
Abstract
Neuroacanthocytosis is a group of rare disorders. We report a 36-year-old right-handed female who presented with gradually progressive abnormal facial movements, generalized weakness, and lower-lip biting starting 4 years ago. On examination, she had lower-lip ulcer, orofacial dyskinesias, and peripheral neuropathy. Her peripheral blood smears showed acanthocytosis and magnetic resonance imaging revealed atrophied head of caudate nuclei and putaminal hyperintensities on T2-weighted and fluid attenuated inversion recovery images. Work-up for autoimmune and metabolic causes was negative. She was diagnosed with chorea-acanthocytosis, an entity under neuroacanthocytosis syndrome and the patient was offered symptomatic treatment.
Video abstract
Point your SmartPhone at the code above. If you have a QR code reader the video abstract will appear. Or use:
Introduction
Neuroacanthocytosis (NA) is a group of very rare disorders estimated to have affected ~1,000 people worldwide. NA are complex, progressive, and incurable disorders and their treatment is symptomatic. The complex presentations that include a hyperkinetic movement disorder, a polyneuropathy, cognitive decline, and elevated creatine kinase (CK) raise the suspicion of a NA syndrome. To our knowledge, this is the first case report of NA from Nepal.
Case report
A 36-year-old right-handed female had presented with gradually worsening abnormal facial movements associated with intermittent rapid brief forceful eyes closure, perioral movements, and lower-lip biting. Her husband reported a history of difficulty in speaking, swallowing, and weakness of all four limbs, and she was wheelchair-bound. She was born to a nonconsanguineous parent and lacked a family history of neurological disease. She was not exposed to long-term medications known to cause extrapyramidal dysfunction.
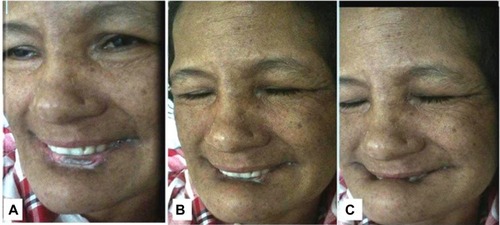
Examination revealed orofaciolingual dyskinesias mimicking suckling and grimacing, and lower-lip ulcer because of repeated biting (). Mini Mental Status Examination (MMSE) could not be performed because of unintelligible speech. She had wasting of all four limbs, diminished power (4/5) and reflexes. Her fundus and sensory examinations were normal. Kayser–Fleicher (KF) ring, cerebellar signs, and autonomic dysfunction were absent.
Figure 1 (A–C) Sequential snapshots from video demonstrating involuntary movements with closure of both eyes and lower-lip biting.
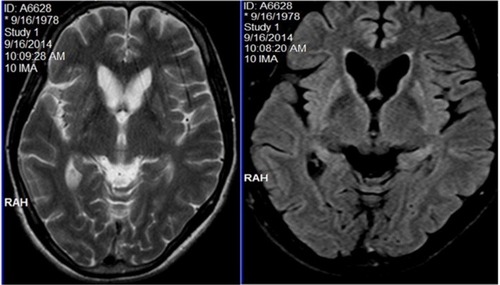
Peripheral smear examination performed following standard protocolCitation1 showed acanthocytes () and magnetic resonance imaging showed degenerative changes in basal ganglia (). Her CK was raised (528 U/L [Normal: up to 165 U/L]). Nerve conduction study showed peripheral mixed axonal neuropathy. Complete blood count, liver function test, random blood sugar, renal function test, antinuclear antibody, double stranded DNA, serum copper, serum ceruloplasmin, HIV, hepatitis B antigen, hepatitis C, cerebrospinal fluid analysis, USG abdomen, ECG, and echocardiography were normal.
Figure 2 Peripheral blood smear showing acanthocytes (red arrows).
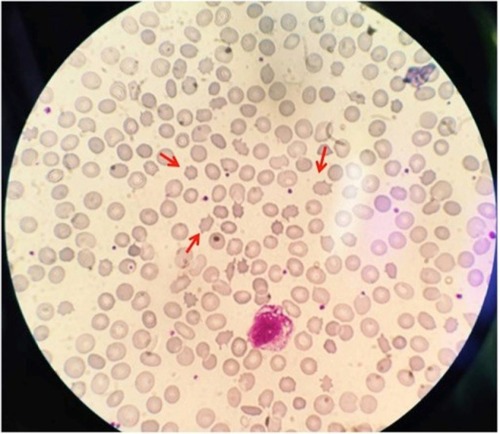
Figure 3 MRI showing caudate atrophy and striatal hyperintensities in both T2WI and FLAIR sequences.
She was treated with haloperidol and clonazepam. On follow-up at 3 months, her condition was static.
Ethics
Written informed consent was obtained from the patient to publish case details and pictures. Ethical clearance was also obtained from the National Institute of Neurological and Allied Sciences Ethical Review Board.
Discussion
NA syndrome disorders are all exceedingly rare, but also very likely to be underdiagnosed.Citation2 It is estimated that 500–1,000 people worldwide have chorea-acanthocytosis (ChAc), an entity under NA syndrome.Citation3 ChAc is more prevalent in Japan.Citation4 As literature search did not reveal any documentation of ChAc from Nepal, we believe that this is the first reported case of ChAc from Nepal.
ChAc is a progressive neurodegenerative autosomal recessive disorder. Its neurological symptoms usually manifest in the twenties.Citation5 NA was first described in 1960 as “Levine–Critchley syndrome”Citation6 and as mentioned earlier, ChAc is one of the entities under NA. Other entities include: McLeod syndrome (MLS), Huntington’s disease like 2 (HDL2), and pantothenate kinase–associated neurodegeneration.
Our diagnosis of ChAc was based on clinical ground and laboratory findings. Clinically, our patient had adult-onset illness, hence, our diagnosis was restricted to either ChAc or MLS because HDL2 and pantothenate kinase–associated neurodegeneration have a childhood or juvenile onset, and HDL2 is usually found in patients with African ancestry. MLS is an X-linked disorder that has predominant cardiovascular manifestations (our patient’s cardiovascular status was normal). Also, lip biting is exceedingly rare in MLS. This feature with female sex in our case rules out MLS.
ChAc consists of choreoathetoid movement disorder with orofacial dyskinesia and progressive cognitive decline. The movement problems worsen with age. In few cases, seizures may precede movement disorders by a decade.Citation7 The characteristic phenotype includes chorea, a very peculiar “feeding dystonia” with tongue protrusion,Citation8 orofacial dyskinesias, involuntary vocalizations, dysarthria, and involuntary tongue- and lip-biting. All these mentioned features were prominent manifestations of our case. The gait of ChAc patients may have a “rubber man” appearance with truncal instability and sudden, violent trunk spasms.Citation9 We could not examine gait because of weakness. Behavioral changes are a common feature and may be the first presenting feature.
Clinical neuromuscular manifestations include areflexia, sensorimotor neuropathy, and variable weakness and atrophy.Citation4 Most ChAc patients have elevated levels of CK.Citation4 Our patient had all of these findings known to be typical manifestation of ChAc.
ChAc is caused by various mutations of a 73 exon gene on chromosome 9, VPS13A, coding for chorein.Citation6,Citation10 Confirmatory DNA analysis of the large VPS13A gene is difficult, due to the large gene size and heterogeneity of mutation sites.Citation11–Citation13 However, absence of chorein in erythrocytes can be demonstrated on Western blot.Citation14 Chorein is implicated in intracellular protein sorting but its physiological functions are not yet known.Citation4 We did not test for chorein because of financial constraints although protein testing is available free of charge.Citation15
We diagnosed the case as ChAc based on history and also because acanthocytes were readily documented in peripheral smear with elevated serum CK level. The elevated CK level suggested presence of myopathy, an important diagnostic clue to NA syndrome, however, her relative declined electromyography test. Acanthocytosis may be absent in peripheral blood smears and a negative screen does not reliably exclude an NA syndrome.Citation16
Neurodegeneration affects predominantly the caudate nucleus, putamen, and globus pallidus. In ChAc, thalamus and substantia nigra are also involved.Citation4 In magnetic resonance imaging, we observed changes in all these mentioned regions. Computed tomography scan did not show calcification in the aforementioned regions.
So far no curative or disease-modifying treatments are available. Recognition of treatable complications such as seizures, and swallowing problems are essential. Neuropsychiatric issues are more amenable to pharmacotherapy. Dopamine antagonists or depleters such as clozapine or tetrabenazine may ameliorate the movement disorders. Results of deep brain stimulation (DBS) in ChAc are mixed.Citation17–Citation19 NA disorders have a relentlessly progressive course and are eventually fatal. Sudden death may be attributable to seizure or autonomic dysfunction. Early identification of the case may avoid the need for invasive and nondiagnostic tests such as muscle, bone marrow, or liver biopsy and help guide the proper management and counseling.
Conclusion
This rare disorder should be considered in the differential diagnosis of atypical cases of epilepsy, peripheral neuropathy, and behavioral disorder associated with movement disorder.
Acknowledgments
The authors would like to thank Prof Madhu Dixit Devkota, Dr Samarth Singh, and all the Magister Chirurgiae Registrars of National Institute of Neurological and Allied Sciences.
Disclosure
The authors report no conflicts of interest in this work.
References
- StorchAKornhassMSchwarzJTesting for acanthocytosis A prospective reader-blinded study in movement disorder patientsJ Neurol2005252849015654559
- JungHHDanekAWalkerRHNeuroacanthocytosis syndromesOrphanet J Rare Dis201166822027213
- Chorea-acanthocytosisGenetic home reference Available from: http://ghr.nlm.nih.gov/condition/chorea-acanthocytosisAccessed May 30, 2015
- UenoSMarukiYNakamuraMThe gene encoding a newly discovered protein, chorein, is mutated in chorea-acanthocytosisNat Genet200128212112211381254
- WalkerRHJungHHDobson-StoneCNeurologic phenotypes associated with acanthocytosisNeurology200768929817210889
- DanekANeuroacanthocytosis SyndromesDordrechtSpringer Verlag200557
- Al-AsmiAJansenACBadhwarAFamilial temporal lobe epilepsy as a presenting feature of choreoacanthocytosisEpilepsia20054681256126316060937
- BaderBWalkerRHVogelMTongue protrusion and feeding dystonia: a hallmark of chorea-acanthocytosisMov Disord201025112712919938148
- SchneiderSALangAEMoroEBaderBDanekABhatiaKPCharacteristic head drops and axial extension in advanced chorea-acanthocytosisMov Disord201025101487149120544815
- RampoldiLDobson-StoneCRubioJPA conserved sorting-associated protein is mutant in chorea-acanthocytosisNat Genet200128211912011381253
- AllenFHKrabbeSMRCorcoranPAA new phenotype (McLeod) in the Kell blood-group systemVox Sang1961655556013860532
- MarshWLTaswellHFØyenRNicholsMEVergeraMSPinedaAAKx antigen of the Kell system and its relationship to chronic granu-lomatous disease. Evidence the Kx gene is X-linkedTransfusion197515527
- MarshWLMarshNJMooreAElevated serum creatine phosphokinase in subjects with McLeod syndromeVox Sang19814064034117197431
- Dobson-StoneCVelayos-BaezaAFilipponeLAChorein detection for the diagnosis of chorea-acanthocytosisAnn Neurol200456229930215293285
- EHDN-Neuroacanthocytosis submodule Available from: https://www.euro-hd.net/edit/na/network/docs/na-blood-sampling-instructions.pdfAccessed August 23, 2015
- SorrentinoGDe RenzoAMinielloSNoriOBonavitaVLate appearance of acanthocytes during the course of chorea-acanthocytosisJ Neurol Sci1999163217517810371080
- WihlGVolkmannJAllertNLehrkeRESturmVFreundHJDeep brain stimulation of the internal pallidum did not improve chorea in a patient with neuro-acanthocytosisMov Disord200116357257511391763
- BurbaudPRougierAFerrerXImprovement of severe trunk spasms by bilateral high-frequency stimulation of the motor thalamus in a patient with chorea-acanthocytosisMov Disord200217120420711835468
- BurbaudPVitalARougierAMinimal tissue damage after stimulation of the motor thalamus in a case of chorea-acanthocytosisNeurology200259121982198412499498