
Abstract
Sturge-Weber syndrome is a rare, sporadic, progressive neurocutaneous condition that presents with congenital hamartomatous malformations, epilepsy, and a variety of facial symptoms. We discussed a rare case of an eighteen-year-old female child who came to our neurology department with status epilepticus, mental impairment, and a port-wine in the lateral left side of her face. We diagnosed Sturge-Weber syndrome after a thorough neurological and radiological evaluation. The purpose of presenting this case is to illustrate both the characteristic presentation and the complications associated with managing Sturge-Weber syndrome.
Keywords:
Introduction
Sturge-Weber syndrome (SWS) is a rare congenital disorder characterized by vascular hamartomas affecting the brain and facial tissues. It’s also known as encephalotrigeminal angiomatosis.Citation1 The most common characteristic that is usually present is the presence of port-wine stains in the ophthalmic and maxillary divisions of the trigeminal nerve.Citation2 The main sign is usually a reddish-purple or dark red color in the maxillary division and ophthalmic division of the trigeminal nerve. There may also be venous angiomas in the leptomeninges over the cerebral cortex and ipsilateral gyriform calcification, which can cause epilepsy and hemiparesis on the opposite side.Citation3 Other related features of this syndrome include glaucoma and mental retardation. The primary brain imaging modality is a magnetic resonance imaging (MRI) scan with gadolinium contrast to show the characteristic features of calcification, leptomeningeal, and leptomeningeal enhancement.Citation4 However the feature which is commonly found in the oral cavity is gingival hemangioma, which mostly affects the ipsilateral area of maxilla, mandible, mouth’s floor, lips, plate, tongue and jugal mucosa.Citation5 Radiological investigations are the most useful, with standard skull radiography, computed tomography, and magnetic resonance imaging all playing important roles in revealing brain changes.Citation6 Other investigative techniques include electroencephalography (EEG) to assess brain function and ocular ultrasonography to discover ocular abnormalities.Citation7 Since Struggle-Weber syndrome has an intractable seizure, treatment mainly consists of seizure control with anticonvulsants and may need multiple anti-seizure combinations.Citation8
Case Presentation
An 18-year-old female patient presented with a history of seizures and weakness in the right side of the body. Seizures initially started on the right side of the body and then became generalized, which was associated with post-ictal confusion. The patient’s past medical history reported that she had developed a convulsive disorder at the age of 7 months old, for which she was taking long-term antiepileptic drugs (carbamazepine 200 mg) for her seizures.
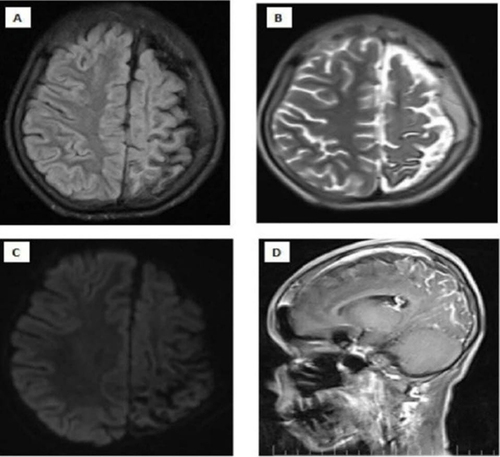
On examination, the patient’s Glasgow coma scale was 11/15 (eye opening: 3, verbal response: 3, motor response: 5), indicating mild disorientation. She had right-sided weakness; on the right side, muscle strength was 3/5. Likewise, other cranial nerves were intact and symmetric. Pupils were equally reactive to light. She had generalized hyporeflexia on examination of the patient’s upper and lower limbs. There was no facial asymmetry at the time of admission, but a red-purple facial skin lesion was visible at the ophthalmic and maxillary divisions of the left side of the face (). Since the patient was confused, a cerebellar examination was not possible. The rest of the neurological examination was normal. The chest was clear for auscultation. The gastrointestinal and cardiovascular systems were unremarkable. The FNA revealed a fibroadenoma, leading to the appearance of a lump at the right breast. The laboratory investigation revealed a normal complete blood picture, but the arterial blood gas (ABG) reveals bicarbonate at 19.3 and carbon dioxide at 48.1, potentially linked to the seizure attack. A computed tomography scan of the brain showed that the left hemisphere of the brain was moderately atrophying and had a lot of gyriform cortical-subcortical (tram-track) calcification ( and ). To confirm further, we conducted a brain MRI with contrast, which revealed prominent atrophic changes at the sulcus and fissures of the parietal lobe, thickening of the adjacent left parietal bone for the left cerebral hemisphere, and gyral-meningeal contrast enhancement in the left parietal lobe (). Based on neurological findings and neuroimaging, we established the diagnosis of SWS. We admitted the patient to the intensive care unit and initiated her on levetiracetam 1000 mg IV twice a day. After stabilization of the patient’s condition, we transferred her to the neurology ward, but she developed focal seizures on the mouth and eyes, and we added Carbamazepine 200 mg twice per day. The patient’s primary issue was poor feeding and two weeks of nasogastric tube feeding. In the neurology ward, we consulted the general surgery department and recommended an open percutaneous endoscopic gastrostomy for feeding. Following this, we discharged the patient with an improved general condition and a seizure.
Figure 1 This is a patient’s image showing the left fronto-maxillary reddish-purple patch, which indicated port-wine of the face.
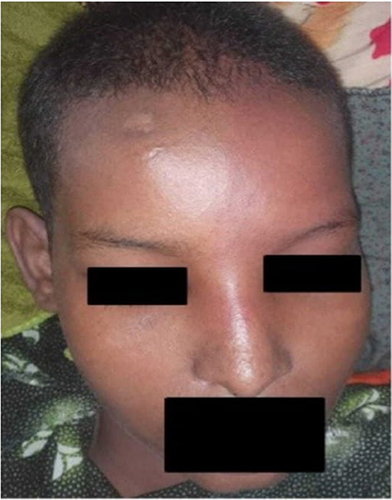
Figure 2 A computed tomography of the brain reveals unilateral left-sided moderate atrophy of the cerebral hemisphere (A) with extensive gyriform cortical-subcortical (tram-track) calcifications (B).
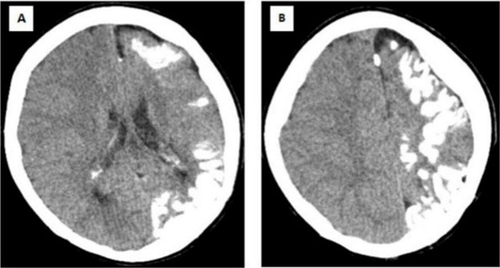
Figure 3 Magnetic Resonance imaging of the brain which shows left cerebral hemisphere, prominent atrophic changes at the sulcus and fissures of the parietal lobe and thickening of the adjacent of the left parietal lobe are noteworthy (A and B). There is no diffusion restriction (C). After Intravenous contrast injection (D). There is gyral-meningeal contrast enhancement in the cortical regions in the areas described of the left parietal lobe.
Discussion
The clinical characteristics of Sturge-Weber syndrome may differ greatly and include ipsilateral gyriform calcification, leptomeningeal angiomas, port-wine stain in the face, various ocular signs, hemiparesthesia, and hemiplegia.Citation9 Port-wine staining in the face establishes the diagnosis, followed by other signs like glaucoma, epilepsy, and mental retardation.Citation10 The precise etiology is not fully understood, however, there are theories. Leptomeningeal and facial angiomata have been hypothesized to be signs of persistently undeveloped sinusoidal vascular channels.Citation11 Another theory is the incomplete development of superficial venous drainage with subsequent compensatory dilation of small venous channels.Citation12 The clinical features of SWS can be neurological or nonneurological. The neurological symptoms include seizures, hemiparesis, headaches, visual field deficits, and cognitive impairments, as seen in the above case illustration.Citation13 Other features include early handedness, gaze preferences, and stroke-like episodes. Behavioral and mental disorders, endocrine disorders, difficulties with learning, and other medical conditions are among the non-neurological symptoms of Sturge-Weber Syndrome.Citation14 In 90% of cases, the initial symptom is typically a seizure that happens within the first year of life.Citation10 They may be in the form of infantile spasms, myoclonic, atonic, or tonic seizures. It is also possible to have localized and generalized seizures. External factors like sleep deprivation, stress, and disease, including infection, typically trigger seizures. The main underlying cause of seizures is believed to be calcification-induced epileptic focus.Citation15 Most Sturge-Weber syndrome cases rarely pose a serious risk to the patient’s life. This is a slow-progressive neurological disease, associated with continuous neurological decline in the patient. Quality of life can be preserved by effectively controlling and treating symptoms such as seizures, vision impairments, and mental disorders.Citation16 In patients with difficult-to-control seizures, neurosurgery treatments like lobectomy or hemispherectomy appear to be the best options. There is no data to conclude that surgical care during infancy improves prognosis; yet delaying surgical treatment may exacerbate cognitive impairment.Citation17 In our case, the patient’s poor cooperation prevented us from obtaining the full ophthalmological examination, and the patient’s condition prevented us from performing the EEG. We could not test the genetic investigation due to a lack of availability in the whole country.
Conclusion
Sturge-Weber syndrome is a neurodevelopmental disease that is rarely observed in clinics. In our case report, we described a young girl who has been experiencing refractory seizures since she was a child. Following a neurological and imaging evaluation, we determined that she had Sturge-Weber syndrome. We recommend that young patients who experience intractable seizures should look into the underlying cause.
Ethical Consideration
Ethical approval is not required for the publication of case reports from our hospital.
Consent for Publication
Verbal informed consent was obtained from the mother of the child for the publication of this case report and its image. The evidence of the verbal consent is available for review by the corresponding author on request. The reason for obtaining verbal consent is: the mother of the child came from far rural area, she is an illiterate, which means she can not write or read, so it is difficult to understand the consent of the case and sign it. Therefore, we recorded verbally for the publication of the case.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors have no conflicts of interest in this work.
References
- Mantelli F, Bruscolini A, La Cava M, Abdolrahimzadeh S, Lambiase A. Ocular manifestations of sturge–weber syndrome: Pathogenesis, diagnosis, and management. Clin Ophthalmol. 2016;871–878. doi:10.2147/OPTH.S101963
- Waelchli R, Aylett SE, Robinson K, Chong WK, Martinez AE, Kinsler VA. New vascular classification of port‐wine stains: Improving prediction of Sturge–Weber risk. Br J Dermatol. 2014;171(4):861–867. doi:10.1111/bjd.13203
- Khambete N, Risbud M, Kshar A. Sturge-Weber syndrome: a case report. Int J Dent Clin. 2011;3(1):79.
- Bar C, Pedespan JM, Boccara O, et al. Early magnetic resonance imaging to detect presymptomatic leptomeningeal angioma in children with suspected Sturge–Weber syndrome. Dev Med Child Neurol. 2020;62(2):227–233. doi:10.1111/dmcn.14253
- Carvalho VA, Dallazen E, Statkievicz C, et al. Oral surgery in patients with Sturge-Weber syndrome. J Craniofacial Surg. 2021;32(1):e85–e88. doi:10.1097/SCS.0000000000007048
- Bálint A. Advanced neuroimaging studies in sturge–weber syndrome: Clinical Correlates; 2012.
- Maslin JS, Dorairaj SK, Ritch R. Sturge-Weber syndrome (encephalotrigeminal angiomatosis): Recent advances and future challenges. Asia-Pac J Ophthalmol. 2014;3(6):361–367. doi:10.1097/APO.0000000000000093
- Gill NC, Bhaskar N. Sturge–Weber syndrome: a case report. Contemp Clin Dentis. 2010;1(3):183–185. doi:10.4103/0976-237X.72789
- Garro SJ, Bradshaw WT. Sturge-Weber syndrome: a case study. Adv Neonatal Care. 2014;14(2):96–102. doi:10.1097/ANC.0000000000000060
- Conceição JGD, Santos LFGD, Bahia TPDS, Silva VDA, Ramos MEB, Israel M. Sturge-Weber syndrome: A case report. RSBO. 2011;8(4):469–472. doi:10.21726/rsbo.v8i4.1103
- Comi AM. Sturge–Weber syndrome. Handbook Clin Neurol. 2015;132:157–168.
- Nguyen V, Hochman M, Mihm MC Jr, Nelson JS, Tan W. The pathogenesis of port wine stain and Sturge Weber syndrome: complex interactions between genetic alterations and aberrant MAPK and PI3K activation. Int J Mol Sci. 2019;20(9):2243. doi:10.3390/ijms20092243
- Sudarsanam A, Ardern-Holmes SL. Sturge–Weber syndrome: from the past to the present. Eur J Paediatr Neurol. 2014;18(3):257–266. doi:10.1016/j.ejpn.2013.10.003
- Jansonius NM, Bennebroek CA, Schalij-Delfos NE, Nielsen CC, van Dijk M, Vehmeijer WB. Visual outcome in Sturge-Weber syndrome: a systematic review and Dutch multicenter cohort. Visual Field Exam Child Brain Dis. 2016;101.
- Govori V, Gjikolli B, Ajvazi H, Morina N. Management of patient with Sturge-Weber syndrome: a case report. Cases J. 2009;2:1–6. doi:10.1186/1757-1626-2-9394
- Higueros E, Roe E, Granell E, Baselga E. Sturge-Weber syndrome: A review. Actas Dermo-Sifiliográficas. 2017;108(5):407–417. doi:10.1016/j.ad.2016.09.022
- Arzimanoglou AA, Andermann F, Aicardi J, et al. Sturge–Weber syndrome: indications and results of surgery in 20 patients. Neurology. 2000;55(10):1472–1479. doi:10.1212/WNL.55.10.1472