
Abstract
Background
The clinical picture of intracerebral calcification is so varied that it constitutes an essential element of a wide range of clinical syndromes of variable expression that continue to be described. In this article, we discuss the diagnostic possibilities of basal ganglia calcification considering the association of failure to thrive and macular degeneration in our patient.
Case
A 17-year-old male patient of Congolese origin consulted us for a pyramidal syndrome consisting of upper limb tremors during mobilization and dysgraphia. The patient also presented with a distance vision disorder for which the ophthalmological examination revealed poor visual acuity in both eyes (2/10) and macular degeneration in the left eye. On physical examination, we noted a short stature with a small head circumference in relation to age. The brain scan revealed the presence of bilateral striato-pallidal calcifications giving the appearance of Fahr's disease. However, the association of delay of stature development with microcrania, macular degeneration with reduced visual acuity and basal ganglia calcifications could suggest a wide range of syndromic hypotheses, the most likely of which is Rajab-type cerebral calcification.
Conclusion
The association of failure to thrive, macular degeneration, and cerebral calcification of the basal ganglia is revealed as a particular phenotype compared to cases reported in the literature. An in-depth analysis would be necessary to identify a possible genetic basis.
Introduction
Cerebral calcinosis or intracerebral calcification is an accumulation of calcium in the brain parenchyma. Many syndromes involving intracerebral calcification have been reported in the literature. The incriminated causes are multiple, divided between hereditary (familial) and acquired conditions. Six main groups are generally described according to their etiopathogenesis: age-related (or physiological), congenital, infectious, endocrine (or metabolic), vascular, and neoplastic.Citation1
In children, intracerebral calcifications are most often acquired. They are associated with antenatal infections, cerebral hemorrhage in the neonatal period, and sometimes neoplasia.Citation2
Several different types of cerebral calcinosis have been reported, distinguished by their association with endocrinopathies with abnormal phosphocalcic metabolism (hypoparathyroidism and pseudohypoparathyroidism: Albright’s hereditary osteodystrophy)Citation3 or metabolic abnormalities (Krabbe’s disease and mitochondriopathies such as mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS) and myoclonic epilepsy with red ragged fibers (MERRF)).Citation4,Citation5
On the other hand, other cases associate intracerebral calcification with systemic disorders and diseases of various kinds. These include systemic lupus erythematosus,Citation6 Aicardi-Goutières syndrome (elevated interferon alpha and lymphocytosis in the cerebrospinal fluid (CSF)),Citation7 Coats plus syndromeCitation8 (cerebroretinal microangiopathy with cysts) and pseudo-TORCH syndrome (microcephaly, convulsion and profound mental retardation).Citation9
A particular calcinosis (Fahr’s disease) is characterized by the predominance of extrapyramidal manifestations with major movement disorders.Citation10,Citation11 The clinical picture of intracerebral calcification presents such a broad phenotype that it is an important component of a wide range of clinical syndromes of varying expression that continue to be described.
We report a clinical case of an adolescent with intracerebral calcifications of the Farh type with extrapyramidal clinical manifestations that are particularly associated with failure to thrive and retinopathy. Contrary to the classic description of Fahr's disease, the growth retardation and retinopathy present in our patient make it a particular phenotype. Although the two signs taken separately can be found in some calcinosis, the association of the two is not described in the literature.
In this case study, we discuss the particularities of our patient and review the different possibilities of differential diagnosis that could explain this association of signs.
Case Report
A 17-year-old boy of Congolese origin, came to the hospital for tremors of the upper limbs occurring when the latter were solicited (eg, lifting a glass of water, bringing his hand to the mouth, and writing). The patient reported that the symptoms had been slowly progressing for approximately 6 months. In addition, the patient reported difficulty in seeing at a distance (decreased distance vision).
The patient is the second of three siblings, the eldest of whom died of acute respiratory infections in early childhood. It should be noted that the patient was the product of a dizygotic full-term twin pregnancy and that his twin was in apparent good health at the time of consultation. No morbid history was reported in the neonatal period or in the first years of life. There was no history of abortion or miscarriage in the mother, who did not acknowledge any consanguinity with her partner (although the couple were monoethnic).
Apart from the abovementioned complaints, the parents report a slowing of growth since the age of 10–11 years characterized by weight and stature stagnation (unlike his twin sister, who shows “normal” growth).
The patient’s general condition was preserved with slight ptosis in both eyes on morphological examination. From an anthropometric point of view, the patient weighed 30 kg with a height of 143 centimeters and a head circumference of 52 centimeters (BMI for age = 14.6). It should be noted that secondary sexual characteristics (external genitalia, pubic hair, and axillary hair) were present and consistent with the declared age (see Tanner stage P5/G5). Ophthalmological examination reported poor visual acuity in both eyes (2 out of 10) and macular degeneration in the left eye (the right eye was normal on the fundus). Neurologically, the patient is oriented in time and space and does not show any tremor of the limbs at rest. However, we note the appearance of tremors of the upper limbs during action, essentially during the finger-nose maneuver, as well as during writing, resulting in significant dysgraphia. The walk is straight and balanced. No nystagmus was noted, and the various clinical neurological tests ruled out dysdiadochokinesia in our patient. A cerebellar syndrome with delayed growth was evoked in a 17-year-old adolescent with no previous neurological history.
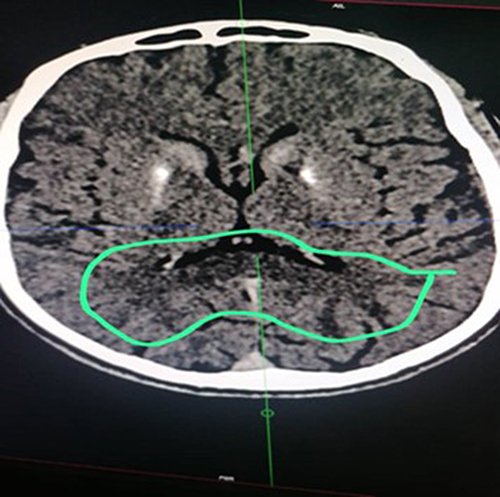
The helical CT scan of the brain without and after injection of iodinated contrast medium revealed bilateral striatopalidal calcifications (). In addition, there are bilateral and symmetrical areas of hypodensity of the subcortical white matter opposite the occipital horns of the lateral ventricles, bridging the splenium of the corpus callosum and extending downward via the midbrain and the protuberance toward the two cerebellar hemispheres where they are more marked (). In addition, there is an enlargement of the basal cisterns with an appearance of moderate bulboprotuberantial atrophy.
Figure 1 Calcifications of the heads of the caudate nucleus and putamen (Striatum) and globi pallidum.
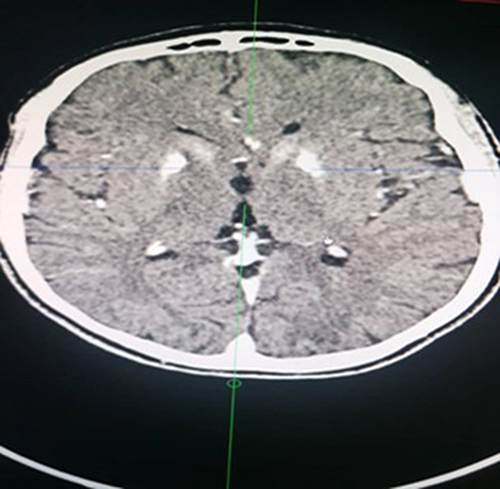
Figure 2 Bilateral and symmetrical hypodensity of the cerebellar peduncles (green arrows).
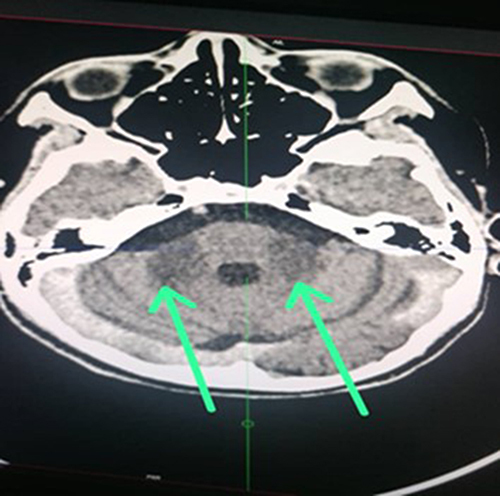
Figure 3 Bilateral and symmetrical hypodensity of the 2 cerebellar hemispheres.
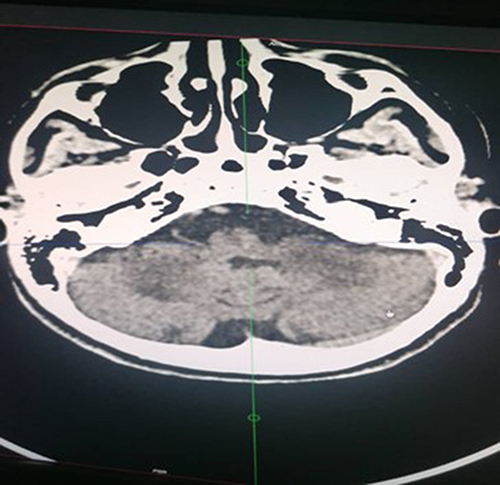
Figure 4 Hypodensity of the subcortical white matter opposite the occipital horns and bridging the splenium of the corpus callosum.
The phosphocalcic balance was normal (calcemia 9.1 mg/100 mL [8.8–10.4]; inorganic phosphorus 4.8 mg/100 mL [2.5–5.0]) as well as negative for syphilis and toxoplasmosis serologies (VDRL negative, positive Toxoplasma IgG 2.2 and positive Toxoplasma IgM 0.5). The anti-nuclear antibody assay was weakly positive (anti-nuclear protein Ac 1/320, anti-nuclear DNA Ac 1/160). Thyroid hormones and cortisol were within normal limits (TSH = 0.98 mIU/L [0.15–5]; T3 = < 0.77 µmol/L; T4 = 114 nmol/L [80–140]; cortisol = 240 nmol/L [110–520]).
In view of the failure to thrive (short stature and microcephaly) and the progressive cerebellar syndrome associated with intracerebral calcification, the diagnostic hypothesis of “Rajab-type cerebral calcification” could also be considered. Genetic testing is essential to confirm the diagnosis.
Discussion
Cerebral calcifications are associated with several pathologies of various etiologies. Many conditions can be responsible for intracerebral calcifications (endocrinopathies, systemic diseases, infection, intoxication, tumor, genetic, and mitochondrial diseases). Fahr's disease, also known as bilateral strio-pallido-dentate calcinosis (or idiopathic basal ganglia calcification (IBGC)), is a genetic form of calcification involving the basal ganglia without associated endocrine disorders.Citation10 The diagnosis is made on the presence of cerebral calcifications involving at least the basal ganglia, without any other cause being identified after an extensive clinical and biological etiology work-up.
It is distinguished from Fahr's syndrome, which is associated with bilateral and symmetrical intracerebral calcifications located mainly in the basal ganglia and serrated nucleus and is associated with disorders of phosphocalcic metabolism such as hypoparathyroidism.Citation11,Citation12 The term “Fahr's disease” is therefore theoretically used for idiopathic forms, whereas the term “Fahr's syndrome” refers to cases of secondary calcinosis.Citation13 However, age-related physiological calcifications are often found on the lenticular and serrated nuclei but are not as prominent as in Fahr's syndrome. Nevertheless, there is no threshold to classify intracerebral basal ganglia calcifications as pathological or not according to age. Several clinical pictures, including basal ganglia calcifications have been reported in the literature ().
Table 1 Reported Cases of Intracranial Calcification Affecting the Basal Ganglia in the Literature
The clinical expression of Fahr's disease is very heterogeneous. Moreover, known as autosomal dominant dystonia, the symptoms of Fahr's disease range from cerebellar syndrome to psychiatric and cognitive disorders.Citation21 Abnormal movements, parkinsonian syndromes, balance disorders, dysarthria, psychiatric manifestations and cognitive disorders have been reported.Citation22,Citation23 As in our case, lesions detected on the brain CT scan are characteristic of Fahr's disease (macrocalcifications of the basal ganglia at the bipallidal level and of the caudate nuclei) (); associated with this, abnormal movements expressed by dysgraphia and the absence of phosphocalcic metabolism disorder. In addition, all other causes, especially infectious ones (toxoplasmosis, syphilis) were excluded as well as probable dysimmunity (antinuclear antibodies and DNA).
However, in our patient, we noted a delay in staturo-ponderal development (short stature, microcephaly, and low BMI for age) and a vision disorder characterized by poor visual acuity in both eyes (2/10) with macular degeneration in the left eye. The combination of failure to thrive (slow growth, short stature, and microcephaly) and retinopathy (macular degeneration) has not been described in many syndromic calcinosis cases. Rajab et alCitation2 reported several cases of cerebral calcinosis associated with delayed growth and microcephaly in the same family. However, the calcification lesions are (contrary to Fahr's disease) diffuse globular, involving a large part of the cortex, the basal ganglia, thalamus and the deep cerebellar nuclei. The familial nature of the cases suggested a genetic origin for which a locus on chromosome 2 was identified. However, in the case of our patient, the fact that the twin sister is apparently in good health means that we cannot formally rule out a possible genetic cause, given the dizygotic nature of the twins (It should also be noted that the 2 brothers are the only siblings).
However, none of the Omani cases described by Rajab et al showed any visual impairment, and the retinal examination of all patients was found to be normal. In addition, most of the patients in Rajab’s series presented with variable cognitive impairment (mild to moderate), which is like cerebroretinal microangiopathy with calcifications and cysts (CRMCC) or “Coats plus” syndrome. However, CRMCC also associates exudative detachment retinopathy with congenital telangiectasia with ante and postnatal growth retardation in some cases.Citation24–26 In our patient, no cognitive deficits were noted, and the level of schooling suggested a normal state of intelligence for his age. In addition, the ocular disorder presented by our patient was unilateral macular degeneration in the left eye leading to a significant decrease in visual acuity.
Conclusion
Intracerebral calcifications in children are a sign found in many entities of various etiologies and phenotypes. The nature of cerebral lesions, the mode of clinical expression and associated signs are indicative of a genetic or acquired character. Great variability in the clinical presentation of the same type of lesions has been described. Our patient had all the features described for Fahr's disease, with the addition of growth retardation and macular degeneration. This phenotype is in addition to the variability of clinical expression in cerebral calcinosis reported in the literature.
Documentation of cases of syndromic association with intracerebral calcinosis would shed light on the gray areas and enrich the understanding of the different phenotypes. Genetic studies are still essential to determine the underlying cause, when all acquired causes seem to have been ruled out.
Ethical Approval and Consent to Participate
Oral and writing consent to participate was granted by the parents. In addition, the approval of the Lubumbashi University Clinics was required to publish the details of the case.
Consent for Publication
Written informed consent was obtained from the patient’s legal guardian for publication of this case report and any accompanying images.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no potential conflicts of interest in this work.
Acknowledgments
We thank Stanislas Wembonyama and Olivier Mukuku for their kind assistance during the writing of this manuscript.
Data Sharing Statement
The data that support the findings of this case report are available from the corresponding author upon reasonable request.
Additional information
Funding
References
- Kiroǧlu Y, Çalli C, Karabulut N, Óncel Ç. Intracranial calcifications on CT. Diagnostic Interv Radiol. 2010;16(4):263–269.
- Rajab A, Aldinger KA, El-Shirbini HA, Dobyns WB, Ross ME. Recessive developmental delay, small stature, microcephaly and brain calcifications with locus on chromosome 2. Am J Med Genet Part A. 2009;149(2):129–137. doi:10.1002/ajmg.a.32630
- Quirúrgica C, Valencia U. Osteodistrofia de A lbright: pseudo-pseudo-hipoparatiroidismo. Rev Esp de Cir Ost. 1978;1959:47–55.
- Moualek D, Bouderba R, Saadi A, et al. Maladie de Krabbe particulière liée au gène PSAP. Rev Neurol. 2021;177:S50. doi:10.1016/j.neurol.2021.02.190
- Kostić VS, Petrović IN. Brain calcification and movement disorders. Curr Neurol Neurosci Rep. 2017;17(1). doi:10.1007/s11910-017-0710-9
- Boisseau W, Bottin L, Frances C, Alamowitch S. Lupus discoïde et calcifications des noyaux gris centraux: à propos de 2 cas. Rev Neurol. 2015;171:A131. doi:10.1016/j.neurol.2015.01.307
- Samanta D, Ramakrishnaiah R, Crary SE, Sukumaran S, Burrow TA. Multiple autoimmune disorders in aicardi-goutières syndrome. Pediatr Neurol. 2019;96:37–39. doi:10.1016/j.pediatrneurol.2019.01.017
- Troumani Y, Ackermann F, Cohen S, et al. Coats Plus: la version systémique de la maladie de Coats. J Fr Ophtalmol. 2016;39(7):e167–70. doi:10.1016/j.jfo.2015.06.015
- Gupta N, Thakur S, Handley MT, Bokaria R, Saxena R, Kohli S. Clinical Vignette A genetic syndrome that mimics congenital TORCH infection. Pati Res. 2016;9(1):2–8.
- Nicolas G, Hannequin D. Idiopathic basal ganglia calcification (Fahr’s disease). In: Pratique Neurologique - FMC. Vol. 4. Elsevier Masson SAS; 2013:143–150.
- Saleem S, Aslam HM, Anwar M, et al. Fahr’s syndrome: literature review of current evidence. Orphanet J Rare Dis. 2013;8(1):1–9. doi:10.1186/1750-1172-8-156
- Richy C, Foulon L, Thiebaud PC. Le syndrome de Fahr. Ann Fr Med d’Urgence. 2017;7(5):330–331. doi:10.1007/s13341-017-0772-y
- Perugula ML, Lippmann S. This column series compares neurological conditions that pose differential challenges in diagnoses. Differential Diagnoses FAHR’S DISEASE OR FAHR’S SYNDROME? Innov Clin Neurosci. 2016;13:1.
- Burn J, Wickramasinghe HT, Harding B, Baraitser M. A syndrome with intracranial calcification and microcephaly in two sibs, resembling intrauterine infection. Clin Genet. 1986;30(2):112–116. doi:10.1111/j.1399-0004.1986.tb00578.x
- Bönnemann CG, Meinecke P, Reich H. Encephalopathy with intracerebral calcification, white matter lesions, growth hormone deficiency, microcephaly, and retinal degeneration: two sibs confirming a probably distinct entity. J Med Genet. 1991;28(10):708–711. doi:10.1136/jmg.28.10.708
- Yoshimara M, Hara T, Maegaki Y, et al. A novel neurological disorder with progressive CNS calcification, deafness, and microcytic anemia. Dev Med Child Neurol. 1997;38:198–201.
- Slee J, Lam G, Walpole I. Syndrome of microcephaly, microphthalmia, cataracts, and intracranial calcification. Am J Med Genet. 1999;84(4):330–333. doi:10.1002/(SICI)1096-8628(19990604)84:4<330::AID-AJMG4>3.0.CO;2-W
- Adderson EE, Viskochil DH, Carey JC, et al. Growth failure, intracranial calcifications, acquired pancytopenia, and unusual humoral immunodeficiency: a genetic syndrome? Am J Med Genet. 2000;95(1):17–20. doi:10.1002/1096-8628(20001106)95:1<17::AID-AJMG5>3.0.CO;2-M
- Mizuno Y, Takahashi K, Igarashi T, Saito M, Mizuguchi M. Congenital infection-like syndrome with intracranial calcification. Brain Dev. 2011;33(6):530–533. doi:10.1016/j.braindev.2010.09.001
- Nakamura K, Kato M, Sasaki A, Kanai M, Hayasaka K. Congenital dysplastic microcephaly and hypoplasia of the brainstem and cerebellum with diffuse intracranial calcification. J Child Neurol. 2012;27(2):218–221. doi:10.1177/0883073811416239
- Geschwind DH, Loginov M, Stern JM. Identification of a locus on chromosome 14q for idiopathic basal ganglia calcification (Fahr disease). Am J Hum Genet. 1999;65(3):764–772. doi:10.1086/302558
- Manyam BV. What is and what is not “Fahr’s disease”. In: Parkinsonism and Related Disorders. Vol. 11. Elsevier BV; 2005:73–80.
- Manyam BV, Walters AS, Narla KR. Bilateral Striopallidodentate calcinosis: clinical characteristics of patients seen in a registry. Mov Disord. 2001;16(2):258–264. doi:10.1002/mds.1049
- Tolmie JL, Browne BH, Mc Gettrick PM, Stephenson JBP. A familial syndrome with coats’ reaction retinal angiomas, hair and nail defects and intracranial calcification. Eye. 1988;2(3):297–303. doi:10.1038/eye.1988.56
- Crow YJ, McMenamin J, Haenggeli CA, et al. Coats’ plus: a progressive familial syndrome of bilateral coats’ disease, characteristic cerebral calcification, leukoencephalopathy, slow pre- and postnatal linear growth and defects of bone marrow and integument. Neuropediatrics. 2004;35(1):10–19. doi:10.1055/s-2003-43552
- Gayatri N, Hughes M, Lloyd I, Wynn R. Association of the congenital bone marrow failure syndromes with retinopathy, intracerebral calcification and progressive neurological impairment. Eur J Paediatr Neurol. 2002;6(2):125–128. doi:10.1053/ejpn.2001.0559