
Abstract
Persistent pain following back surgery remains a major treatment challenge. The primary objective of this open-label exploratory study was to investigate the analgesic effectiveness of pulsed electromagnetic field therapy administered twice daily over a 45-day period in 34 subjects (68% female) with persistent or recurrent pain following back surgery. A secondary goal was to guide the design of future randomized controlled trials that could target responsive subpopulations. All predefined primary and secondary outcomes, including change in pain intensity (PI), physical function (Oswestry Disability Index), analgesic consumption, and overall well-being (Patient Global Impression of Change), are reported. A responder analysis (≥30% reduction in PI versus baseline) was added as a post hoc evaluation. Safety outcomes, as well as results of a cost-avoidance survey, are also summarized. Of the 30 per-protocol subjects who completed the study, 33% reported a clinically meaningful (≥30%) reduction in PI. A higher response rate (60%) was reported for subjects who had undergone discectomy prior to the trial compared to subjects who had undergone other types of surgical interventions (decompression or fusion) without discectomy. Improvements in PI were paralleled by improvements in secondary outcomes. Relative to baseline, responders reported an average 44% and 55% reduction in back PI and leg PI (respectively), and an average 13% improvement in Oswestry Disability Index scores. In the per-protocol population, 50% of responders and 12% of nonresponders reported less analgesia consumption at the end of treatment versus baseline. Sixty-seven percent of per-protocol responders and 0% of nonresponders reported clinically meaningful improvement in overall well-being on the Patient Global Impression of Change scale.
Introduction
“Failed back surgery syndrome” (FBSS) is a broad term used to describe persistent or recurrent back and/or radiating leg pain after spinal surgery,Citation1–Citation5 including anatomically successful surgery.Citation4 Etiological factors for FBSS include misdiagnosis, inappropriate patient selection, wrong-level surgery, surgical technical failure (eg, pseudoarthrodesis), spinal instability, spinal stenosis, epidural fibrosis, recurrent disc herniation, adjacent segment disease, sacroiliac joint pain, and piriformis syndrome.Citation1,Citation5–Citation7 Considering the high volume of lumbar spine surgeries performed annuallyCitation8 and the high incidence of persistent pain following back surgeryCitation1,Citation5 (estimated at 30%–46% following lumbar fusion and 19%–25% following microdiscectomy),Citation1 the unmet need for effective FBSS treatment interventions is an important healthcare concern.Citation2,Citation3
Conservative and minimally invasive interventions are preferred for FBSS prior to undertaking repeat spinal surgeries,Citation5 as there is a diminishing success rate with increasing number of surgeries.Citation1 Despite widespread use of opioids in this population,Citation9,Citation10 there is a lack of data supporting their efficacy in long-term studies of FBSS pain.Citation1,Citation2,Citation5 Due to possible adverse side effects, the benefit-to-risk ratio must be evaluated when considering pharmacological interventions (opioids, nonsteroidal anti-inflammatory drugs, antidepressants, gabapentinoids, epidural steroids, or local anesthetics),Citation1,Citation5,Citation11,Citation12 particularly for long-term use. Complications associated with implantable devices, such as spinal cord stimulators and intrathecal drug pumps, are common,Citation2 limiting their applicability.
In light of the unmet need for safe, effective, noninvasive interventions for FBSS pain, we conducted an open-label, single-arm pilot study to evaluate the analgesic effectiveness of pulsed electromagenetic field (PEMF) therapy administered twice daily over a 45-day period to subjects with persistent or recurrent pain following anatomically successful lumbar spine surgery. The study was intended as a pilot to guide the design of future randomized controlled trials that would target responsive subpopulations.
PEMF therapy is a noninvasive therapy used for adjunctive treatment of postoperative pain and edema in superficial soft tissues.Citation13,Citation14 The therapy is self-administered at home using a portable medical device that delivers nonthermal, nonionizing pulsed electromagnetic energy to the treatment area, without the need for surface or deep electrodes. In vitro and in vivo evidence suggests PEMF may induce a localized analgesic effect by modulating factors involved in pain signaling and the inflammatory response,Citation15–Citation17 and clinical studies have reported that PEMF is effective as an adjuvant for postoperative pain relief after several types of surgeries.Citation14,Citation17–Citation22
Materials and methods
Study design
The trial was designed as an open-label, single-arm pilot study, conducted at four geographically distributed investigative sites with subjects enrolled between November 2013 and February 2014. Enrollment varied from four to 13 subjects per study site in the intent-to-treat population. The trial protocol was approved by a central institutional review board.
Study participants
Study participants were identified and recruited from the clinical practices of the study investigators. Inclusion and exclusion criteria are provided in . Written informed consent was obtained from all study participants. The informed consent documents and process complied with applicable requirements (US Code of Federal Regulations Title 21 Parts 50 and 56) and were reviewed and overseen by the institutional review board.
Table 1 Inclusion and exclusion criteria
Study intervention
All study participants were treated with individual at-home PEMF therapy devices (Provant Therapy System Model 4201; Regenesis Biomedical Inc., Scottsdale, AZ, USA). Provant uses a solid-state, fixed-power output radio frequency generator and transmitter operating at 27.12 MHz and is US Food and Drug Administration (FDA) cleared for adjunctive use in the palliative treatment of postoperative pain and edema in superficial soft tissue. The device delivers imperceptible pulsed electromagnetic field energy to the desired treatment area via a spiral antenna in the device’s treatment applicator. Pulse durations are 42±4 microseconds repeated every 1,000±25 microseconds. In this study, each subject was instructed to administer the treatment for 30 minutes, twice a day, for 45 days (either self-administered or administered by a family member or caregiver).
Screening, enrollment, and follow-up visits
Screening
Eligibility was assessed during an initial screening visit. Consent was obtained from all candidates, after which demographic data (age, weight, height, sex, race/ethnicity), medical, surgical, and medication history, and a baseline Oswestry Disability Index (ODI) score were obtained. A directed physical exam, and a pregnancy test in the case of females of child-bearing potential, were also performed. Consented eligible subjects received an electronic patient-reported outcome (ePRO) diary, along with training in its use and in the 0–10 numerical pain rating scale (NPRS). Subjects were instructed to record current pain intensity (PI) scores using the ePRO diary at 8.00 am and 8.00 pm (±2 hours) for 5 consecutive days (the run-in phase) beginning the day of the screening visit, in order to establish baseline PI.
Enrollment
Subjects who completed the run-in phase and who met the requirements for low back pain and/or leg pain (mean ≥4 NPRS) during this phase, as well as continued to meet all of the inclusion criteria and none of the exclusion criteria, were enrolled in the study. Enrolled study participants were dispensed one study device (Provant Therapy System), and instructed in proper device positioning and operation. Subjects were instructed to self-administer treatment with the device twice daily at 8.00 am and 8.00 pm (±2 hours) for 45 days.
Follow-up
Adverse events and concomitant medications were reviewed and recorded at all follow-up visits. A follow-up telephone call was performed 3 days following the enrollment visit in order to assess adherence to device treatment regimen and confirm consistent entry of PI scores into the ePRO diary. A posttherapy return clinic visit was conducted on or between Days 46–51, during which subjects completed the ODI questionnaire and returned the study device and the ePRO diary device. A further follow-up telephone call was performed at Day 60 (±3 days), at which time NPRS pain intensity was assessed and a health economics questionnaire was administered. NPRS pain intensity and interval history were reassessed during a follow-up telephone call 3 months after completion of treatment (Day 136±3 days).
Concomitant medications
Subjects continued their baseline analgesic medications from the time of the screening visit through and including the Day 60 follow-up visit. They were not allowed to introduce new analgesic therapies, but they could choose to reduce or discontinue opioid or other drug therapies as their pain diminished. Allowed medications included opioids, nonsteroidal anti-inflammatory agents, antidepressants, and muscle relaxants, taken either on an “as needed” basis or on a prescribed schedule. Use of transcutaneous electrical neurostimulators, implanted neurostimulators, physical therapy, local injections, intrathecal infusion, acupuncture, spinal manipulation, and revisional surgery were not allowed during the study.
Outcome measurements
PI
Subjects used a validated 11-point (0–10) NPRS to report “pain right now” (PI),Citation23 immediately before and immediately after each treatment session (0 for no pain and 10 for the worst pain imaginable) using an ePRO diary. Separate scores were recorded for low back PI and leg PI at each time point. Morning and evening scores were analyzed separately and then averaged to generate an overall percent reduction in PI.
A subject was considered a “responder” if he or she reported a decrease in pain scores (back and/or leg) of 30% or greater versus baseline at end of treatment (Day 45). Baseline PI values were calculated as the mean of all PI scores reported during the 5-day baseline phase for low back and leg (separately).
ODI
Level of physical function was assessed using the ODI, a self-assessment tool designed and validated for use in subjects with low back pain and/or radicular pain.Citation24 ODI scores were reported at the screening (baseline) and Day 46 follow-up visits. The ODI assesses disturbance to activities of daily living. There are ten sections that address specific aspects of physical function, each of which is analyzed on a 0–5 scale indicating relative level of disability. ODI scores (point total/50×100) range from 0%–100% disability, with scores of 41%–60% considered “severe disability” and 61%–80% considered “crippled”. A change in ODI of 6% is considered clinically meaningful.Citation24
Patient Global Impression of Change
Participants recorded self-rated change in overall status using a 7-point, validated, Patient Global Impression of Change (PGIC) scale.Citation25 Scores were self-reported on Day 2, Day 7, Day 14, Day 21, Day 28, Day 35, and Day 45 using the ePRO diary. Subjects were asked to rate their overall status compared to the start of the study as either: very much worse, much worse, minimally worse, no change, minimally improved, improved, or much improved. PGIC incorporates pain, other physical symptoms, sleep, function, and drug side effects. “Improved” and “much improved” are considered clinically meaningful results.Citation23
Analgesic consumption
Participants recorded analgesic consumption relative to baseline (consumption reported during screening visit) using a 3-point categorical scale (less, same, more). Analgesic consumption was recorded by each study subject using the ePRO diary on Day 2, Day 7, Day 14, Day 21, Day 28, Day 35, and Day 45.
Safety
Safety was assessed through a review of adverse event reports, concomitant treatments, and medication usage, which were assessed at the Day 46 follow-up visit and at the Day 60 and Day 136 telephone contacts.
Cost-avoidance survey
A health economic cost-avoidance survey was conducted during the Day 60 follow-up telephone call.
Statistical methods
All estimations were based on the intrasubject change from baseline. Demographic and subject characteristics are summarized using descriptive statistics. Pain measurements measured on a numerical scale are summarized descriptively using the mean and standard deviation. Variables that were measured on a categorical scale are summarized as a proportion of the population. Adverse events coded using Medical Dictionary for Regulatory Activities (MedDRA) version 16.0 are summarized by category and severity. Because this was an exploratory study, all efficacy and safety data are based on observed values; no imputation was used for missing data.
Results
Subject characteristics and baseline data
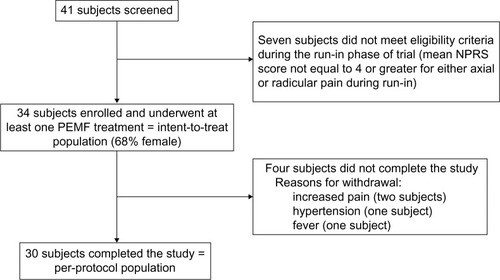
Between November 2013 and February 2014, a total of 41 subjects were consented and were provided with an ePRO diary in order to record daily pain scores during the run-in phase (). Seven subjects did not meet eligibility criteria following the run-in phase of the study. The remaining 34 subjects underwent at least one PEMF treatment (the intent-to-treat population). Thirty subjects completed the 45-day treatment protocol (the per-protocol population); the other four subjects withdrew and did not complete the protocol (). One per-protocol subject stopped treatment and ePRO entries prematurely at Day 38 due to an ePRO software error, but this subject complied with follow-up assessments and was included in the analyses. Of the 30 per-protocol subjects, 25 provided PEMF device-usage meter data, resulting in a mean and median compliance rate of 86.4% and 96.7%, respectively (total hours of treatment per subject: mean =38.9±9.5 hours, median =43.5 hours). The usage meter for five subjects malfunctioned and failed to record data.
Figure 1 Flow chart of study participants.
Baseline characteristics of the intent-to-treat and per-protocol populations were similar (). Per-protocol subjects were predominantly female (67%), mildly obese, and 16.9 months postsurgery on average, and reported mean baseline PI at moderate/severe levels on the 0–10 scale (back pain =6.0±1.6; leg pain =5.3±2.5). Most-recent surgeries included discectomy in 27% of per-protocol subjects (8/30), a decompression procedure (eg, laminectomy, foraminotomy) in 47% (14/30), spinal fusion with metal implantation in 20% (6/30), and a combination of decompression and discectomy in 7% (2/30). Twenty-seven percent (8/30) of per-protocol subjects had a history of two failed lumbar spine surgeries. Subjects had failed multiple nonsurgical postoperative therapeutic interventions for FBSS prior to participation in the study (per-protocol population: mean =1.3 interventions, median =1.0 intervention, range =0–5 interventions), primarily physical therapy (63.3%), epidural injection (23.3%), and transcutaneous electrical neurostimulators (20.0%); overall, 73% of the per-protocol population had one or more failed prior postoperative interventions.
Table 2 Subject baseline characteristics
Primary endpoint
The primary endpoint was decrease in PI at Day 45 versus baseline. In a post hoc analysis, decreases in pain scores of 30% or greater were considered clinically meaningful.Citation23 Subjects meeting this criterion for back PI and/or leg PI were considered “responders”. Subjects not meeting this criterion were considered “nonresponders”. All 30 per-protocol subjects complied and provided data at the first posttherapy follow-up. By this measure, 33% (10/30) of per-protocol subjects responded to PEMF therapy. When categorized by surgical history, a much higher proportion of subjects whose most-recent back surgery was discectomy were responders (60%, 6/10 subjects), compared to subjects who had undergone decompression (laminectomy/foraminotomy; 21%, 3/14) or fusion (17%, 1/6) without discectomy (). Further, 40% of subjects in the discectomy subpopulation (4/10) reported ≥50% improvement in back and/or leg pain scores. No difference was noted in the response rates of subjects who were on opioids at study entry versus those not on opioids, nor in subjects with predominant axial low-back pain versus predominant radicular leg pain at study entry. In terms of subject age, per-protocol subjects older than 50 years of age were more frequently responders (42%, 8/19), relative to the entire per-protocol population. The mean decreases in back and leg PI scores were 44%±28% and 55%±27%, respectively, for responders in the per-protocol group (). A similar mean decrease in back and leg PI was reported for per-protocol group responders in the discectomy subpopulation (44%±29% and 54%±30%, respectively) (). In terms of timing of response, most responders showed a substantial early response, and all responders demonstrated the response in PI within the first 3 weeks of treatment ().
Figure 2 Mean weekly numerical pain rating scores for responders.
Abbreviation: NPRS, numerical pain rating scale score.
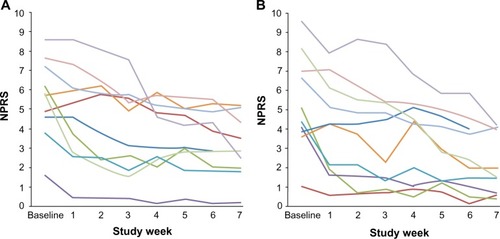
Table 3 Responder analysis for complete per-protocol population and procedure-based subpopulations
At Day 60 (2 weeks following the end of treatment), 70% (7/10) of responders still had ≥30% reduction in PI from baseline in back and/or leg PI. An additional five subjects who were nonresponders at Day 45 reached this success criterion by Day 60. Three months after the end of treatment (Day 136), 63% of per-protocol responders (5/8 reporting data) and 75% of discectomy responders (3/4 reporting data) still met the success criterion of ≥30% reduction in radicular PI. For axial PI, the proportion of responders that still met the success criterion was 38% (3/8) per-protocol and 25% (1/4) discectomy.
Secondary endpoints
Fifty percent (15/30) of subjects in the per-protocol group showed clinically meaningful improvement in physical functioning based on 6% or greater improvement in ODI scores at the Day 46 follow-up relative to baseline ().Citation24 In terms of magnitude of improvement, per-protocol responders reported an average improvement in ODI scores of 13.1% improvement (). Discectomy responders showed a slightly greater improvement in ODI (average 16.5%, ). Of subjects reporting PGIC scores at the end of treatment (Day 45), 17% (4/23) of subjects in the per-protocol group reported “improved” or “much improved” PGIC scores at the end of treatment (Day 45) relative to baseline (), including 67% (4/6) of responders (), and 100% (3/3) of discectomy responders (). Reduced analgesic consumption was reported by 22% (5/23) of per-protocol subjects who provided analgesic consumption data at end of treatment (Day 45) relative to baseline (), with 50% (3/6) of per-protocol responders reporting reduced analgesic consumption at the end of treatment (Day 45) (). Clinically meaningful outcomes for all four outcome measures were most frequently reported by subjects in the discectomy subpopulation ().
Table 4 Percentage of subjects with clinically meaningful outcomes (end of treatment versus baseline)
Safety
Six of 34 subjects in the intent-to-treat population (17.6%) experienced eleven treatment-emergent adverse events (). Of these six subjects, four experienced a single adverse event, one experienced two adverse events, and one experienced five adverse events. All of the adverse events were mild to moderate in severity, and none was attributed to the study device by the study Principal Investigator. There were no serious adverse events. Two subjects withdrew from the study during the treatment period due to worsening of their underlying condition.
Table 5 Treatment-emergent adverse events
Cost-avoidance survey
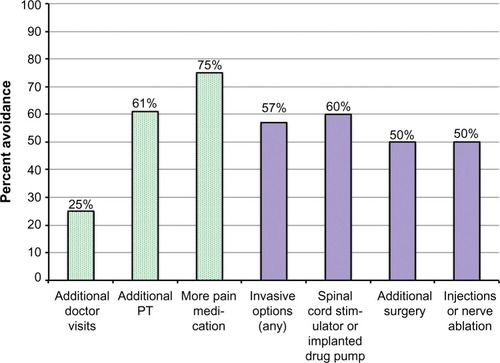
Two weeks following the end of the treatment period, subjects were questioned regarding the need for further therapy for FBSS. Of the 28 subjects who responded, 25% (7/28) felt they would need no further doctor visits, 61% (17/28) felt they would need no additional physical therapy, and 75% (21/28) felt they would need no additional analgesic medications during the 6 months following the survey (). Of those who had been discussing with their doctor the possibility of using further invasive interventions (spinal cord stimulator, implanted drug pump, injections, nerve ablations, repeat surgery), 57% (8/14) felt they could avoid such procedures.
Figure 3 Cost-avoidance survey results for per-protocol survey respondents (28/30).
Abbreviation: PT, physical therapy.
Discussion
In this exploratory clinical trial of 30 subjects with failed back surgery who were treated with PEMF therapy twice daily over a 45-day period, one-third of subjects completing the trial reported a clinically meaningful reduction in back and/or leg pain. For responders, pain intensity dropped by 44% (back) and 55% (leg) during the course of treatment, and the response to PEMF was noted early, within 3 weeks of starting therapy. Overall, at completion of treatment (Day 45), 57% (13/23) of per-protocol subjects considered themselves “improved” (any level of improvement), 22% (5/23) decreased analgesic consumption, and 50% (15/30) had a clinically meaningful improvement in physical function. In terms of surgical history, there was a higher proportion of responders in the discectomy subpopulation (60%), than there was in either the decompression (21%) or fusion (17%) subpopulations. A high proportion of subjects in the discectomy subpopulation also achieved results that were clinically meaningful across all four outcome measures.
It is interesting to consider why subjects who had undergone discectomy reported a better response to PEMF therapy than subjects who had undergone either fusion or decompression without discectomy. It is possible that poorer outcome in subjects in the latter two categories was due to greater underlying baseline disease burden – for example, due to increased stress on adjacent structures (eg, facet joints, discs) following fusion or due to introduction of instability following bone removal in the case of laminectomy. Such anatomical changes might also result in more scar tissue, which might further hinder the effects of the therapy.
In terms of the criterion used to define a “responder”, several reports in the literature cite ≥30% improvement in pain scores to be indicative of a clinically meaningful change. In a landmark study of 2,724 subjects from ten published placebo-controlled studies in a variety of chronic pain conditions, Farrar et alCitation23 demonstrated that a reduction of approximately 30% (or 2 points) on the NPRS is considered to be a clinically important difference. In clinical trials where the test treatment is considered to be highly effective, a continuous responder analysis comparing the efficacy levels of placebo and active drug groups provides a graphical representation of the proportion of subjects who achieve pain reductions over an entire range of cut-off values ranging from 0%–100%.Citation26 Efficacious treatments will show a parallel shift in the proportion of subjects reaching key endpoints (eg, 30% reduction, 50% reduction, 70% reduction in pain) that are considered clinically meaningful by patients and clinicians alike. A 2008 IMMPACT (Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials) consensus statement on interpreting data from chronic pain trials also found that ≥30% and ≥50% improvement in pain scores as reflective of at least moderate and substantial changes of clinical importance.Citation27 Contemporary reports on the effectiveness of novel analgesic therapies often report the proportion of subjects achieving 30% and 50% pain reductions in tabular format.Citation28
Several factors suggest that this was a challenging population to treat. Twenty-seven percent (8/30) of per-protocol study participants had a history two failed back surgeries. Seventy-three percent (22/30) of the study participants had at least one failed nonsurgical postoperative intervention prior to participation in the study, with a majority (77%, 23/30) on opioids at enrollment. Further, the baseline ODI (mean score =43.1) indicates that the average subject was “severely disabled” by their pain. Finally, the timing of the treatment was remote from surgery (mean 16.9 months postsurgery), with 10 of the 30 per-protocol subjects enrolled in the trial 24 months or longer after surgery, suggesting that this may be a potentially recalcitrant population with substantial centralized pain.
Limitations of the trial design included small sample size, lack of a control group, and use of subjective endpoints. Due to such limitations, which are common to small exploratory studies, it is not possible to rule out a possible placebo effect, nor to draw firm conclusions due to lack of statistical power. In addition, the use of subjective endpoints is subject to possible bias. The period used for baseline data collection may have been too brief to identify subjects who were slowly improving (or getting worse) prior to beginning therapy. Generalizability of the study is limited by the small sample size, and due to exclusion criteria, study results cannot be generalized to all types of subjects.
Caution should be used regarding interpretation of trial outcomes in light of these limitations. It is, nonetheless, encouraging that seriously disabled subjects with FBSS pain who previously did not respond to other interventions reported improvement in pain intensity that was rapid, substantial, and sustained, along with meaningful improvement in physical function and overall well-being, as well as reduced pain medication consumption in response to PEMF treatment.
Although PEMF therapy has been reported effective as an adjunctive therapy for postoperative pain and edema following other types of surgeries, to our knowledge this is the first study of this size evaluating its efficacy for persistent pain following back surgery. A 2014 reportCitation29 found PEMF therapy effective at reducing back pain in a population of subjects with persistent lumbar muscle pain, reporting a significant reduction in pain in subjects receiving PEMF therapy but not in sham-device–treated subjects. Although the study excluded subjects who had prior spinal surgery, as well as those with radicular pain, the improvement in back pain reported in the study is comparable to the results of the current study. In terms of underlying mechanism, recent data suggest that PEMF may mediate analgesia by promoting changes in gene expression, both for genes involved in opioid signaling as well as for modulators of inflammatory pain. For example, in in vitro and in vivo studies, PEMF treatment has been associated with a decrease in messenger RNA (mRNA) and protein levels of the proalgesic, proinflammatory cytokine interleukin-1β.Citation15,Citation17 In vitro, PEMF treatment has also been associated with increased mRNA levels of the anti-inflammatory cytokine interleukin-1015 and increased endogenous opioid precursor mRNAs for proenkephalin, pro-opiomelanocortin, prodynorphin, as well as opioid peptides.Citation15
Conclusion
Data from the current study suggest that the effect of PEMF in patients with failed back surgery syndrome may vary depending on type of surgery, with greatest benefit in patients who have undergone discectomy. In light of these clinically meaningful improvements, the use of PEMF therapy for the management of FBSS pain following discectomy merits further study in a large randomized controlled trial.
Acknowledgments
We would like to acknowledge Bruce Stouch, PhD and Gary Stevens, PhD for biostatistical support.
Disclosure
The study was funded by Regenesis Biomedical, Inc., Scottsdale, AZ, USA. WLH served as Principal Investigator for this study. He has otherwise had no financial relationship with the sponsor. NJK and WKS are paid consultants for Regenesis Biomedical, Inc. and were paid during manuscript preparation. RAI is an employee of Regenesis Biomedical, Inc. The authors report no other conflicts of interest in this work.
References
- ChanCWPengPFailed back surgery syndromePain Med201112457760621463472
- ChouRHuffmanLGuideline for the Evaluation and Management of Low Back PainEvidence ReviewGlenview, ILAmerican Pain Society2009
- ChouRLoeserJDOwensDKAmerican Pain Society Low Back Pain Guideline PanelInterventional therapies, surgery, and interdisciplinary rehabilitation for low back pain: an evidence-based clinical practice guideline from the American Pain SocietySpine (Phila Pa 1976)200934101066107719363457
- TaylorRSTaylorRJThe economic impact of failed back surgery syndromeBr J Pain201264174181
- HussainAErdekMInterventional pain management for failed back surgery syndromePain Pract2014141647823374545
- BenzonHTKatzJABenzonHAIqbalMSPiriformis syndrome: anatomic considerations, a new injection technique, and a review of the literatureAnesthesiology20039861442144812766656
- CohenSPSacroiliac joint pain: a comprehensive review of anatomy, diagnosis, and treatmentAnesth Analg200510151440145316244008
- RajaeeSSBaeHWKanimLEDelamarterRBSpinal fusion in the United States: analysis of trends from 1998 to 2008Spine (Phila Pa 1976)2012371677621311399
- KumarKBishopSFinancial impact of spinal cord stimulation on the healthcare budget: a comparative analysis of costs in Canada and the United StatesJ Neurosurg Spine200910656457319558289
- TurnerJAHollingworthWComstockBADeyoRASpinal Cord Stimulators (SCS) for Injured Workers with Chronic Back and Leg Pain after Lumbar Surgery – A Prospective Study to Describe Costs, Complications, and Patient Outcomes: Final ReportOlympia, WAWashington State Department of Labor and Industries2008 Available from: http://www.lni.wa.gov/ClaimsIns/Files/OMD/finalReportSCS.pdfAccessed October 23, 2014
- HaanpääMLGourlayGKKentJLTreatment considerations for patients with neuropathic pain and other medical comorbiditiesMayo Clin Proc201085Suppl 3S15S2520194144
- ManchikantiLBenyaminRDattaSVallejoRSmithHOpioids in chronic noncancer painExpert Rev Neurother201010577578920420496
- GuoLKubatNJIsenbergRAPulsed radio frequency energy (PRFE) use in human medical applicationsElectromagn Biol Med2011301214521554100
- GuoLKubatNJNelsonTRIsenbergRAMeta-analysis of clinical efficacy of pulsed radio frequency energy treatmentAnn Surg2012255345746722301609
- MoffettJFrayLMKubatNJActivation of endogenous opioid gene expression in human keratinocytes and fibroblasts by pulsed radiofrequency energy fieldsJ Pain Res2012534735723055776
- MoffettJGriffinNERitzMCGeorgeFRPulsed radio frequency energy field treatment of cells in culture results in increased expression of genes involved in the inflammation phase of lower extremity diabetic wound healingThe Journal of Diabetic Foot Complications2010235764
- RohdeCChiangAAdipojuOCasperDPillaAAEffects of pulsed electromagnetic fields on interleukin-1 beta and postoperative pain: a double-blind, placebo-controlled, pilot study in breast reduction patientsPlast Reconstr Surg201012561620162920527063
- AronofskyDHReduction of dental postsurgical symptoms using nonthermal pulsed high-peak-power electromagnetic energyOral Surg Oral Med Oral Pathol19713256886965315479
- CortesJKubatNJapourCPulsed radio frequency energy therapy use for pain relief following surgery for tendinopathy-associated chronic pain: two case reportsMil Med20131781e125e12923764157
- HedénPPillaAAEffects of pulsed electromagnetic fields on postoperative pain: a double-blind randomized pilot study in breast augmentation patientsAesthetic Plast Surg200832466066618506512
- KaplanEGWeinstockREClinical evaluation of diapulse as adjunctive therapy following foot surgeryJ Am Podiatry Assoc19685852182214870782
- RaweIMLowensteinABarceloCRGenecovDGControl of postoperative pain with a wearable continuously operating pulsed radiofrequency energy device: a preliminary studyAesthetic Plast Surg201236245846322037572
- FarrarJTYoungJPJrLaMoreauxLWerthJLPooleRMClinical importance of changes in chronic pain intensity measured on an 11-point numerical pain rating scalePain200194214915811690728
- FritzJMIrrgangJJA comparison of a modified oswestry low back pain disability questionnaire and the quebec back pain disability scalePhys Ther200181277678811175676
- KamperSJMaherCGMackayGGlobal rating of change scales: a review of strengths and weaknesses and considerations for designJ Man Manip Ther200917316317020046623
- FarrarJTDworkinRHMaxMBUse of the cumulative proportion of responders analysis graph to present pain data over a range of cut-off points: making clinical trial data more understandableJ Pain Symptom Manage200631436937716632085
- DworkinRHTurkDCWyrwichKWInterpreting the clinical importance of treatment outcomes in chronic pain clinical trials: IMMPACT recommendationsJ Pain20089210512118055266
- RiceASDworkinRHMcCarthyTDEMA401-003 study groupEMA401, an orally administered highly selective angiotensin II type 2 receptor antagonist, as a novel treatment for postherpetic neuralgia: a randomised, double-blind, placebo-controlled phase 2 clinical trialLancet201438399291637164724507377
- ParkWHSunSHLeeSGEffect of pulsed electromagnetic field treatment on alleviation of lumbar myalgia; a single center, randomized, double-blind, sham-controlled pilot trial studyJ Magnetics2014192161169