
Abstract
Background
Immunoglobulin A deficiency (IgAD) is the most common form of primary immunodeficiency in western countries. It can be associated with the development of autoimmune diseases both in adults and in children even though the exact pathophysiology is not fully defined.
Case Presentation
We report here a case of a young patient who developed nephrotic syndrome secondary to membranoproliferative glomerulonephritis associated with the incidental finding of IgAD. We began corticosteroid therapy and angiotensin-converting enzyme inhibitor, and we observed partial remission of the nephrotic syndrome after about nine months; nonetheless, in the following follow-up visits, a progressive decline of renal function was found.
Conclusion
Our case extends the spectrum of hitherto described glomerulonephritides associated with IgAD which were described until now.
Introduction
Selective IgAD is the most common primary immunoglobulin deficiency in western countries. It can be associated with the development of autoimmune diseases being more prevalent among adults though the exact pathophysiology is not fully defined. In this paper, we report the case of a young patient suffering from nephrotic syndrome secondary to membranoproliferative glomerulonephritis associated with IgAD. To the best of our knowledges, this is the first report of such association in a young adult.
Case Presentation
On December 2013, a 19-year-old man was referred to our Unit due to the sudden development of nephrotic syndrome manifesting with leg edema and ascites. Blood arterial pressure was 140/70 mmHg, heart rate 70 bpm, and body weight 74.5 kg. Renal ultrasound showed kidney enlargement and cortical hyperechogenicity. Laboratory tests revealed reduced renal function (serum creatinine 1.5 mg/dL, urea 38 mg/dL), hypoproteinemia (4.1 g/dl), hypoalbuminaemia (1.96 g/dL), and dyslipidemia (total cholesterol 290 mg/dL, triglycerides 250 mg/dL). Urinalysis documented heavy proteinuria (300 mg/dL) with microhematuria and leucocyturia. Immunological investigations showed an isolated reduction of complement factor (C) 3 (26.7 mg/dl) and of immunoglobulin (Ig) G (447 mg/dL) and IgA (<7.8 mg/dL); C4, IgM, IgG subclasses, and IgE in the normal range; negative search for ANA, ENA, c-ANCA, p-ANCA, thyroid peroxidase, thyroglobulin antibodies, HBV and HCV serologic analyses. We performed a percutaneous kidney biopsy that revealed membranoproliferative glomerulonephritis () associated with a marked immunostaining positivity for C3 in a diffuse granular manner and mild mesangial segmental focal total IgG positivity. The patient was treated with methylprednisolone, 60 mg i.v. for 1 week, followed by 64 mg per os, and with the angiotensin-converting enzyme inhibitor (ACEi) lisinopril (20 mg/day), until February 2014, when he was readmitted due to weight gain (79 kg). We resumed methylprednisolone iv therapy for 7 days, followed by methylprednisolone 32 mg per os. During hospitalization, normalization of kidney function was achieved (serum creatinine 1.1 mg/dL, serum urea 35 mg/dL) with persistence of proteinuria (dipstick 300 mg/dL). Partial remission was observed in June 2014, and methylprednisolone therapy was gradually tapered until suspension in September 2014. Molecular analyses of the complement CFH, CFI, MCP, C3, CFB genes were also performed. As regards to factor H (which is composed of 20 SCR, Short Consensus Repeat), mutation screening was carried out by next-generation sequencing: no mutations were identified in any SCRs 1–20, in the gene promoter or in the signal peptide. As regards factor B, screening for mutations by next-generation sequencing was carried out in the 18 exons that make up the CFB gene and no mutation was identified. Regarding factor I, screening was conducted in the 13 exons that make up the CFI gene and no mutation was identified, as was found for the 41 exons that make up the C3 gene.
From December 2014 until September 2016, at follow-up checks we found normal kidney function (creatinine 1.2 mg/dL, urea 32 mg/dL), mild hypoproteinemia (6.2 g/dL), a mild C3 reduction (78.6 mg/dl), and proteinuria (30–50 mg/dL). At that time the patient was on therapy with ACEi only.
In October 2016, we again observed reduced kidney function (creatinine 1.5 mg/dL, urea 26 mg/dL) associated with an increase in proteinuria (at dipstick, 150 mg/dL), hypoproteinemia (5.9 g/dL) and C3 reduction (79 mg/dL). Thus, in the light of these data, methylprednisolone 32 mg per os was started again, and gradually tapered until August 2017. However, despite the steroid therapy, we observed a further deterioration of kidney function (creatinine values progressively increasing from 1.5 mg/dl to 2.2 mg/dl) associated with persistence of proteinuria and C3 reduction. Hence, we readmitted our patient and in January 2019 performed a second kidney biopsy (), which disclosed disease progression with presence of sclerosis (4/14 glomeruli) and a marked diffuse immunostaining positivity for C3, like the previous biopsy. Electron microscopy confirmed the diagnosis of membranoproliferative glomerulonephritis (). We decided to restart methylprednisolone therapy 32 mg, which the patient continued for six months before it was progressively reduced until definitive suspension. The latest kidney function values show creatinine 2.5 mg/dL, proteinuria (100 mg/dl).
Since this was a retrospective case report in which the patient was not identified, the requirement for institutional approval to publish the case details was waived. Written informed consent from the patient for publication of the study was obtained.
Figure 1 Kidney biopsy performed in 2013. Marked mesangial proliferation and diffuse simplification of the glomeruli. Hematoxylin eosin, x20.
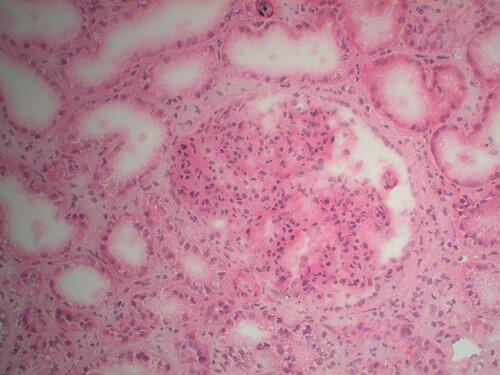
Figure 2 Kidney biopsy performed in 2019. Glomerular enlargement with marked mesangial proliferation and diffuse lobular simplification of the glomeruli. Compared to the first biopsy, the glomeruli showed an accentuated lobular configuration and tubulointerstitial damage (tubular atrophy and fibrosis and inflammation). Hematoxylin eosin, x10.
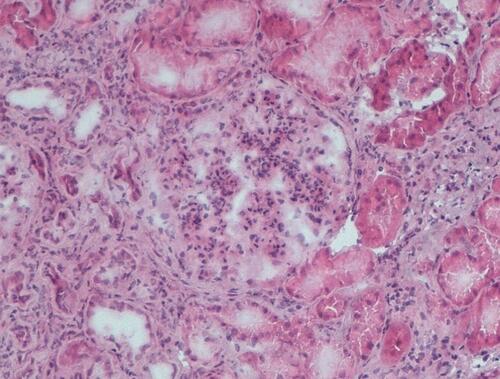
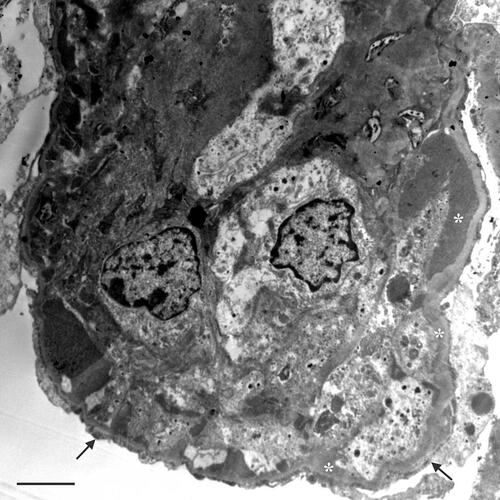
Figure 3 Kidney biopsy performed in 2019: electron micrograph showing an extensive circumferential electron-dense deposits (asterisks), and visceral epithelial foot process effacement (arrows). Scale bar 2.7 μm.
Discussion
IgAD is defined as serum IgA levels below or equal to 7 mg/dL in subjects older than 4 years after exclusion of other causes of hypogammaglobulinemia and in the presence of normal serum levels of IgG and IgM.Citation1 IgAD can be primary or secondary to several conditions (drugs, infections, chromosopathies, monogenic diseases) which were however reasonably excluded in the case reported. IgA deficiency patients are often asymptomatic and diagnosis is incidental. However, the defect may be associated with respiratory and gastrointestinal tract infections, malignancies (particularly of gastrointestinal tract and lymphoid) and autoimmune diseases.Citation1 Autoimmunity is more prevalent in adults (median age 29 years) and in females. There is reported to be an increased percentage of autoantibodies in children suffering from IgAD without necessarily any clinical manifestations.Citation2 However, preexisting selective IgAD significantly contribute to the development of two different autoimmune disorders (type 1 diabetes, Guillain-Barré syndrome) complicating SARS-Cov-2 infection.Citation3 Moreover, Naito et al observed that the frequency of selective IgAD has a strong positive correlation with the prevalence of COVID-19 per population.Citation4
Our discussion focuses on kidney diseases, especially glomerular diseases, associated with IgAD.
IgAD and Glomerulonephritis in Children and Adolescents
In children and adolescents, there are few reports about glomerulonephritis and IgAD. Liu et al described a child with nephrotic syndrome secondary to diffuse and generalized mesangiopathic glomerulonephritis and a medical history of asthma, chronic otitis media and IgAD.Citation5 Kawasaki et al described a child suffering from nephrotic syndrome secondary to diffuse membranous glomerulonephritis stage 1 and IgAD.Citation6 Ichikawa et al reported a probably multimodal autoimmune disorder producing juvenile alopecia, autoimmune encephalitis (Rasmussen syndrome) and membranous nephropathy, based on congenital IgAD.Citation7 Again, Martini et al reported the case of a 9-year-old girl with clinical signs and symptoms resembling Henoch-Schonlein syndrome and IgAD, the renal biopsy indicating mesangial proliferative glomerulonephritis with diffuse granular deposits of C3 on immunofluorescence without IgA. However, the authors believed that this case represented an unusual variant of acute post-streptococcal glomerulonephritis characterized by mild systemic vasculitis.Citation8
Recently, Di Genova et al reported 2 cases of children suffering from nephrotic syndrome and IgAD, not biopsied due to ethical issues associated with severe relapses during corticosteroid therapy. However, these patients obtained a positive outcome with human monoclonal anti-CD20 antibodies (rituximab and ofatumumab, respectively).Citation9 The most recent case refers to a 5-year-old girl with selective IgAD and membranoproliferative glomerulonephritis related to streptococcal infection and treated with benefit multidrug therapy.Citation10
IgAD and Glomerulonephritis in Adults
The association between glomerulonephritis and IgAD is better documented in adults. It is supposed that IgAD, which predisposes to infections, may contribute to the onset of glomerulonephritis, especially in chronic infectious patients.Citation11 There are also reports of elevated levels of circulating immune complexes containing IgG and IgM, which may mediate some cases of vasculitis. A review by Huang et al reported 18 cases of glomerulonephritis associated with IgAD: the most common renal pathology associated with IgAD is mesangioproliferative glomerulonephritis.Citation12 Camilleri et al reported the case of a 59 years old woman with IgAD associated with oligoarthritis and glomerulonephritis mediated by immune complexes.Citation13 John et al reported a case of mesangioproliferative glomerulonephritis in a 55-year-old woman with IgAD and serum antinuclear antibodies who presented with nephrotic syndrome without a diagnosis of systemic lupus erythematosus.Citation14 Also described is a case of anti-neutrophil cytoplasmic antibody (ANCA)-associated glomerulonephritis in a Japanese woman who had been diagnosed with IgAD 12 years previously; however, the patient had also been diagnosed with generalized epilepsy and received phenytoin and valproic acid for at least 15 years, which are known to cause IgAD.Citation15
The pathogenesis of glomerular diseases in IgAD is still poorly understood. In our case, we did not find direct evidence of a pathogenic linkage between IgAD and membranoproliferative glomerulonephritis. One may only speculate on an association with circulating immune complexes, which are present in 50–60% of patients with selective IgAD.Citation16
Conclusions
As far as we know, there is one sole case in the literature reporting an association between immunoglobulin deficiency and a mesangiocapillary glomerulonephritis pattern: this is a 55-year-old man presenting with a fainting attack.Citation17 By contrast, our patient presented only IgAD, IgG reduction being related to nephrotic syndrome. Our patient has no causes of secondary IgAD; moreover, we twice sampled IgA titers, in 2013 and 2019, and we found them very low on both occasions. He had a mild response to corticosteroid therapy but no infectious diseases during this therapy, as one might expect due to IgAD.
Disclosure
The authors report no conflicts of interest in this work.
References
- Yel L. Selective IgA deficiency. Clin Immunol. 2010;30(1):10–16. doi:10.1007/s10875-009-9357-x
- Barka N, SHen GQ, Shoenfeld Y, et al. Multireactive pattern of serum autoantibodies in asymptomatic individuals with immunoglobulin a deficiency. Clin Diagn Lab Immunol. 1995;2(4):469–472. doi:10.1128/CDLI.2.4.469-472.1995
- Pfeuffer S, Pawlowski M, Joos GS, et al. Autoimmunity complicating SARS-Cov-2 infection in selective IgA-deficiency. Neurol Neuroimmunol Neuroinflamm. 2020;7(6):e 881. doi:10.1212/NXI.0000000000000881
- Naito Y, Takagi T, Yamamoto T, Watanabe S. Association between selective IgA deficiency and COVID-19. J Clin Biochem Nutr. 2020;67(2):122–125. doi:10.3164/jcbn.20-102
- Liu K, Wigfall DR, Harland RC, Sanfilippo FP, Howell DN. Identification of selective immunoglobulin A deficiency by renal biopsy. Am J Kidney Dis. 1995;26(3):520–526. doi:10.1016/0272-6386(95)90501-4
- Kawasaki Y, Suzuki J, Onishi N, Takahashi A, Isome M, Suzuki H. IgA deficiency and membranous glomerulonephritis presenting as nephrotic syndrome. Pediatr Nephrol. 2005;20(5):662–664. doi:10.1007/s00467-004-1720-9
- Ichikawa K, Takeshita S, Ito S, Nezu A. Rasmussen syndrome combined with IgA deficiency and membranous nephropathy. Pediatr Nephrol. 2009;40(6):468–470. doi:10.1016/j.pediatrneurol.2008.12.008
- Martini A, Ravelli A, Notarangelo LD, Burgio VL, Plebani A. Henoch-Schönlein syndrome and selective IgA deficiency. Arch Dis Child. 1985;60(2):160–162. doi:10.1136/adc.60.2.160
- Di Genova L, Ceppi S, Stefanelli M, Esposito S. IgA deficiency and nephrotic syndrome in children. Int J Environ Res Public Health. 2018;15(8):1702. doi:10.3390/ijerph15081702
- Sugimoto K, Enya T, Miyazaki K, Mijazawa T, Takemura T, Okada M. Membranoproliferative glomerulonephritis related to a streptococcal infection in a girl with IgA deficiency: a case report. BMC Nephrol. 2020;21(1):68.
- Sleasman JW. The association between immunodeficiency and the development of autoimmune disease. Adv Dental Res. 1996;10(1):57–61. doi:10.1177/08959374960100011101
- Huang JB, Yang WC, Hu CC, Yang AH, Lin CC. IgA deficiency with membranous glomerulonephritis: a case report and review. J Nephrol. 2003;16(1):154–158.
- Camilleri JP, Moore RH, Griths DS, Williams BD. Selective IgA deficiency associated with glomerulonephritis and oligoarthritis. Ann Rheum Dis. 1992;51(1):123–125. doi:10.1136/ard.51.1.123
- John M, Lam M, Latham B, Saker B, French MA. Nephrotic syndrome in a patient with IgA deficiency-associated mesangioproliferative glomerulonephritis. Pathology. 2000;32(1):56–58. doi:10.1080/003130200104600
- Sugiyama M, Banno Y, Nozaki Y, Kinoshita K, Funauchi M. A case of selective immunoglobulin A deficiency with anti-neutrophil cytoplasmic antibody-associated glomerulonephritis. Int J Rheum Dis. 2014;17(3):341–343. doi:10.1111/1756-185X.12157
- Kwitko AO, McKenzie PE, Shearman DJC, Gormly AA, Woodroffe AJ. Circulating immune complexes in IgA deficiency. Clin Exp Immunol. 1979;38:45–51.
- Rashid HU, Biswas CK, Morley AR, Kerr DN. Mesangiocapillary glomerulonephritis type I associated with immunoglobulin deficiency. Br Med J (Clin Res Ed). 1981;283(6288):407–408. doi:10.1136/bmj.283.6288.407