Abstract
Osteogenesis imperfecta (OI) is a rare inherited disorder with a broad spectrum of clinical and genetic variability. The genetic diversity involves, in the majority of the cases, mutations in one of the genes that encodes the type 1 collagen protein (COL1 A1 and COL1 A2), but it is not a requirement for the diagnosis. The most benign form is OI type I. The authors present a case report of a 25-year-old woman who had severe low back pain associated with incapacity to walk and breast-feed post-partum. Symptoms developed 2 weeks after delivery. The radiological examination revealed severe osteoporosis with no abnormalities in the laboratory findings. The clinical signs and a positive personal and family history of multiple fractures in childhood suggested OI type I, although other diagnosis, such as pregnancy-associated osteoporosis, was also considered. The atypical presentation of this rare disorder in adulthood calls attention to the need for early diagnosis for prompt treatment. Treatment of OI is never curative, but it improves the quality of the patient’s life.
Introduction
Osteogenesis Imperfecta (OI), also known as the “brittle bone disease”, is a heritable connective tissue disorder, characterized by bone fragility due to low bone mass, with an increased propensity to fractures and bone deformity. It is a rare disorder with an incidence of up to 1/30,000 and a prevalence of 6 to 7/100,000 births.Citation1,Citation2
Patients with OI present small, slender bones and the fractures affect mainly the spine and the extremities. The most frequent extraskeletal clinical features are the eye (blue sclera), hyperlaxity of ligaments and skin, hearing impairment, dentinogenesis imperfecta, and the presence of wormian bones on skull radiographs.Citation3,Citation4
In recent decades there has been a tendency to view OI as an autosomal dominant disorder of type I collagen, a major structural protein of the extracellular matrix of the bone. This suggested that most of the cases would have aroused from a mutation in one of the two genes, which encoded the α-chains of the collagen type I (COL1 A1 and COL1 A2 genes).Citation5 However, in recent years, the molecular understanding of this disease recognized newer autosomal recessive forms of OI (types V, VI, and VII) not associated with type I collagen gene defects. This allowed the modification, of the original Sillence classification (), in a large number of subgroups, based on new clinical, radiological and histological features and type of mutation involved.Citation1,Citation3
Diagnosis of OI is straightforward in individuals with a positive family history. However, there are some forms of OI (such as OI type I) with mild clinical expression where other possible diagnosis, such as idiopathic juvenile osteoporosis, must be considered. In these cases cultured skin fibroblasts and/or genetic analysis may be helpful in confirming the diagnosis.Citation6,Citation7 Alternatively, analysis of the DNA with the screening of the coding region of the COL1 A1 and the COL1 A2 genes may identify the mutation.Citation3,Citation6 Genomic DNA sequencing is more sensitive than fibroblast cultures and allows screening the mutation in other family members. Both approaches are thought to detect 90% of the collagen type 1 mutations.Citation3,Citation8–Citation10
A consistent genotype–phenotype correlation is still not fully defined. Therefore, it is not possible to predict with certainty the phenotypic effect of a particular mutation.Citation3,Citation4,Citation6,Citation11,Citation12 Because of the highly variable expression in OI, individuals with the same genotype have different phenotype, which is an interesting feature of many dominant disorders.Citation12 Moreover mutations are absent or located in a different gene in a considerable number of individuals.Citation4 For these reasons, the definition of OI should remain on signs and symptoms outlined above.Citation3,Citation4
Genetic counseling is an important component of the diagnostic approach, since individuals who are affected with dominant OI trait have a 50% risk of transmission with each pregnancy.
Treatment of OI involves a multidisciplinary approach. Physiotherapy, rehabilitation, and orthopedic surgery are the mainstays of treatment. In recent years there have been developments in the medical management of OI. Biphosphonates have been shown to increase bone turnover, decrease fracture rates, and relieve symptoms in patients with OI.Citation4
The following case report calls attention to the challenging diagnosis of OI type I, whose clinical features may overlap the symptoms of other disorders. The diagnosis of the milder forms of OI requires a high index of suspicion, along with proper follow up and counseling to prevent further complications.
Case report
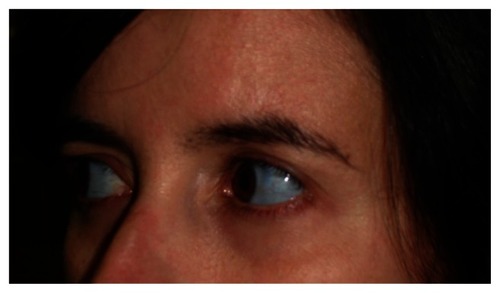
A previously healthy 25-year-old woman was admitted to the emergency department complaining of severe low back pain. The patient was unable to carry her 3-month-old baby and she was having difficulties in walking and doing her daily activities, because of back pain. There was tenderness in all spinous process with restricted spinal range motion and spasm on vertebral muscles. She denied recent trauma or intense exercise. Neurological examination was unremarkable. The low back pain began in the third trimester of pregnancy, but was not as debilitating as after delivery. On physical examination the patient presented a triangular-shaped face, blue sclera (), and short stature. She did not have discolored teeth. Her height was 150 cm and weight 51 kg (before pregnancy her weight was around 42 kg). Both pregnancy and labor were carried out without any other intervention. She denied taking any drugs and she was a non-smoker.
Figure 1 Blue sclera.
A renal ultrasound did not reveal any abnormality. The spine X-ray and the lumbar CT-scan showed diffuse low bone density (). Laboratory assessment included erythrocyte sedimentation rate, serum creatinine, calcium, phosphorus, liver enzymes (alanine aminotransferase [ALT], aspartate aminotransferase), thyroid hormones, parathyroid hormone, osteocalcin, and 25-hydroxyvitamin D (25-OHD), and urinary excretion of calcium and hydroxyproline were within the normal range, except for an elevated ALT (260 U/L, N: 36–101). Antibodies for the celiac disease were negative. The protein electrophoresis was also in normal range. Because of the intensity of the symptoms and the laboratory and radiological negativity for arthritis, a bone scintigraphy was done revealing signs suggestive of “old” fractures in the 7th, 8th and 9th right ribs, and lumbar vertebras (). Bone mineral density (BMD) was measured by using dual-energy X-ray absorptiometry (DEXA) and the results showed a total T score of –4.1 for the lumbar spine (L1–L4) (), which indicated severe bone loss (osteoporosis). Further review of the patient’s past medical history revealed multiple episodes of fractures during childhood and adolescence: 1 tibial and 3 rib fractures. There was also a positive family history of premature osteoporosis in her mother and history of fractures in 2 aunts. These findings suggested the diagnosis of a mild form OI type I.
Figure 2 Lumbar spine X-ray (A) and computed tomography scan (B, sagittal view and C, axial view) of the spine showing diffuse low bone density.
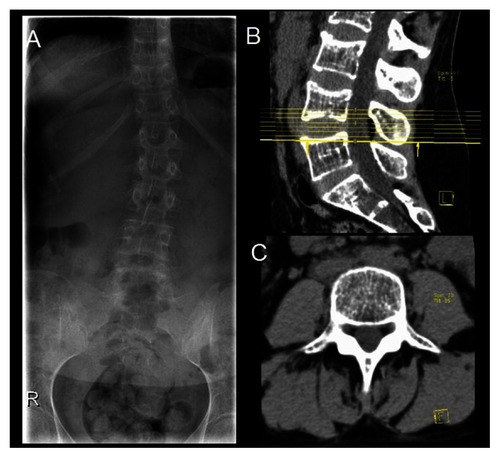
Figure 3 Bone scintigraphy (A, anterior view, B, posterior view) showing increased uptake in the 7th, 8th and 9th right ribs (arrows).
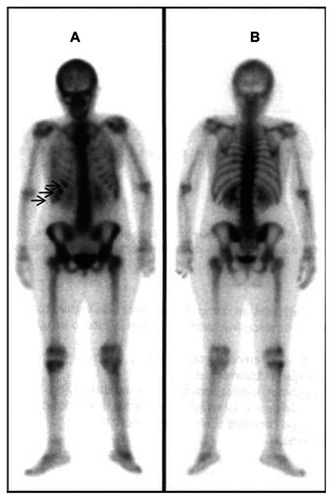
Table 2 Dual-energy X-ray absorptiometry results revealing an increase of bone mineral density (BMD) of the lumbar spine in the 3 years after treatment with alendronate
The patient began analgesic treatment with acetaminophen 1 g 3 times a day and also intranasal calcitonin (200 IU/day), alendronate (70 mg/week), elementary calcium (1000 mg/day), and vitamin D. A thoracolumbosacral orthosis (TLSO) was prescribed and she was advised to bottle-feed her baby. After 6 months’ treatment her symptoms improved. Low back pain disappeared completely and she was able to walk and carry her baby. The TLSO was then discontinued. After 1-year follow-up she denied back pain and was able to carry out her daily activities at work and at home. Three years after starting treatment with oral alendronate, the lumbar spine BMD increased by 31.6% (). Recently, alendronate was substituted by strontium ranelate and she is currently on antiosteoporotic treatment.
The patient’s son was sent to a metabolic and genetic diseases outpatient clinic of the pediatric hospital. When he was 1 year old he had a fracture of the 7th rib after a minor trauma. Laboratory workup excluded metabolic and endocrine abnormalities. Based on clinical findings (blue sclera and fractures) and positive family history, he was also diagnosed with OI. He was treated with pamidronate given in cycles of 3 days, repeated every 2 months for 9 months, and since then he has had no other episode of fractures.
Discussion
OI type I is the most prevalent form of OI and is often undiagnosed during childhood because children rarely develop significant physical deformities. It is caused by quantitative defects in collagen production, although the collagen produced is structurally normal. Patients typically have blue sclera, short stature and a triangular face shape. They tend to present early osteoporosis. Dentinogenesis imperfecta is uncommon, but hearing impairment is possible ().
The present patient had been experiencing low back pain that, during pregnancy, was attributed to a mechanical condition. This resulted in increasing pain and inability to perform her daily activities and breast-feeding after pregnancy. At physical examination the patient presented blue sclera, a triangular- shaped face, short stature, but no physical deformity and no dentinogenesis imperfecta. Radiological findings were suggestive of osteoporosis. Clinical history and laboratory assessment excluded metabolic, endocrine (ie, hyperparathyroidism), and infectious causes for her symptoms. There were no eating disorders and she was not overweight.
Although clinical features were in favor of a diagnosis of a mild form of OI, pregnancy-associated osteoporosis (PAO) was also considered in the differential diagnosis.
PAO is a rare and transient form of osteoporosis. Its incidence is underestimated, since back pain in a pregnant or lactating woman is normally attributed to mechanical conditions. Patients complain of low back, ankle and hip pain developing in the last trimester or after delivery, and these conditions are often interpreted as normal for pregnancy.Citation13,Citation14 If the disease is recognized early and treated, the prognosis is good for subsequent pregnancies. The risk of fracture recurrence is low even in the absence of pregnancy, but not negligible. Citation15 The PAO is characterized as a localized osteoporosis of unknown etiology in contrast to the diffuse generalized osteoporosis of the post-menopausal women.
In the presenting case we could not exclude the existence of genetic predisposition, since the patient’s mother and aunt suffered from early osteoporosis with a past medical history of multiple fractures. However, genetic analysis of COL1 A1 could not be done. Scintigraphy revealed the existence of old fractures and DEXA results confirmed the existence of severe osteoporosis. These findings, in conjunction, with the clinical features and a positive personal and family history, supported the diagnosis of OI type I.
Analgesic treatment with acetaminophen and intranasal calcitonin allowed fast pain relief. Antiresorptive agents such as biphosphonates are considered the first-line treatment for osteoporosis. In this case, because of severe osteoporosis, the patient was treated with oral alendronate, calcium, and vitamin D. Breastfeeding was stopped because of the negative effect on BMD and calcium metabolism.Citation5 After 6 months’ treatment, symptoms improved and after a 3-year-treatment with alendronate, there was an increase in bone mass density (). The recent substitution of oral alendronate for strontium ranelate was based on scientific evidence that support its efficacy for BMD and improvements in bone cortical microarchitecture that decrease fracture rate in osteoporotic women,Citation16 and because current studies have demonstrated the long-term use of biphosphonates is associated with an increased risk of uncommon side-effects, such as osteonecrosis of the jaw and atypical fractures.Citation17
Surgical interventions, which are alternative treatment options for OI when medical therapy fails, were not necessary since there was a good response to medical treatment. No other fractures have occurred. In OI type I there is a 50% risk of transmitting the disease and in the presenting case it was also diagnosed on our patient’s son.Citation2 He was also treated with biphosphonates. There have been some concerns about the use of bisphosphonates in children, because of possible interference with the rapid remodeling process, which is necessary in growing bones, and which can result in bone deformity or serious disruption of the normal mineralization process. However, to date, studies have shown that the linear growth of children on biphosphonate treatment is not affected.Citation5,Citation18
This case presentation illustrates the importance of early recognition of the different possible factors for a low back pain in young pregnant women. OI should always be kept in mind as a possible cause.
Conclusion
OI is a rare disorder that should always be kept in mind as possible cause of chronic low back pain and osteoporosis in a pregnant or lactating woman. In our case report the clinical features and positive personal and family history were very suggestive of OI type I. Because of subtle clinical features this disorder may remain undiagnosed until adulthood. OI is associated with bone fragility and increased risk for fractures, and therefore prompt medical management is mandatory. Biphosphonates are the cornerstone therapy and the response to treatment is measured with BMD.
The goal in OI type I patients is to promote general physical well-being, reduce the risk of fracture, and increase bone density. Medical treatment is not curative, but it improves the patient’s quality of life.
Acknowledgment
The authors gratefully acknowledge all the staff of the Department of Nuclear Medicine for kindly providing the scintigraphy and DEXA images.
Disclosure
The authors declare no conflicts of interest.
References
- MariniJCCabralWABarnesAMNull mutations in LEPRE and CRTAP cause severe recessive osteogenesis imperfectCell Tissue Res20103391597019862557
- MartinEShapiroJROsteogenesis Imperfecta: epidemiology and pathophisiologyCurr Osteoporos Rep200753919717925189
- RaunchFGlorieuxFHOsteogenesis imperfecta. (Seminar)Lancet200436394181377138515110498
- GlourieuxFHOsteogenesis ImperfectaBest Pract Res Clin Rheumatol20082218510018328983
- ByersPHSteinerRDOsteogenesis ImperfectaAnnu Rev Med1992432692821580589
- RoughleyPJRaunchFGlorieuxFHOsteogenesis imperfecta – Clinical and molecular diversityEur Cell Mater20035414714562271
- PaceJMWieseMDrenguisASDefective C-propeptides of the proalpha2(I) chain of type I procollagen impede molecular assembly and result in osteogenesis imperfectaJ Biol Chem200828323160611606718375391
- ShapiroJRSponsellorPDOsteogenesis Imperfecta: questions and answersCurr Opin Pediatr200921670971619907330
- KorkooJAla-KokkoLDe PaepeANuytinckLEarleyJProckopDJAnalysis of the COL1 A1 and COL1 A2 genes by PCR amplification and scanning by conformation-sensitive gel electrophoresis identifies only COL1 A1 mutations in 15 patients with osteogenesis imperfect type I: identification of common sequences null-allele mutationsAm J Hum Genet1998621981109443882
- MarloweAPepinMGByersPHTesting for osteogenesis imperfecta in cases of suspected non-accidental injuryJ Med Genet200239638238612070242
- CheungMSGlorieuxFOsteogenesis Imperfecta: Update on presentation and managementRev Endocr Metab Disord20089215316018404382
- TrummerTBrennerRJustWVogelWKennerknechtIRecurrent mutations in COL1 A2 in patients with Osteogenesis ImperfectaClin Genet200159533834311359465
- KhovidhunkitWEpsteinSOsteoporosis in PregnancyOsteoporos Int1996653453548931028
- GregorioSDDanilowiczKRubinZMautalenCOsteoporosis with Vertebral Fracture Associated with Pregnancy and LactationNutrition20001611–121052105511118824
- SullivanSMGreyABSinghRReidIRBiphosphonates in pregnancy and lactation-associated osteoporosisOsteoporos Int20061771008101216758139
- ReginsterJYBruyèreOSawickiALong-term treatment of postmenopausal osteoporosis with strontium ranelate: results at 8 yearsBone20094561059106419679207
- WattsNBDiabDLLong-term use of biphosphonates in osteoporosisJ Clin Endocrinol Metab20109541555156520173017
- BianchiMLCimazRBardareMEfficacy and safety of alendronate for the treatment of osteoporosis in diffuse connective tissue diseases in childrenClin Pediatr (Phila)2002415362363