
Abstract
Objective
The deletion of the short arm of chromosome 18 is thought to be one of the rare chromosomal aberrations. Here, we report a case to review this disease.
Case report
The proband is a five-and-a-half-year-old girl who has had phenotypes manifested mainly by ptosis, broad face, broad neck with low posterior hairline, mental retardation, short stature, and other malformations. Chromosomal analysis for her mother showed a normal karyotype. Her father and younger brother were phenotypically normal.
Result
Phenotypical features were quite similar throughout other cases and in accordance with the usual phenotype of del(18p) suggested within the same cases and among the del(18p) cases described. She underwent blepharoplasty, which improved her appearance.
Conclusion
18p deletion syndrome is diagnosed by gene analysis. Plastic surgeries for improving the appearance might be an option for these patients.
Keywords:
Introduction
The deletion of part or the entire short arm of chromosome 18 is a rare chromosomal aberration, which is estimated to affect ~1 in 50,000 live births, with a female to male ratio of 3:2.Citation1–Citation4 Since its first description by Jean de Grouchy in 1963,Citation5 >150 cases have been reported.
The clinical features of this syndrome vary from one patient to another, depending on the deletion size and specific breakpoints. The larger the deletion and the closer to the centromeric region of the chromosome, the more severe the anomalies.Citation3 The clinical abnormalities include growth deficiency, mental retardation, facial deformity (eg, ptosis, epicanthal folds, low nasal bridge, and large protruding ears), microcephaly, holoprosencephaly, and clinodactyly of the fifth finger. Besides, the discrepancy between verbal and nonverbal performance, as well as dystonias, has also been reported.Citation6,Citation7 Though there are diverse phenotypic manifestations, the diagnosis depends mostly on cytogenetic findings.Citation4
Two-thirds of del(18p) cases were supposed to originate from de novo pure terminal deletion, while one-third of cases were caused by a de novo unbalanced translocation, malsegregation of a parental translocation or inversion, and a ring chromosome or isochromosome 18q.Citation8–Citation11 Here, we report a girl with del(18p) with variable clinical features, whose facial appearance improved after blepharoplasty.
Case report
This study complied with the tenets of the Declaration of Helsinki. The consent form for the publication of this report and the accompanying images has been signed by the parents of the child.
The proband was born with a birth weight of 2,200 × g, following a normal gestation and delivery without complications, the first child of a healthy couple (mother: 37 years, 152 cm; father: 39 years, 170 cm). Developmental milestones were retarded; she can sit at 7 months, walk at 18 months, and speak at 27 months. At the age of five-and-a-half years, she was brought to medical attention because of ptosis. At that time, she could only count numbers within 10 and her height was 98 cm (less than third centile), weight was 13.5 kg (less than third centile), and head circumference was 46 cm (less than third centile). The width of palpebral fissure was 7–8 mm in the right eye and 2–3 mm in the left eye when raising her eyebrows. The axial length was 19.89 mm in the right eye and 19.74 mm in the left eye. She was not cooperative when examining visual acuity, ocular movement, anterior segment, and fundus of the eyes. But both eyes had pupil size and light reaction were normal. Other phenotypic manifestations were as followings (): rectangle face, epicanthal folds, low nasal bridge, broad neck with a low posterior hairline, and mental retardation (intelligence within the level of 2-year-old child). Auscultation revealed no heart murmur and normal respiratory sounds. Neither epileptic seizure nor dystonia was noted. Her cerebral imaging was performed and revealed no abnormalities. The results of laboratory evaluations, including complete blood count, electrolytes, liver function, renal function, blood level of thyroid hormone, and gonadal hormone, were all normal.
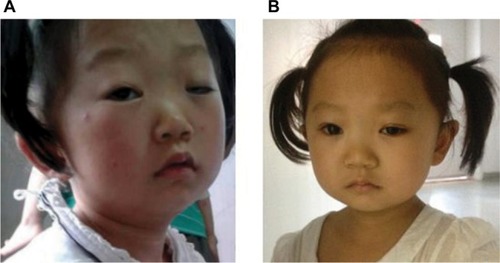
Figure 1 Phenotypical characteristics of the proband before (A) and after (B) blepharoplasty.
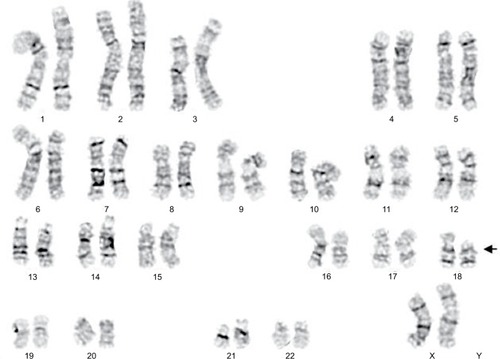
Chromosomal analysis of peripheral blood cells was carried out as follows: 0.3 mL of whole blood was used to inoculate each 5 mL cell culture fluid. Then, 68–69 hours after culture initiation, the blood culture was synchronized with colchicine and returned to the incubator for another 3–4 hours. Then, the blood cells were separated, fixed, and dyed. The procedure was also performed on the proband’s mother. The proband analysis revealed a 46, XX, del (18) (p11.2) complement (). The proband’s mother revealed normal 46-XX karyotype. Her father and younger brother (2 years old) were phenotypically normal. No family history of hereditary disease or mental retardation was noted.
Figure 2 Chromosomal characteristics of the proband showing 18p deletion.
Blepharoplasty was performed because of ptosis in both eyes, and she had a good postoperative recovery, with the width of both palpebral fissures being 9 mm ().
Discussion
This patient has a major clone with deletion of part of the p-arm (18p11.2), resulting in 18p deletion syndrome. The poor intellectual performance was in accordance with the usual phenotype of del(18p). Tsukahara et alCitation12 reported about a Japanese child also bearing del (18p11.2) with an IQ of 74 with significant speech delay, placing him on a borderline intellectual functioning level. Maranda et alCitation13 reported about a mother and her two daughters who had mild-to-moderate mental retardation, and all of them had the same chromosomal deletion: 46, XX, del (18) (p11.2). Peng et alCitation14 studied a girl with mosaic 46, XX, del (18) (p11.2)/46, XX, i (18q), and mild mental retardation and communication disorders were observed.
Epilepsy, together with mental retardation, commonly occurs in chromosomal aberrations.Citation15 However, this girl has no history of epilepsy. Grosso et alCitation16 reported that the short arm of chromosome was less likely to be associated with epilepsy. Hasi-Zogaj et alCitation17 reviewed 106 18p deletions and found that seizures were not particularly common.
Short stature is another feature of this patient. There were reports suggesting that growth hormone (GH) deficiency occurs with deletions of the distal region of 18p and is involved in the main clinical features of the syndrome.Citation18 The growth delay of our patient could be the result of the region of deletion involved. Though several cases of 18p monosomy with GH deficiency reported that the patients showed excellent catch-up growth after GH treatment;Citation19–Citation20 however, there is no indication of GH treatment according to the guideline of US Food and Drug Administration and Growth Hormone Research Society.Citation21,Citation22 Additionally, there is controversy regarding GH treatment of patients with gene defectsCitation23; thus, after fully understanding the possible consequences of the GH application, the parents of our case are cautious and taking a “wait-and-see” approach.
Other phenotypic features were quite similar with other cases of del(18p). It commonly includes a microcephaly, ptosis, a broad, flat nose, a “carp-shaped” mouth, large, protruding ears, widely-spaced eyes, and/or other abnormalities. Other neurologic findings and/or extremely variable midline facial defects include the presence of maxillary incisor, hypotelorism, an abnormal groove in the upper lip, cleft palate, and/or in severe cases, absence of the nose and/or cyclopia. For some of the abnormal appearances, plastic surgery might be an option. In our case, the girl’s facial appearance greatly improved after blepharoplasty.
Detailed breakpoints location and deleted genes identification help to pick up the del(18p). There are 19 genes related to this syndrome, contributing to the variable clinical pictures. USP 14 gene encodes a member of proteases, and mice with a mutation were retarded for growth.Citation24 Gripp et alCitation25 reported that TGIF1 links to NODAL signal pathway to the bifurcation of the human forebrain and the establishment of ventral midline structures. However, the information about breakpoints or genotype–phenotype correlations is limited. This report presents a case deletion of part of the p-arm (18p11.2). It is usually caused by spontaneous (de novo) errors very early in the development of the embryo that appear to occur randomly for unknown reasons. Thus, further study is needed to clarify the etiology and the correlation between genes and symptoms. Besides, the patient should be followed up for a long term.
Conclusion
This report sheds new lights on the del(18p) syndrome. Plastic surgery may be an option for some of the abnormalities to improve the appearance, as well as the life quality. Genetic counseling for these patients should be taken into account.
Acknowledgments
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Disclosure
The authors report no conflicts of interest in this work.
References
- Babovic-VuksanovicDJenkinsSCEnsenauerRNewmanDCJalalSMSubtelomeric deletion of 18p in an adult with paranoid schizophrenia and mental retardationAm J Med Genet A2004124A331832214708108
- ThompsonRWPetersJESmithSDIntellectual, behavioral, and linguistic characteristics of three children with 18p-syndromeJ Dev Behav Pediatr198671173949985
- WesterUBondesonMLEdebyCAnnerénGClinical and molecular characterization of individuals with 18p deletion: a genotype-phenotype correlationAm J Med Genet A2006140111164117116691587
- TurleauCMonosomy 18pOrphanet J Rare Dis20083418284672
- De GrouchyJLamyMThieffrySDysmorphie complexe avec oligophrenie: deletion des bras courts d’un chromosome 17–18. [Deletion of the short arms of chromosome 17-18: complex deformities with oligophrenia]Arch Fr Pediatr19632074074513980940
- de RavelTJThiryPFrynsJPFollow-up of adult males with chromosome 18p deletionEur J Med Genet200548218919316053911
- KleinCPageCELeWittPGenetic analysis of three patients with an 18p-syndrome and dystoniaNeurology199952364965110025808
- ChenCPChernSRWangWPrenatal diagnosis of partial monosomy 18p(18p11.2–>pter) and trisomy 21q(21q22.3–>qter) with alobar holoprosencephaly and premaxillary agenesisPrenat Diagn200121534635011360273
- ChenCPLinSPTsaiFJWangTHChernSRWangWCharacterization of a de novo unbalanced Y;autosome translocation in a 45,X mentally retarded male and literature reviewFertil Steril2008904e11e18
- ChenCPKuoYTLinSPMosaic ring chromosome 18, ring chromosome 18 duplication/deletion and disomy 18: perinatal findings and molecular cytogenetic characterization by fluorescence in situ hybridization and array comparative genomic hybridizationTaiwan J Obstet Gynecol201049332733221056319
- ChenCPKuoYKSuYNPrenatal diagnosis and molecular cytogenetic characterization of a derivative chromosome der(18;18) (q10;q10)del(18)(q11.1q12.1)del(18)(q22.1q22.3) presenting as apparent isochromosome 18q in a fetus with holoprosencephalyTaiwan J Obstet Gynecol201150218218721791305
- TsukaharaMImaizumiKFujitaKTateishiHUchidaMFamilial Del(18p) syndromeAm J Med Genet2001991676911170097
- MarandaBLemieuxNLemyreEFamilial deletion 18p syndrome: case reportBMC Med Genet200676016842614
- PengDLongPPWenBYuRHA study of a rare chromosomal disorder: mosaic 46, XX, del (18)(p11.2)/46, XX, i(18q)J Genet201392361161524371185
- SchinzelANiedristDChromosome imbalances associated with epilepsyAm J Med Genet2001106211912411579431
- GrossoSPucciLDi BartoloRMChromosome 18 aberrations and epilepsy: a reviewAm J Med Genet A2005134A1889415690352
- Hasi-ZogajMSeboldCHeardPA review of 18p deletionsAm J Med Genet C Semin Med Genet2015169325126426250845
- PortnoïMFGruchyNMarlinSMidline defects in deletion 18p syndrome: clinical and molecular characterization of three patientsClin Dysmorphol200716424725217786116
- SchoberEScheibenreiterSFrischH18p monosomy with GH-deficiency and empty sella: good response to GH-treatmentClin Genet19954752542567554351
- ArtmanHGMorrisCAStockAD18p-syndrome and hypopituitarismJ Med Genet19922996716721404301
- Growth Hormone Research SocietyConsensus guidelines for the diagnosis and treatment of growth hormone (GH) deficiency in childhood and adolescence: summary statement of the GH Research Society. GH Research SocietyJ Clin Endocrinol Metab200085113990399311095419
- WilsonTARoseSRCohenPLawson Wilkins Pediatric Endocrinology Society Drug and Therapeutics CommitteeUpdate of guidelines for the use of growth hormone in children: the Lawson Wilkins Pediatric Endocrinology Society Drug and Therapeutics CommitteeJ Pediatr2003143441542114571209
- SavageMOCamacho-HübnerCDavidAIdiopathic short stature: will genetics influence the choice between GH and IGF-I therapy?Eur J Endocrinol2007157suppl 1S33S3717785695
- WilsonSMBhattacharyyaBRachelRASynaptic defects in ataxia mice result from a mutation in Usp14, encoding a ubiquitin-specific proteaseNat Genet200232342042512368914
- GrippKWWottonDEdwardsMCMutations in TGIF cause holoprosencephaly and link NODAL signalling to human neural axis determinationNat Genet200025220520810835638