antral ovarian follicle count makes the diagnosis of POI less likely. For those women with diminished ovarian function, options to assess ovarian reserve include cycle day 3 FSH levels, AMH, inhibin B, and transvaginal ultrasound-determined antral follicle count. Elevated FSH levels drawn on day 3 imply a poor ovarian reserve.Citation31,Citation32
As a consequence of decreased estrogen levels, women with POI often do not achieve peak bone density and may experience loss of bone mass. If hormone therapy is initiated and the woman has not experienced fractures, it is not necessary to do bone mineral density testing.
Management of POI
A multidimensional approach should be undertaken to manage POI (see ). This includes initiation and management of hormone replacement therapy, contraception or fertility management, psychosocial support, and annual screening to assess thyroid function, adrenal function, and routine preventive health care.
Table 5 Treatment of primary ovarian insufficiency
Hormone replacement therapy
Hormone replacement therapy (HRT) is advised not only to provide relief from menopause-related symptoms (ie, vasomotor symptoms and vaginal atrophy) but also to maintain bone density and reduce the risk of cardiovascular disease.Citation33 Starting estrogen doses should be equivalent to the mid-follicular phase menstrual cycle estrogen concentrations, ie, providing the equivalent of 50–100 μg transdermal estradiol daily.Citation30,Citation31 To reduce the risk of endometrial hyperplasia, 5–10 mg of medroxyprogesterone acetate should be given for 12 days of the month, provided that the uterus is present. Although there is no clinical trial to lend support to an optimal length of time HRT should continue for women with POI, it is generally recommended that this regimen be continued at least until the average age of natural menopause (age 51 years).Citation32 The risks of HRT are low and do not appear to be significantly different from those women with ongoing ovarian function at a perimenopausal age. If HRT is contraindicated or declined, weight-bearing exercises, increased calcium and vitamin D intake, and avoidance of tobacco and alcohol should be recommended. It must be emphasized that these later strategies have been shown to be inadequate at maintaining bone density in the reproductive-aged population. In addition, bone density should be monitored in those women who are not on hormone therapy.
Follow-up visits should be scheduled at 6–12-month intervals with periodic testing of thyroid-stimulating hormone, calcium, and cortisol levels. If antiadrenal antibodies are present, an adrenocorticotropic hormone stimulation test to assess adrenal reserve or referral to an endocrinologist is advised.
Contraception/fertility management
It is important to establish plans for conception soon after the diagnosis of POI is made. There is the distinct possibility of spontaneous and unpredictable ovulation. For those who are not interested in achieving a pregnancy, contraception should be offered. Combined oral contraceptives have the added benefit of providing HRT but intrauterine devices or barrier methods are also acceptable alternatives. For those opting for an intrauterine device or barrier methods, supplemental HRT should be advised.
Fertility preservation
Ideally, counseling regarding future childbearing options will be with a specialist in fertility prior to initiation of chemotherapy or radiation treatment. Women undergoing these treatments are in a unique position in that there is a potential for fertility preservation. Options for fertility preservation include ovarian hyperstimulation with oocyte retrieval followed by oocyte or embryo cryopreservation, ovarian tissue cryopreservation, or ovarian suppression with a gonadotropin-releasing hormone agonist. It should be noted that not all of these options are available for women at all centers, and some techniques such as ovarian tissue cryopreservation are still considered experimental. To date, the fertility treatment with the highest success rate has been in vitro fertilization with use of donor oocytes. This has been associated with a live birth rate of approximately 30%–40% per embryo transferred. Other alternatives include cryopreservation of embryos and or oocytes for use in future in vitro fertilization cycles. However, the current evidence indicates inferiority in pregnancy success rates when compared with use of donor oocytes. This is due to our limited ability to reliably stimulate mature follicles from stored ovarian tissue, frozen oocytes, and the likelihood that follicles harvested from women with underlying POI would be of lower quality. Embryo donation and adoption are other alternatives that should be discussed.
Psychologic support
Women undergoing POI may experience significant psychologic disturbances.Citation34 Some will experience a range of emotions, and providers should offer support regarding infertility, altered self-image, and sexual dysfunction.Citation35 Patients may benefit from referral to a psychologist and support groups, such as the International Premature Ovarian Failure Association.Citation35
Summary
POI represents a continuum from impaired ovarian function with intermittent ovulation to premature menopause as the defining end point, characterized by permanent loss of ovarian function. Because of the potential long-term consequences of hypoestrogenism, including cardiovascular disease, neurocognitive decline, menopausal symptoms, osteoporosis, and infertility, it is important to establish a correct diagnosis expeditiously and identify associated medical conditions. Management should reflect a comprehensive approach, including providing hormone replacement therapy, fertility management, and physical or emotional support. The need for long-term follow-up of this population with preventive maintenance therapy and periodic surveillance cannot be overemphasized.
Disclosure
The authors report no conflicts of interest in this work.
References
- McKinlaySMBrambillaDJPosnerJGThe normal menopause transitionMaturitas1992141031151565019
- MiroFParkerSWAspinallLJColeyJPerryPWEllisJESequential classification of endocrine stages during reproductive aging in women: the FREEDOM studyMenopause20051228129015879917
- CoulamCBBustilloMSchulmanJDEmpty follicle syndromeFertil Steril198646115311553781029
- NelsonLMClinical practice. Primary ovarian insufficiencyN Engl J Med200936060661419196677
- RafiqueSSterlingELawrenceNA new approach to primary ovarian insufficiencyObstet Gynecol Clin N Am201239567586
- PersaniLRossettiRCacciatoreCGenes involved in human premature ovarian failureJ Mol Endocrinol201045257279
- SimpsonJLGonadal dysgenesis and abnormalities of the human sex chromosomes: current status of phenotypic-karyotypic correlationsBirth Defects Orig Artic Ser1975112359
- WillardHFThe sex chromosomes and X chromosome inactivationScriverCRBeaudetALSlyWSThe Metabolic and Molecular Bases of Inherited DiseaseNew York, NY, USAMcGraw-Hill20018
- FoudilaTSoderstrom-AntillaVHovattaOTurner’s syndrome and pregnancies after oocyte donationHum Reprod199914532535
- WillemsenRLevengaJOostraBCGG repeat in the FMR1 gene: size mattersClin Genet20118021422521651511
- SimpsonJLGonadal dysgenesis and sex chromosome abnormalities Phenotypic/karyotypic correlationsValletHLPeterIHGenetic Mechanisms of Sexual DevelopmentNew York, NY, USAAcademic Press1994
- SimpsonJLChristakosACHorwithMSilvermanFSGonadal dysgenesis associated with apparently normal chromosomal complementsBirth Defects197112215218
- BoczkowskiKPure gonadal digenesis and ovarian dysplasia in sistersAm J Obstet Gynecol19701066266285412860
- SimpsonJLRajkovicAOvarian differentiation and gonadal failureAm J Med Genet199989186200
- SkreHBassöeHHBergKFrövigAGCerebellar ataxia and hypergonadotropic hypogonadism in two kindreds. Chance concurrence, pleiotropism or linkage?Clin Genet19769234244
- MaximilianCIonescaBBucurATwo sisters with major gonadal dysgenesis, dwarfism, microcephaly, arachnodactyly, and normal karyotype 46, XXJ Genet Hum197018365378 French
- QuayleSACopelandKC46,XX gonadal dysgenesis with epibulbar dermoidAm J Med Genet19914075761909490
- PoberBRZemelSHisamaFM46, XX gonadal dysgenesis, short stature and recurrent metabolic acidosis in two sistersAm J Hum Genet199863A117
- KaufmanFRKogutMDDonnellGNGoebelsmannUMarchCKochRHypergonadotropic hypogonadism in female patients with galactosemiaN Engl J Med1981304994998
- BjursellCStiblerHWahlströmJFine mapping of the gene for carbohydrate-deficient glycoprotein syndrome, type 1 (CDG1): linkage disequilibrium and founder effect in Scandinavian familiesGenomics1997392472539119361
- YanaseT17α-hydroxylase/17,20-lyse defectsJ Steroid Biochem Mol Biol199553153157
- MullisPEYoshimuraNKuhlmannBLippunerKJaegerPHaradaHAromatase deficiency in a female who is compared heterozygote for two new point mutations in the P450arom gene: impact of estrogens on hypergonadotropic hypogonadism, multicystic ovaries, and bone densitometry in childhoodJ Clin Endocrinol Metab19978217391745
- RebarREvaluation of amenorrhea, anovulation, and abnormal bleeding Available from: http://www.endotext.org/chapter/evaluation-of-amenorrhea-anovulation-and-abnormal-bleeding?singlepage=trueAccessed October 10, 2013
- OhlJPartisaniMDemangeatCBinder-FoucardFNisandILangJMAlterations of ovarian reserve tests in human immunodeficiency virus (HIV)-infected womenGynecol Obstet Fertil201038313317 French20430670
- ClowseMEBeheraMAAndersCKOvarian preservation by GnRH agonists during chemotherapy; a meta-analysisJ Womens Health (Larchmt)200918311319
- FleischerRVollenhovenBWestonGThe effects of chemotherapy and radiotherapy on fertility in premenopausal womenObstet Gynecol Surv20116624825421756407
- De Moraes-RuehsenMJonesGPremature ovarian failureFertil Steril1967184404616028784
- FarquharCMSadlerLHarverySAStewartAWThe association of hysterectomy and menopause: a prospective cohort studyBJOG2005112956962
- SiddleNSarrelPWhiteheadMThe effect of hysterectomy on the age at ovarian failure: identification of a subgroup of women with premature loss of ovarian function and literature reviewFertil Steril19874794100
- SeiferDBGolubETLambert-MesserlianGVariations in serum Mullerian inhibiting substance between white, black and Hispanic womenFertil Steril2009921674167818930217
- RebarRWPremature ovarian failureObstet Gynecol200911313551363
- ParkerWHBroderMSChangEOvarian conservation at the time of hysterectomy and long-term health outcomes in the Nurses’ Health StudyObstet Gynecol20091131027103719384117
- RiveraCMGrossardtBRhodesDIncreased cardiovascular mortality after early bilateral oophorectomyMenopause200916152319034050
- van der StegeJGGroenHvan ZadelhoffSJDecreased androgen concentrations and diminished general and sexual well-being in women with premature ovarian failureMenopause2008152331
- International Premature Ovarian Failure AssociationAlexandria, VA, USA Available from: http://www.pofsupport.orgAccessed August 10, 2013
- SimpsonJOvarian dysgenesis and premature ovarian failure caused by X chromosomal abnormalitiesThe Global Library of Women’s Medicine2011 Available from: http://www.glowm.com/section_view/heading/Ovarian%20Dysgenesis%20and%20Premature%20Ovarian%20Failure%20Caused%20by%20X%20Chromosomal%20Abnormalities/item/353Accessed August 10, 2013