
Abstract
Rheumatoid arthritis (RA) is a progressive autoimmune disease that is characterized by inflammation of the synovial joints leading to cartilage and bone damage. The pathogenesis is sustained by the production of pro-inflammatory cytokines including tumor necrosis factor (TNF), interleukin (IL)-1 and IL-6, which can be targeted therapeutically to alleviate disease severity. Several innate immune receptors are suggested to contribute to the chronic inflammation in RA, through the production of pro-inflammatory factors in response to endogenous danger signals. Much research has focused on toll-like receptors and more recently the nucleotide-binding domain and leucine-rich repeat pyrin containing protein-3 (NLRP3) inflammasome, which is required for the processing and release of IL-1β. This review summarizes the current understanding of the potential involvement of these receptors in the initiation and maintenance of inflammation and tissue damage in RA and experimental arthritis models.
Rheumatoid Arthritis
Rheumatoid arthritis (RA) affects 0.5–1% of the population. It is a systemic disease, characterized by an erosive symmetrical polyarthritis, where widespread synovial inflammation affects both large and small peripheral joints. In addition to joint destruction, the accompanying systemic inflammation can lead to comorbidities including pulmonary inflammation, vasculitis and an increased risk of cardiovascular disease.Citation1 RA is regarded as a classic polygenic autoimmune disease, primarily on the basis that 70–80% of patients have autoantibodies such as rheumatoid factor (RF) and anti-citrullinated protein antibodies (ACPA). However, not all patients express these autoantibodies, and they are neither necessary nor sufficient to cause disease, but are predictive of a more aggressive disease course with greater joint erosion.Citation2 The heritability of RA is estimated to be ~50% in ACPA positive patients, while seronegative RA is much lower at ~20%.Citation3 However, the disease concordance in identical twins is around 12–15%, suggesting a role for environmental factors. To date, over 100 genetic loci have been associated with RA, though the exact relationship of many of these loci to the disease remains to be determined.Citation4
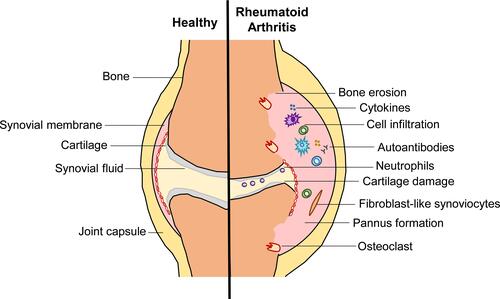
Within the RA joint, peripheral blood mononuclear cells infiltrate the synovial fluid and the synovial membrane, alongside expansion of tissue resident fibroblast-like synoviocytes (FLS) leading to the formation of a pannus (). These cells are highly activated releasing pro-inflammatory factors, such as Interleukin (IL)-1, IL-6, IL-17, tumor necrosis factor (TNF), vascular endothelial growth factor (VEGF) and matrix metalloproteases (MMPs).Citation1 Furthermore, neutrophils that accumulate in the synovial fluid undergo NETosis releasing citrullinated proteins that can be recognized by ACPA.Citation5 This sustained inflammatory environment, leads to the recruitment of further cells into the joint space, whilst FLS invade the cartilage matrix alongside activated osteoclasts, degrading the surrounding cartilage and bone.Citation1
Figure 1 Pathological changes in a rheumatoid arthritis joint. In established RA, the inflamed synovial membrane forms a pannus, due to infiltration of peripheral blood cells and proliferation of fibroblast-like synoviocytes. These cells are highly activated releasing pro-inflammatory mediators and autoantibodies within the joint sustaining the inflammatory process. This is accompanied by cartilage damage and osteoclast-mediated bone erosion leading to invasion of the pannus tissue and irreversible deformation of the joint.
In clinical practice, the most widely used and effective therapies are designed to dampen down inflammatory processes. Historically, non-steroidal anti-inflammatory drugs and corticosteroids were used. However, for the last 20 years, the mainstay of therapy in RA have been biological therapies targeting pro-inflammatory cytokines or their receptors, eg anti-TNF antibodies or IL-6 receptor antibodies. Although modestly effective, those that target IL-1 are not frequently used due to the superior performance of the other biologicals.Citation6 Anti-cytokine activity can also be mediated by a number of oral Janus Kinases (JAK) inhibitors that have recently been approved for the treatment of RA.Citation7 However, all of these anti-cytokine therapies target inflammation in RA downstream in the inflammatory process, none are effective in all patients, many lose their efficacy with time and all have significant side effects. Thus, there is great interest in exploring upstream inflammatory mechanisms, with a view to the identification of new therapeutic targets. Over the past two decades there has been a considerable focus on understanding the contribution of toll-like receptors (TLRs) and more recently the nucleotide-binding domain and leucine-rich repeat pyrin containing protein-3 (NLRP3) inflammasome in sustaining inflammation and joint destruction in RA.
Toll-Like Receptors
TLRs are a family of innate pattern recognition receptors that induce pro-inflammatory cytokines in response to both microbial-associated molecular patterns (MAMPs) and endogenous danger signals termed damage-associated molecular patterns (DAMPs). In humans, there are 10 TLRs that are differentially expressed on both immune and non-immune cells. TLRs 1, 2, 4, 5 and 6 are predominantly expressed at the plasma membrane, whereas TLRs 3, 7, 8 and 9 are localized to the endosome.Citation8 TLR10 is the least characterized member of the family and has been suggested to function both at the cell surface and within the endosomal compartment.Citation9
TLRs are type 1 integral membrane receptors that share a common structure, consisting of an ectodomain of leucine-rich repeats (LRR) where they engage their ligands and a cytoplasmic toll-interleukin-1 receptor homology (TIR) domain, also shared by the IL-1 receptor, from where they initiate signaling. Upon activation, TLRs form homo or heterodimers bringing their TIR domains into close proximity, permitting the recruitment of the TLR adaptor proteins, myeloid differentiation response protein 88 (MyD88), MyD88 adaptor-like (MAL), TIR-domain-containing adapter-inducing interferon-β (TRIF) and TRIF-related adaptor molecule (TRAM). Generally, MyD88 is engaged by all TLRs except TLR3, MAL by TLR2 and TLR4, TRIF by TLR3 and TLR4 and TRAM by TLR4.Citation10 However, TLR adaptor proteins have been shown to signal from TLRs outside this general consensus in a cell type dependent manner. For example, in murine bone marrow-derived macrophages (BMDM), TLR7 and TLR9 require MAL with TLR7 also suggested to use TRAM.Citation11,Citation12 Furthermore, TRAM may also function as an adaptor protein for TLR2 in primary human FLS, human umbilical vein endothelial cells and murine embryonic fibroblasts.Citation13
Dependent on the adaptor proteins recruited, various signaling pathways are engaged that culminate in the activation of transcription factors that include nuclear factor-κB (NF-kB), activator protein-1 (AP-1) and interferon regulatory factors (IRFs) to induce pro-inflammatory cytokines such as TNF, IL-1β, IL-6 and type I interferon.Citation8 Following TLR activation, IL-1β is translated as a biologically inactive 31kDa precursor requiring proteolytic cleavage by caspase-1 to an active mature 17kDa molecule before being released from cells. This process requires the formation of the inflammasome, a cytosolic multi-protein complex to first activate pro-caspase-1.Citation14,Citation15
The NLRP3 Inflammasome
Several types of inflammasome have been identified, of which the nucleotide-binding domain and leucine-rich repeat pyrin containing protein-3 (NLRP3) inflammasome is the most extensively studied. It consists of a LRR domain at the C-terminus considered to be the sensing domain, a central nucleotide-binding domain (NBD or NACHT domain) and a pyrin domain (PYD) at the N-terminus.Citation16 Upon activation, NLRP3 oligomerizes with the adaptor protein apoptosis-associated speck-like protein containing CARD (a caspase activation and recruitment domain) (ASC), which then recruits and activates pro-caspase-1 to form the inflammasome ().Citation17 Once activated, caspase-1 cleaves pro-IL-1β and gasdermin D (GSDMD) into two fragments. The GSDMD-N terminus fragments then oligomerize forming pores in the cell membrane which facilitate IL-1β release. This also initiates pyroptosis, an inflammatory form of cell death characterized by cell swelling and rupture, leading to the release of the cytoplasmic contents.Citation18,Citation19
Figure 2 Two signal model for classical NLRP3 inflammasome activation by TLR4. During the priming stage, activation of TLR4 by MAMPs or DAMPs upregulates NLRP3 and pro-IL-1β expression through NF-κB activation. This is closely followed by activation and assembly of the NLRP3 inflammasome, which can be induced by various stimuli including K+ efflux, Ca2+ signaling, mitochondrial dysfunction, and lysosomal rupture. Upon activation, caspase-1 cleaves pro-IL-1β and GSDMD resulting in pyroptosis and IL-1β release. Created with BioRender.com.
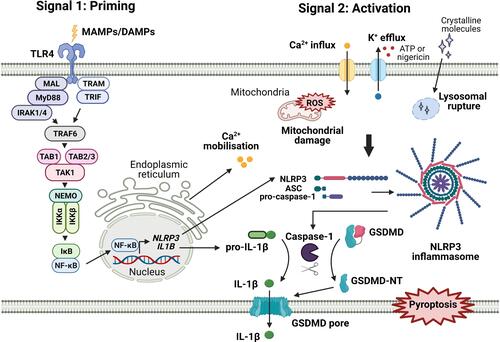
To produce mature IL-1β, most cells require two distinct signals. A priming step is required to to activate NF-κB to initiate the transcription of pro-IL-1β and NLRP3, which is expressed at low levels under resting conditions.Citation20 A second signal is then needed to stimulate the assembly of the NLRP3 inflammasome to enable the processing and release of IL-1β (). Priming has been demonstrated following activation of several different receptors. Ligands that activate TLR2, TLR3, TLR4, TLR7, TLR8 and TLR9 all induce IL-1β release.Citation21,Citation22 Upregulation of NLRP3 expression has been observed in murine macrophages following activation of TLR2, 3 and 4.Citation23 In addition, activation of TLR2, TLR3, TLR4, TLR7, NOD2 or stimulation with TNF, IL-1α or IL-1β results in the cleavage of caspase-1 in the presence of ATP, where ATP alone is not sufficient; further demonstrating the requirement for priming.Citation20,Citation24 NLRP3 is also activated by a diverse range of stimuli, suggesting a role as a sensor of cellular stress. These stimuli include reactive oxygen species, mitochondrial dysfunction, ion fluxes due to K+ or Cl− efflux, Na+ influx and Ca2+ mobilization and lysosomal damage due to uptake of crystalline molecules, such as monosodium urate and cholesterol crystals.Citation25 In contrast, primary human monocytes engage an alternative pathway; TLR4 can induce IL-1β release in the absence of a separate NLRP3 activation signal or the induction of pyroptosis.Citation26
As a potent inducer of inflammation and cell death, NLRP3 activity needs to be tightly regulated. In addition to a low expression in resting cells, further regulation can be achieved through post-translational modifications including phosphorylation, ubiquitination, nitrosylation and sumoylation.Citation27–Citation30 Furthermore, several proteins are suggested to interact with NLRP3 to regulate inflammasome assembly.Citation25 Most recently, a member of the TIR domain protein family, sterile alpha and TIR motif containing 1 (SARM1) was demonstrated to regulate NLRP3 through its TIR domain, inhibiting ASC oligomerization and caspase-1 activation.Citation31
Together, the ability of TLRs and NLRP3 to respond to DAMPs and stimuli induced by cellular stress makes them key candidates for sustaining inflammation in sterile inflammatory diseases such as RA. Moreover, induction of pyroptosis following NLRP3 activation would potentially release further DAMPs with the potential to activate TLRs sustaining a chronic cycle of inflammation.
TLRs in RA
In early studies of TLRs in RA, their potential involvement in RA pathophysiology became evident from studies of arthritis models using TLR deficient mice. In addition, we also demonstrated a role for the TLR adaptor proteins MyD88 and MAL in spontaneous production of cytokines and MMPs from human RA synovial membrane cultures.Citation32 Since then, a wealth of research studies has demonstrated the upregulation of potential endogenous TLR ligands within the serum and synovial joints of RA patients (), with all members of the TLR family having been associated with RA in some way (). However, for several TLRs, it is yet to be determined whether changes in expression or function are a cause or a consequence of inflammation in RA.
Table 1 Endogenous Toll-Like Receptor Ligands Associated with Rheumatoid Arthritis
Table 2 Summary of Some of the Main Associations of TLRs with RA Pathogenesis in Human and Animal Disease Models
TLR2
To signal, TLR2 forms a heterodimer with TLR1 or TLR6 and possibly TLR10, which will be discussed later. TLR2 is highly expressed in RA blood and synovial fluid monocytes and synovial lining macrophages.Citation33,Citation34 Compared with osteoarthritis (OA), TLR2 is also elevated at the mRNA level in RA synovial tissue, with the highest expression associated with patients that do not respond to anti-TNF treatment.Citation35 Correspondingly, TLR1, 2 and 6 mRNA levels in whole blood are reduced in patients that respond to anti-TNF therapy.Citation35 However, within the synovial tissue TLR6 mRNA is not upregulated and TLR1 expression is mainly increased in seropositive RA.Citation35,Citation36 In addition to increased expression of TLR2, several endogenous TLR2 ligands are also present within RA serum and synovial tissue indicating the potential for TLR2 activation in RA pathogenesis (). Indeed, High-Mobility-Group-Protein B1 (HMGB-1) can stimulate TLR2 on RA monocytes to induce IL-23, IL-6 and IL-17 promoting the differentiation of Th17 cells.Citation37,Citation38 Also, extracellular heat shock protein 96 within the RA synovium correlates with inflammation and synovial lining thickness.Citation39
Additionally, RA patient monocytes produce higher levels of cytokines compared to healthy donors upon activation of TLR1/2 and TLR2/6.Citation33,Citation40,Citation41 However, despite increased TLR1 in seropositive RA, we found no association of RF or ACPA status with the level of TLR1/2 cytokine production; although TLR1/2 induced IL-6 did correlate with DAS28.Citation36,Citation40 TLR2 activation of RA FLS also strongly induces Receptor activator of nuclear factor kappa-Β ligand (RANKL) promoting osteoclastogenesis and TLR2 activated M2 macrophages derived from RA patient monocytes exhibit an impaired anti-inflammatory activity.Citation42,Citation43 In addition to cytokine production, TLR2 has also been demonstrated to promote cell invasion and migration in RA synovial explants.Citation44 Indeed, inhibition of TLR2 in RA synovial explants with the anti-TLR2 antibody OPN301 led to a decrease in spontaneous cytokines, MMPs and FLS migration in response to explant conditioned media.Citation45
Similar to RA, experimental arthritis models also report increased TLR2 expression, which is decreased in studies where anti-inflammatory agents are used to ameliorate disease.Citation35,Citation46–Citation48 However, variable results have been described for TLR2 in the pathogenesis. In the IL-1Ra−/- spontaneous arthritis model, mice develop a more severe disease in the absence of TLR2 due to a modulation of T cell balance from T helper (Th)2 and regulatory T cells (Tregs) towards Th1 cells, suggesting a protective role for TLR2.Citation49 However, TLR2 has been shown to be important in the development of arthritis induced by intra-articular injection of streptococcal cell wall fragments, with TLR2−/- mice having a reduced disease severity.Citation50 Furthermore, in the murine collagen induced arthritis (CIA) model, TLR2 becomes elevated in blood samples during the pre-onset stage and then falls during early arthritis, suggesting a possible role in disease induction.Citation35
TLR3
TLR3 is also highly expressed in the RA synovium in both early and established disease, where it is potentially activated by dsRNA released from cells within the joint.Citation51,Citation52 In culture, RA FLS release RNA under hypoxic conditions, as would be present in the pannus and correspondingly extracellular RNA has been detected within the RA synovial lining layer of patient samples.Citation53 Additionally, RNA released from necrotic synovial fluid cells has been shown to stimulate TLR3 on FLS in culture and significantly increased levels of dsRNA are present in the synovial fluid of RA patient with an erosive disease course.Citation54,Citation55 Upon activation of TLR3, FLS induce IL-6, MMPs, B cell activating factor (BAFF) and VEGF that support inflammation, cartilage damage, angiogenesis, B cell activation and can enhance Th1 and Th17 cell expansion.Citation56,Citation57 In addition, TLR3 activation of monocytes induces osteoclast differentiation, which is further enhanced by TLR3 induced RANKL released from FLS.Citation58
Elevated TLR3 expression has also been observed in the CIA model and in rat pristane induced arthritis (PIA), where treatment with methotrexate to suppress disease also prevented TLR3 induction.Citation59 Likewise, suppression of TLR3 with the microRNA mimic miRNA-26a ameliorates disease in the PIA model.Citation60 This increase in TLR3 expression may also be connected with T cell activation, as co-culture of pristane primed T cells or their conditioned media upregulated TLR3 on FLS.Citation61 This effect may in part be associated with IL-17, a pathogenic cytokine released by Th17 cells, which increases TLR3 expression in FLS in culture.Citation62
TLR4
Increased TLR4 is evident in synovial fluid cells of patients with early and longstanding RA, as well as in RA peripheral blood monocytes and CD8+ T cells.Citation35,Citation51,Citation63–Citation66 Furthermore, RA synovial fluid macrophages have an increased cytokine response upon TLR4 stimulation.Citation65 In particular, seropositive RA patients are reported to have higher levels of synovial TLR4, which positively correlates with synovitis.Citation36 Within the synovium, TLR4 may be upregulated in RA FLS due to overexpressed histone methyltransferase mixed-lineage leukemia 1, which in turn upregulates TLR4 expression.Citation67 However, miRNA regulation may also be important, for example, RA FLS have reduced expression of miR-506 which is suggested to limit TLR4 expression.Citation68
Elevated TLR4 alongside the presence of a considerable number of TLR4 DAMPs such as HMGB-1 and ACPA immune complexes containing citrullinated fibrinogen within the serum and synovial fluid of RA patients, suggests TLR4 may play an active role in RA ().Citation37,Citation69 Indeed, in murine CIA, disease development leads to the upregulation of multiple endogenous TLR4 ligands, which are associated with CIA pathogenesis and promote osteoclast differentiation.Citation70 Furthermore, in CIA and the K/BxN serum transfer model, TLR4 deficient mice are protected from joint destruction with reduced cell infiltration.Citation71,Citation72 Additionally, the naturally occurring LPS from Bartonella Quintana that antagonizes TLR4, can also therapeutically suppress disease severity in both CIA and the spontaneous IL-1Ra−/- model.Citation73 Similarly, the TLR4 antagonist TAK-242 can suppress the expression of inflammatory cytokines from FLS and reduce local joint inflammation and bone damage in a complete Freund’s adjuvant (CFA)-induced arthritis (AIA) rat model.Citation74 However, despite these encouraging results from experimental models, inhibition of TLR4 with a monoclonal antibody NI-0101 in RA patients produced no benefit in a recent clinical trial.Citation75
TLR5
Similar to TLR2 and TLR4, TLR5 recognizes HMGB-1 as its endogenous ligand ().Citation76 TLR5 was initially associated with RA pathogenesis due to increased expression in RA synovial tissue lining and sublining macrophages and endothelial cells.Citation77 The expression of TLR5 on peripheral blood monocytes has since been correlated with DAS28, where this elevated expression is reduced in patients receiving anti-TNF treatment, suggesting a possible regulatory effect of TNF.Citation77,Citation78 Further influence on expression may also come from miRNAs, as miR-3926 which limits TLR5 expression is down-regulated in FLS where TLR5 is accordingly upregulated.Citation79
Functionally, a connection between TLR5 and RA pathogenesis may arise through its ability to promote angiogenesis and osteoclastogenesis. RA synovial fluid can induce endothelial cell migration and tube formation and also monocyte chemotaxis in a TLR5 dependent manner.Citation78,Citation80 Activation of TLR5 on RA peripheral blood mononuclear cells (PBMCs) can also synergise with TNF to facilitate osteoclast precursor cell differentiation.Citation78 Furthermore, the activation of TLR5 on RA monocytes with flagellin can also induce higher levels of IL-6 and IL-10 than healthy donor monocytes irrespective of RF or ACPA status; however, this did not correlate with DAS28.Citation40
TLR7 and TLR8
TLR7 and TLR8 both recognise ssRNA and are expressed at higher levels in RA synovial tissue lining and sublining macrophages, synovial fluid macrophages and peripheral blood monocytes.Citation36,Citation81 In particular, TLR8 expression is notably raised within the synovial tissue of seropositive RA patients.Citation36 However, when comparing mRNA levels within RA synovial tissue with OA samples, a strong trend towards increased TLR8 was observed but no difference was detected for TLR7.Citation35 Interestingly, RA patients carrying the M1V variant of TLR8 that induces lower cytokine levels upon TLR8 stimulation of monocytes exhibit a reduced disease severity.Citation82 In agreement with this, we have demonstrated that inhibition of endosomal TLRs and in particular inhibitors that target TLR8 can suppress spontaneous cytokine production from human RA synovial membrane cultures.Citation83–Citation85 However, it is expression of TLR7 but not TLR8 in RA monocytes that is reported to be associated with DAS28 and TNF. Additionally, this study demonstrated that RNA present in RA synovial fluid could stimulate RA monocytes to produce TNF.Citation81 Although RNA released from cells is quite unstable, LL-37 which is upregulated within the RA synovium can protect it from degradation to enable activation of TLR7 and TLR8.Citation86 Furthermore, FLS from ACPA+ patients release extracellular vesicles containing miR-574-5p, which activates TLR7 and TLR8 to induce osteoclastogenesis.Citation87 Similarly, miR-let-7b can activate TLR7 on monocytes to induce TNF and IL-6 and promote differentiation to M1 macrophages when released in extracellular vesicles by synovial fluid macrophages.Citation88
Experimental arthritis models have also indicated a pathogenic role for TLR7 and TLR8. We have demonstrated that inhibitors of the endosomal TLRs therapeutically suppress disease in the murine CIA model.Citation85,Citation89,Citation90 Moreover, mice deficient of TLR7, exhibit reduced disease severity in the CIA model following disease onset. This was associated with decreased IL-17 and elevated levels of Tregs, suggesting a role for TLR7 in regulating T cell responses.Citation89 In agreement with this data, intra-articular knockdown of TLR7 also improves disease activity in the rat CIA model.Citation91 In contrast, the investigation of TLR8 has proven more complicated, as unlike in human cells, murine TLR8 does not respond to stimulation with ssRNA.Citation92 However, transgenic mice expressing human TLR8 have been generated and found to be more susceptible to CIA with TLR8 expression correlated with pro-inflammatory cytokines within the joints.Citation93
TLR9
As a receptor for unmethylated CpG motifs within DNA, TLR9 also has the potential to be activated in RA, where patients have elevated levels of circulating immune complexes containing cell-free DNA compared to healthy controls.Citation94 In addition, TLR9 is upregulated in FLS, B-cells, monocytes and neutrophils of RA patients.Citation33,Citation56,Citation95 However, there are few studies of TLR9 in RA pathology, instead, most data have been generated in experimental arthritis models. Although an anti-inflammatory effect has been reported in the CIA model, where addition of apoptotic cells reduced the arthritis score in a DNA and TLR9 dependent manner, most studies indicate a pro-inflammatory role for TLR9.Citation96 Indeed, intra-articular injection of bacterial DNA containing CpG motifs in C57BL/6 mice induces arthritis.Citation97 Furthermore, co-activation of TLR9 and the B cell receptor with DNA containing immune complexes can stimulate RF autoreactive B cells.Citation98 In more recent studies, TLR9 was suggested to participate in the T cell-dependent phase of inflammatory arthritis models. In the rat PIA model, inhibition of TLR9 before the onset of disease reduced the severity of disease, serum IL-6, osteoclast formation and cartilage degradation, whereas therapeutic inhibition had no effect. In addition, TLR9−/- mice demonstrated a reduction in the T cell-dependent phase of streptococcal cell wall-induced arthritis. Whereas TLR9 deficiency had no effect on the T cell-independent K/BxN serum transfer model.Citation99
TLR10
Currently, TLR10 is the least understood of the human TLRs. It has been suggested to form homodimers or heterodimers with TLR1, 2 or 6, permitting engagement with a diverse range of ligands including dsRNA and the TLR1/2 ligand Pam3Cys. Furthermore, depending on the type of dimer formed, TLR10 is suggested to be able to produce a pro-inflammatory or an inhibitory effect.Citation100 Similarly, mixed results have emerged for the role of TLR10 in RA. In line with TLR10 having an anti-inflammatory role, TLR10 mRNA is expressed at lower levels in PBMCs of RA patients with active disease, whilst a missense mutation (I473T) has been associated with increased disease severity and a lower response to the anti-TNF biological infliximab.Citation101,Citation102 However, TLR10 is conversely upregulated in RA natural killer cells compared to healthy controls and in B cell subsets where a correlation with disease activity was observed.Citation103,Citation104 Thus, the function of TLR10 in RA may be complex and cell type dependent.
The NLRP3-Mediated Immune Response in Rheumatoid Arthritis
In addition to the induction of proinflammatory cytokines by TLRs in RA, the NLRP3 inflammasome is likely to have a key role in the processing and release of IL-1β. This was first demonstrated in the CIA model, where NLRP3 expression is increased within synovial tissue and correlates with disease severity.Citation105 Furthermore, when treated with the NLRP3 inhibitor MCC950, CIA mice exhibit a reduction in disease severity, synovial inflammation and cartilage erosion.Citation106 NLRP3 is also elevated in RA synovial tissue, as well as whole blood and CD4 T cells from RA patients with active disease.Citation106–Citation108 In addition, active caspase-1 in CD4 T cells correlates with DAS28 and IL-17A in patient sera.Citation108
The activation of NLRP3 in RA could be triggered by several different pathways. As discussed previously, TLR activation by DAMPs can induce NLRP3 and pro-IL-1β expression. However, several DAMPs are suggested to additionally activate NLRP3 assembly. Extracellular heat shock protein 96 which is elevated in RA has a dual action activating TLR2 and NLRP3 in murine macrophages where 2 signals are required for IL-1β release.Citation39,Citation109 Also, ACPA that can activate TLR4 when complexed with citrullinated fibrinogen can also indirectly stimulate NLRP3 induced IL-1β release in macrophages, due to activation of pannexin channels releasing ATP which then activates P2X7 receptors resulting in K+ efflux.Citation69,Citation110 Accordingly, higher levels of IL-1β are detected in the synovial tissue of ACPA+ compared to ACPA- patients and OA patients.Citation110 In addition, the uptake of colloidal calciprotein particles by RA monocytes at sites of bone erosion has been suggested to activate the NLRP3 inflammasome.Citation111
Further enhancement of NLRP3 activity may arise in RA due to dysregulation of molecular regulators of inflammatory signaling. Mice deficient in the RA susceptibility gene A20, also known as tumour necrosis factor-α inducible protein 3 (TNFAIP3), develop a spontaneous erosive arthritis associated with enhanced NLRP3 expression and IL-1β secretion, similar to that observed in RA patients. Moreover, A20 deficient murine BMDM demonstrates hyperactivation of NLRP3 inflammasome, IL-1β release and pyroptosis, suggesting a negative regulatory role for A20 on NLRP3.Citation112 In addition, the PTPN22 R620W gain-of-function variant associated with RA susceptibility, has also been shown to regulate NLRP3 dephosphorylation and subsequent activation.Citation113
Interestingly, the vitamin D receptor has also been suggested to negatively regulate NLRP3 inflammasome assembly through suppressing BRCC3-mediated deubiquitination of NLRP3, which corresponds with the finding that RA patients frequently have low Vitamin D levels that correlate with disease activity.Citation114,Citation115 More recently, we demonstrated in RA monocytes that a reduced expression of SARM, which negatively regulates NLRP3, was associated with elevated TLR1/2-induced IL-1β and DAS28. Furthermore, RA patients responsive to anti-TNF therapy then displayed a transient increase in the expression of SARM in their monocytes, which was not observed in non-responders.Citation116
Further compounding effects on NLRP3 activation have been suggested in the presence of key RA cytokines. TNF can prime cells such as FLS to upregulate NLRP3 and pro-IL-1β. However, FLS require an additional signal to induce inflammasome activation but this can be achieved by extracellular calreticulin which is elevated in RA joint and serum where it correlates with disease activity leading to an increase in IL-1β release.Citation117–Citation119 In addition, IL-6 can enhance monocyte NLRP3 overactivation and pyroptosis, induced by pentraxin-3 (PTX3) and C1q which are elevated in RA serum.Citation120 Furthermore, inhibition of IL-6 in the CIA model reduces NLRP3 activation and IL-1β release.Citation121
Conclusion
Although there is a wealth of information supporting a contribution from both TLRs and the NLRP3 inflammasome in RA pathophysiology, there are still significant gaps in our understanding. Despite two decades of research, therapeutic interventions targeting these pathways have yet to be successfully translated into the clinic. Mechanistic insights have been forthcoming from experimental arthritis models; however, these do not always translate to the human disease, as can be seen with the recent clinical trial of NI-0101 to inhibit TLR4.Citation75 In recent years, numerous inhibitors targeting TLR activation have entered clinical trials for other inflammatory conditions or Phase I safety trials in healthy volunteers, but other than NI-0101, none have yet entered clinical trials for RA.Citation122 However, several trials have commenced with inhibitors that target IRAK4 or Bruton’s tyrosine kinase (Btk) which lie downstream of many TLRs. A Phase II trial with Fenebrutinib (GDC-0853) a Btk inhibitor showed a moderate improvement in disease activity compared to placebo and a phase IIb trial of PF-06650833 an IRAK4 inhibitor, produced a significant clinical improvement in moderate and severe RA patients compared to placebo control.Citation123,Citation124 For the TLRs, it will now be important to determine which receptors are pivotal in the disease process rather than simply dysregulated as a downstream consequence of the inflammatory environment. With so many TLRs potentially contributing to RA, it will also be important to gain a better understanding of how their expression and function is affected by LncRNA, miRNAs and shared downstream signaling regulators. This may provide insights into novel ways to limit inflammation. Several small molecular weight drugs have already been developed to inhibit the NLRP3 inflammasome. A phase II clinical trial for RA using CP-456,773 (later renamed MCC950) was discontinued due to liver toxicity; however, several new inhibitors that target NLRP3 activation are in development either at the preclinical stage, in early clinical trials in healthy volunteers or trials for other inflammatory conditions.Citation125,Citation126 Although inhibition of IL-1 is not as effective as suppressing other cytokines such as TNF and IL-6 in RA, NLRP3 inhibitors may still have a place alongside these biological therapies. Indeed, with the potential for many different pathways driving inflammation in parallel within the joint, blocking a single pathway may not be sufficient.
Disclosure
The authors report no conflicts of interest to declare.
Additional information
Funding
References
- Smolen JS, Aletaha D, Barton A, et al. Rheumatoid arthritis. Nat Rev Dis Primers. 2018;4:18001.
- Malmstrom V, Catrina AI, Klareskog L. The immunopathogenesis of seropositive rheumatoid arthritis: from triggering to targeting. Nat Rev Immunol. 2017;17(1):60–75. doi:10.1038/nri.2016.124
- Frisell T, Holmqvist M, Källberg H, Klareskog L, Alfredsson L, Askling J. Familial risks and heritability of rheumatoid arthritis: role of rheumatoid factor/anti-citrullinated protein antibody status, number and type of affected relatives, sex, and age. Arthritis Rheum. 2013;65(11):2773–2782. doi:10.1002/art.38097
- Okada Y, Eyre S, Suzuki A, Kochi Y, Yamamoto K. Genetics of rheumatoid arthritis: 2018 status. Ann Rheum Dis. 2019;78(4):446–453. doi:10.1136/annrheumdis-2018-213678
- Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med. 2013;5(178):178ra140. doi:10.1126/scitranslmed.3005580
- Law ST, Taylor PC. Role of biological agents in treatment of rheumatoid arthritis. Pharmacol Res. 2019;150:104497. doi:10.1016/j.phrs.2019.104497
- Taylor PC. Clinical efficacy of launched JAK inhibitors in rheumatoid arthritis. Rheumatology. 2019;58(Suppl 1):i17–i26. doi:10.1093/rheumatology/key225
- Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. 2014;5:461. doi:10.3389/fimmu.2014.00461
- Lee SM, Yip TF, Yan S, et al. Recognition of double-stranded RNA and regulation of interferon pathway by toll-like receptor 10. Front Immunol. 2018;9:516. doi:10.3389/fimmu.2018.00516
- Jenkins KA, Mansell A. TIR-containing adaptors in toll-like receptor signalling. Cytokine. 2010;49(3):237–244. doi:10.1016/j.cyto.2009.01.009
- Shevlin E, Miggin SM, Wang T. The TIR-domain containing adaptor TRAM is required for TLR7 mediated RANTES production. PLoS One. 2014;9(9):e107141. doi:10.1371/journal.pone.0107141
- Leszczyńska E, Makuch E, Mitkiewicz M, et al. Absence of Mal/TIRAP results in abrogated imidazoquinolinones-dependent activation of IRF7 and suppressed IFNβ and IFN-I activated gene production. Int J Mol Sci. 2020;21(23):8925. doi:10.3390/ijms21238925
- Sacre SM, Lundberg AM, Andreakos E, Taylor C, Feldmann M, Foxwell BM. Selective use of TRAM in lipopolysaccharide (LPS) and lipoteichoic acid (LTA) induced NF-kappaB activation and cytokine production in primary human cells: TRAM is an adaptor for LPS and LTA signaling. J Immunol. 2007;178(4):2148–2154. doi:10.4049/jimmunol.178.4.2148
- Hazuda DJ, Lee JC, Young PR. The kinetics of interleukin 1 secretion from activated monocytes. Differences between interleukin 1α and interleukin 1β. J Biol Chem. 1988;263(17):8473–8479. doi:10.1016/S0021-9258(18)68502-3
- Martinon F, Burns K, Tschopp J. The Inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol Cell. 2002;10(2):417–426. doi:10.1016/S1097-2765(02)00599-3
- Martinon F, Tschopp J. NLRs join TLRs as innate sensors of pathogens. Trends Immunol. 2005;26(8):447–454. doi:10.1016/j.it.2005.06.004
- Fernandes-Alnemri T, Wu J, Yu JW, et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007;14(9):1590–1604. doi:10.1038/sj.cdd.4402194
- He W-T, Wan H, Hu L, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015;25(12):1285–1298. doi:10.1038/cr.2015.139
- Liu X, Zhang Z, Ruan J, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535(7610):153–158. doi:10.1038/nature18629
- Bauernfeind FG, Horvath G, Stutz A, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183(2):787–791. doi:10.4049/jimmunol.0901363
- He Y, Franchi L, Núñez G. TLR agonists stimulate Nlrp3-dependent IL-1β production independently of the purinergic P2X7 receptor in dendritic cells and in vivo. J Immunol. 2013;190(1):334–339. doi:10.4049/jimmunol.1202737
- Dietsch GN, Lu H, Yang Y, et al. Coordinated activation of toll-like receptor 8 (TLR8) and NLRP3 by the TLR8 agonist, VTX-2337, ignites tumoricidal natural killer cell activity. PLoS One. 2016;11(2):e0148764–e0148764. doi:10.1371/journal.pone.0148764
- Fernandes-Alnemri T, Kang S, Anderson C, Sagara J, Fitzgerald KA, Alnemri ES. Cutting edge: TLR signaling licenses IRAK1 for rapid activation of the NLRP3 inflammasome. J Immunol. 2013;191(8):3995–3999. doi:10.4049/jimmunol.1301681
- Franchi L, Eigenbrod T, Núñez G. Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J Immunol. 2009;183(2):792–796. doi:10.4049/jimmunol.0900173
- Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. 2019;20(13):3328. doi:10.3390/ijms20133328
- Gaidt Moritz M, Ebert TS, Chauhan D, et al. Human monocytes engage an alternative inflammasome pathway. Immunity. 2016;44(4):833–846. doi:10.1016/j.immuni.2016.01.012
- Sandall CF, MacDonald JA. Effects of phosphorylation on the NLRP3 inflammasome. Arch Biochem Biophys. 2019;670:43–57. doi:10.1016/j.abb.2019.02.020
- Shao L, Liu Y, Wang W, et al. SUMO1 SUMOylates and SENP3 deSUMOylates NLRP3 to orchestrate the inflammasome activation. FASEB J. 2020;34(1):1497–1515. doi:10.1096/fj.201901653R
- Mishra BB, Rathinam VA, Martens GW, et al. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1β. Nat Immunol. 2013;14(1):52–60. doi:10.1038/ni.2474
- Lopez-Castejon G. Control of the inflammasome by the ubiquitin system. Febs j. 2020;287(1):11–26. doi:10.1111/febs.15118
- Carty M, Kearney J, Shanahan KA, et al. Cell survival and cytokine release after inflammasome activation is regulated by the toll-IL-1R protein SARM. Immunity. 2019;50(6):1412–1424.e1416. doi:10.1016/j.immuni.2019.04.005
- Sacre SM, Andreakos E, Kiriakidis S, et al. The toll-like receptor adaptor proteins MyD88 and Mal/TIRAP contribute to the inflammatory and destructive processes in a human model of rheumatoid arthritis. Am J Pathol. 2007;170(2):518–525. doi:10.2353/ajpath.2007.060657
- Lacerte P, Brunet A, Egarnes B, Duchene B, Brown JP, Gosselin J. Overexpression of TLR2 and TLR9 on monocyte subsets of active rheumatoid arthritis patients contributes to enhance responsiveness to TLR agonists. Arthritis Res Ther. 2016;18:10. doi:10.1186/s13075-015-0901-1
- Iwahashi M, Yamamura M, Aita T, et al. Expression of toll-like receptor 2 on CD16+ blood monocytes and synovial tissue macrophages in rheumatoid arthritis. Arthritis Rheum. 2004;50(5):1457–1467. doi:10.1002/art.20219
- Clanchy FIL, Borghese F, Bystrom J, et al. TLR expression profiles are a function of disease status in rheumatoid arthritis and experimental arthritis. J Autoimmun. 2021;118:102597. doi:10.1016/j.jaut.2021.102597
- Abdelwahab A, Palosaari S, Abdelwahab SA, et al. Differential synovial tissue expression of TLRs in seropositive and seronegative rheumatoid arthritis: a preliminary report. Autoimmunity. 2021;54(1):23–34. doi:10.1080/08916934.2020.1864729
- Yu M, Wang H, Ding A, et al. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock. 2006;26(2):174–179. doi:10.1097/01.shk.0000225404.51320.82
- He Z, Shotorbani SS, Jiao Z, et al. HMGB1 promotes the differentiation of Th17 via up-regulating TLR2 and IL-23 of CD14+ monocytes from patients with rheumatoid arthritis. Scand J Immunol. 2012;76(5):483–490. doi:10.1111/j.1365-3083.2012.02759.x
- Huang QQ, Sobkoviak R, Jockheck-Clark AR, et al. Heat shock protein 96 is elevated in rheumatoid arthritis and activates macrophages primarily via TLR2 signaling. J Immunol. 2009;182(8):4965–4973. doi:10.4049/jimmunol.0801563
- Thwaites RS, Unterberger S, Chamberlain G, Walker-Bone K, Davies KA, Sacre S. TLR1/2 and 5 induce elevated cytokine levels from rheumatoid arthritis monocytes independent of ACPA or RF autoantibody status. Rheumatology. 2020;59(11):3533–3539. doi:10.1093/rheumatology/keaa220
- Thwaites RS, Unterberger S, Chamberlain G, et al. Expression of sterile-α and armadillo motif in rheumatoid arthritis monocytes correlates with TLR2 induced IL-1β and disease activity. Rheumatology. 2021. doi:10.1093/rheumatology/keab162
- Kim K-W, Cho M-L, Lee S-H, et al. Human rheumatoid synovial fibroblasts promote osteoclastogenic activity by activating RANKL via TLR-2 and TLR-4 activation. Immunol Lett. 2007;110(1):54–64. doi:10.1016/j.imlet.2007.03.004
- Quero L, Hanser E, Manigold T, Tiaden AN, Kyburz D. TLR2 stimulation impairs anti-inflammatory activity of M2-like macrophages, generating a chimeric M1/M2 phenotype. Arthritis Res Ther. 2017;19(1):245. doi:10.1186/s13075-017-1447-1
- McGarry T, Veale DJ, Gao W, Orr C, Fearon U, Connolly M. Toll-like receptor 2 (TLR2) induces migration and invasive mechanisms in rheumatoid arthritis. Arthritis Res Ther. 2015;17(1):153. doi:10.1186/s13075-015-0664-8
- Ultaigh SN, Saber TP, McCormick J, et al. Blockade of toll-like receptor 2 prevents spontaneous cytokine release from rheumatoid arthritis ex vivo synovial explant cultures. Arthritis Res Ther. 2011;13(1):R33. doi:10.1186/ar3261
- Zhu W, Meng L, Jiang C, et al. Induction of toll-like receptor 2 positive antigen-presenting cells in spleen of pristane-induced arthritis in rats. Mol Biol Rep. 2012;39(4):3667–3673. doi:10.1007/s11033-011-1141-3
- Roome T, Aziz S, Razzak A, et al. Opuntioside, opuntiol and its metallic nanoparticles attenuate adjuvant-induced arthritis: novel suppressors of toll-like receptors −2 and −4. Biomed Pharmacother. 2019;112:108624. doi:10.1016/j.biopha.2019.108624
- Bai L, Bai Y, Yang Y, et al. Baicalin alleviates collagen-induced arthritis and suppresses TLR2/MYD88/NF‑κB p65 signaling in rats and HFLS‑RAs. Mol Med Rep. 2020;22(4):2833–2841.
- Abdollahi-Roodsaz S, Joosten LA, Koenders MI, et al. Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J Clin Invest. 2008;118(1):205–216. doi:10.1172/JCI32639
- Joosten LA, Koenders MI, Smeets RL, et al. Toll-like receptor 2 pathway drives streptococcal cell wall-induced joint inflammation: critical role of myeloid differentiation factor 88. J Immunol. 2003;171(11):6145–6153. doi:10.4049/jimmunol.171.11.6145
- Ospelt C, Brentano F, Rengel Y, et al. Overexpression of toll-like receptors 3 and 4 in synovial tissue from patients with early rheumatoid arthritis: toll-like receptor expression in early and longstanding arthritis. Arthritis Rheum. 2008;58(12):3684–3692. doi:10.1002/art.24140
- Roelofs MF, Joosten LA, Abdollahi-Roodsaz S, et al. The expression of toll-like receptors 3 and 7 in rheumatoid arthritis synovium is increased and costimulation of toll-like receptors 3, 4, and 7/8 results in synergistic cytokine production by dendritic cells. Arthritis Rheum. 2005;52(8):2313–2322. doi:10.1002/art.21278
- Zimmermann-Geller B, Köppert S, Fischer S, et al. Influence of extracellular RNAs, released by rheumatoid arthritis synovial fibroblasts, on their adhesive and invasive properties. J Immunol. 2016;197(7):2589–2597. doi:10.4049/jimmunol.1501580
- Brentano F, Schorr O, Gay RE, Gay S, Kyburz D. RNA released from necrotic synovial fluid cells activates rheumatoid arthritis synovial fibroblasts via toll-like receptor 3. Arthritis Rheum. 2005;52(9):2656–2665. doi:10.1002/art.21273
- Bokarewa M, Tarkowski A, Lind M, Dahlberg L, Magnusson M. Arthritogenic dsRNA is present in synovial fluid from rheumatoid arthritis patients with an erosive disease course. Eur J Immunol. 2008;38(11):3237–3244. doi:10.1002/eji.200838362
- Hu F, Li Y, Zheng L, et al. Toll-like receptors expressed by synovial fibroblasts perpetuate th1 and th17 cell responses in rheumatoid arthritis. PLoS One. 2014;9(6):e100266. doi:10.1371/journal.pone.0100266
- Bombardieri M, Kam NW, Brentano F, et al. A BAFF/APRIL-dependent TLR3-stimulated pathway enhances the capacity of rheumatoid synovial fibroblasts to induce AID expression and Ig class-switching in B cells. Ann Rheum Dis. 2011;70(10):1857–1865. doi:10.1136/ard.2011.150219
- Kim KW, Cho ML, Oh HJ, et al. TLR-3 enhances osteoclastogenesis through upregulation of RANKL expression from fibroblast-like synoviocytes in patients with rheumatoid arthritis. Immunol Lett. 2009;124(1):9–17. doi:10.1016/j.imlet.2009.02.006
- Zhu W, Meng L, Jiang C, et al. Overexpression of toll-like receptor 3 in spleen is associated with experimental arthritis in rats. Scand J Immunol. 2012;76(3):263–270. doi:10.1111/j.1365-3083.2012.02724.x
- Jiang C, Zhu W, Xu J, et al. MicroRNA-26a negatively regulates toll-like receptor 3 expression of rat macrophages and ameliorates pristane induced arthritis in rats. Arthritis Res Ther. 2014;16(1):R9. doi:10.1186/ar4435
- Zhu W, Meng L, Jiang C, et al. Arthritis is associated with T-cell-induced upregulation of toll-like receptor 3 on synovial fibroblasts. Arthritis Res Ther. 2011;13(3):R103. doi:10.1186/ar3384
- Lee SY, Yoon BY, Kim JI, et al. Interleukin-17 increases the expression of toll-like receptor 3 via the STAT3 pathway in rheumatoid arthritis fibroblast-like synoviocytes. Immunology. 2014;141(3):353–361. doi:10.1111/imm.12196
- Radstake TRDJ, Roelofs MF, Jenniskens YM, et al. Expression of toll-like receptors 2 and 4 in rheumatoid synovial tissue and regulation by proinflammatory cytokines interleukin-12 and interleukin-18 via interferon-γ. Arthritis Rheum. 2004;50(12):3856–3865. doi:10.1002/art.20678
- De Rycke L, Vandooren B, Kruithof E, De Keyser F, Veys EM, Baeten D. Tumor necrosis factor α blockade treatment down-modulates the increased systemic and local expression of toll-like receptor 2 and toll-like receptor 4 in spondylarthropathy. Arthritis Rheum. 2005;52(7):2146–2158. doi:10.1002/art.21155
- Huang Q, Ma Y, Adebayo A, Pope RM. Increased macrophage activation mediated through toll-like receptors in rheumatoid arthritis. Arthritis Rheum. 2007;56(7):2192–2201. doi:10.1002/art.22707
- Tripathy A, Khanna S, Padhan P, Smita S, Raghav S, Gupta B. Direct recognition of LPS drive TLR4 expressing CD8(+) T cell activation in patients with rheumatoid arthritis. Sci Rep. 2017;7(1):933. doi:10.1038/s41598-017-01033-7
- Zhang Y, Ji T, Ma S, Wu W. MLL1 promotes migration and invasion of fibroblast-like synoviocytes in rheumatoid arthritis by activating the TRIF/NF-κB signaling pathway via H3K4me3 enrichment in the TLR4 promoter region. Int Immunopharmacol. 2020;82:106220. doi:10.1016/j.intimp.2020.106220
- Li D, Zhou Q, Hu G, Wang G. MiRNA-506 inhibits rheumatoid arthritis fibroblast-like synoviocytes proliferation and induces apoptosis by targeting TLR4. Biosci Rep. 2019;39(5):BSR20182500. doi:10.1042/BSR20182500
- Sokolove J, Zhao X, Chandra PE, Robinson WH. Immune complexes containing citrullinated fibrinogen costimulate macrophages via toll-like receptor 4 and Fcγ receptor. Arthritis Rheum. 2011;63(1):53–62. doi:10.1002/art.30081
- Kiyeko GW, Hatterer E, Herren S, et al. Spatiotemporal expression of endogenous TLR4 ligands leads to inflammation and bone erosion in mouse collagen-induced arthritis. Eur J Immunol. 2016;46(11):2629–2638. doi:10.1002/eji.201646453
- Pierer M, Wagner U, Rossol M, Ibrahim S. Toll-like receptor 4 is involved in inflammatory and joint destructive pathways in collagen-induced arthritis in DBA1J mice. PLoS One. 2011;6(8):e23539–e23539. doi:10.1371/journal.pone.0023539
- Kim HS, Chung DH. TLR4-mediated IL-12 production enhances IFN-γ and IL-1β production, which inhibits TGF-β production and promotes antibody-induced joint inflammation. Arthritis Res Ther. 2012;14(5):R210–R210. doi:10.1186/ar4048
- Abdollahi-Roodsaz S, Joosten LAB, Roelofs MF, et al. Inhibition of toll-like receptor 4 breaks the inflammatory loop in autoimmune destructive arthritis. Arthritis Rheum. 2007;56(9):2957–2967. doi:10.1002/art.22848
- Samarpita S, Kim JY, Rasool MK, Kim KS. Investigation of toll-like receptor (TLR) 4 inhibitor TAK-242 as a new potential anti-rheumatoid arthritis drug. Arthritis Res Ther. 2020;22(1):16. doi:10.1186/s13075-020-2097-2
- Monnet E, Choy EH, McInnes I, et al. Efficacy and safety of NI-0101, an anti-toll-like receptor 4 monoclonal antibody, in patients with rheumatoid arthritis after inadequate response to methotrexate: a phase II study. Ann Rheum Dis. 2020;79(3):316–323. doi:10.1136/annrheumdis-2019-216487
- Das N, Dewan V, Grace PM, et al. HMGB1 activates proinflammatory signaling via TLR5 leading to allodynia. Cell Rep. 2016;17(4):1128–1140. doi:10.1016/j.celrep.2016.09.076
- Chamberlain ND, Vila OM, Volin MV, et al. TLR5, a novel and unidentified inflammatory mediator in rheumatoid arthritis that correlates with disease activity score and joint TNF-alpha levels. J Immunol. 2012;189(1):475–483. doi:10.4049/jimmunol.1102977
- Kim SJ, Chen Z, Chamberlain ND, et al. Ligation of TLR5 promotes myeloid cell infiltration and differentiation into mature osteoclasts in rheumatoid arthritis and experimental arthritis. J Immunol. 2014;193(8):3902–3913. doi:10.4049/jimmunol.1302998
- Fu D, Xiao C, Xie Y, Gao J, Ye S. MiR-3926 inhibits synovial fibroblasts proliferation and inflammatory cytokines secretion through targeting toll like receptor 5. Gene. 2019;687:200–206. doi:10.1016/j.gene.2018.11.014
- Kim SJ, Chen Z, Chamberlain ND, et al. Angiogenesis in rheumatoid arthritis is fostered directly by toll-like receptor 5 ligation and indirectly through interleukin-17 induction. Arthritis Rheum. 2013;65(8):2024–2036. doi:10.1002/art.37992
- Chamberlain ND, Kim SJ, Vila OM, et al. Ligation of TLR7 by rheumatoid arthritis synovial fluid single strand RNA induces transcription of TNFα in monocytes. Ann Rheum Dis. 2013;72(3):418–426. doi:10.1136/annrheumdis-2011-201203
- Torices S, Alvarez-Rodríguez L, Varela I, et al. Evaluation of toll-like-receptor gene family variants as prognostic biomarkers in rheumatoid arthritis. Immunol Lett. 2017;187:35–40. doi:10.1016/j.imlet.2017.04.011
- Mullen L, Ferdjani J, Sacre S. Simvastatin inhibits toll-like receptor 8 (TLR8) signaling in primary human monocytes and spontaneous tumor necrosis factor production from rheumatoid synovial membrane cultures. Mol Med. 2015;21(1):726–734. doi:10.2119/molmed.2015.00154
- Sacre SM, Lo A, Gregory B, et al. Inhibitors of TLR8 reduce TNF production from human rheumatoid synovial membrane cultures. J Immunol. 2008;181(11):8002–8009. doi:10.4049/jimmunol.181.11.8002
- Sacre S, Medghalchi M, Gregory B, Brennan F, Williams R. Fluoxetine and citalopram exhibit potent antiinflammatory activity in human and murine models of rheumatoid arthritis and inhibit toll-like receptors. Arthritis Rheum. 2010;62(3):683–693. doi:10.1002/art.27304
- Neregård P, Engström M, Agerberth B, Catrina AI. LL-37 is expressed in the inflamed synovium in patients with rheumatoid arthritis and downregulated by TNF inhibitors. Ann Rheum Dis. 2012;71(Suppl 1):A12–A12. doi:10.1136/annrheumdis-2011-201230.26
- Hegewald AB, Breitwieser K, Ottinger SM, et al. Extracellular miR-574-5p induces osteoclast differentiation via TLR 7/8 in rheumatoid arthritis. Front Immunol. 2020;11:585282. doi:10.3389/fimmu.2020.585282
- Kim S-J, Chen Z, Essani AB, et al. Identification of a novel toll-like receptor 7 endogenous ligand in rheumatoid arthritis synovial fluid that can provoke arthritic joint inflammation. Arthritis Rheumatol. 2016;68(5):1099–1110.
- Alzabin S, Kong P, Medghalchi M, Palfreeman A, Williams R, Sacre S. Investigation of the role of endosomal toll-like receptors in murine collagen-induced arthritis reveals a potential role for TLR7 in disease maintenance. Arthritis Res Ther. 2012;14(3):R142–R142. doi:10.1186/ar3875
- Sacre S, Lo A, Gregory B, et al. Oligodeoxynucleotide inhibition of toll-like receptors 3, 7, 8, and 9 suppresses cytokine production in a human rheumatoid arthritis model. Eur J Immunol. 2016;46(3):772–781. doi:10.1002/eji.201546123
- Chen SY, Shiau AL, Li YT, et al. Suppression of collagen-induced arthritis by intra-articular lentiviral vector-mediated delivery of TOLL-like receptor 7 short hairpin RNA gene. Gene Ther. 2012;19(7):752–760. doi:10.1038/gt.2011.173
- Heil F, Hemmi H, Hochrein H, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303(5663):1526–1529. doi:10.1126/science.1093620
- Guiducci C, Gong M, Cepika AM, et al. RNA recognition by human TLR8 can lead to autoimmune inflammation. J Exp Med. 2013;210(13):2903–2919. doi:10.1084/jem.20131044
- Zhong X-Y, von Mühlenen I, Li Y, et al. Increased concentrations of antibody-bound circulatory cell-free DNA in rheumatoid arthritis. Clin Chem. 2007;53(9):1609–1614. doi:10.1373/clinchem.2006.084509
- Szodoray P, Alex P, Frank MB, et al. A genome-scale assessment of peripheral blood B-cell molecular homeostasis in patients with rheumatoid arthritis. Rheumatology. 2006;45(12):1466–1476. doi:10.1093/rheumatology/kel095
- Miles K, Heaney J, Sibinska Z, et al. A tolerogenic role for toll-like receptor 9 is revealed by B-cell interaction with DNA complexes expressed on apoptotic cells. Proc Natl Acad Sci. 2012;109(3):887–892. doi:10.1073/pnas.1109173109
- Deng G-M, Nilsson I-M, Verdrengh M, Collins LV, Tarkowski A. Intra-articularly localized bacterial DNA containing CpG motifs induces arthritis. Nat Med. 1999;5(6):702–705. doi:10.1038/9554
- Viglianti GA, Lau CM, Hanley TM, Miko BA, Shlomchik MJ, Marshak-Rothstein A. Activation of autoreactive B cells by CpG dsDNA. Immunity. 2003;19(6):837–847. doi:10.1016/S1074-7613(03)00323-6
- Fischer A, Abdollahi-Roodsaz S, Böhm C, et al. The involvement of Toll-like receptor 9 in the pathogenesis of erosive autoimmune arthritis. J Cell Mol Med. 2018;22(9):4399–4409. doi:10.1111/jcmm.13735
- Su SB, Tao L, Deng ZP, Chen W, Qin SY, Jiang HX. TLR10: insights, controversies and potential utility as a therapeutic target. Scand J Immunol. 2021;93:e12988.
- Petrackova A, Horak P, Radvansky M, et al. Revealed heterogeneity in rheumatoid arthritis based on multivariate innate signature analysis. Clin Exp Rheumatol. 2020;38(2):289–298.
- Torices S, Julia A, Muñoz P, et al. A functional variant of TLR10 modifies the activity of NFkB and may help predict a worse prognosis in patients with rheumatoid arthritis. Arthritis Res Ther. 2016;18(1):221. doi:10.1186/s13075-016-1113-z
- Elemam NM, Hachim MY, Hannawi S, Maghazachi AA. Differentially expressed genes of natural killer cells can distinguish rheumatoid arthritis patients from healthy controls. Genes (Basel). 2020;11(5):492. doi:10.3390/genes11050492
- Zhang Y, Cao R, Ying H, et al. Increased expression of TLR10 in B cell subsets correlates with disease activity in rheumatoid arthritis. Mediators Inflamm. 2018;2018:9372436. doi:10.1155/2018/9372436
- Zhang Y, Zheng Y, Li H. NLRP3 inflammasome plays an important role in the pathogenesis of collagen-induced arthritis. Mediators Inflamm. 2016;2016:9656270. doi:10.1155/2016/9656270
- Guo C, Fu R, Wang S, et al. NLRP3 inflammasome activation contributes to the pathogenesis of rheumatoid arthritis. Clin Exp Immunol. 2018;194(2):231–243. doi:10.1111/cei.13167
- Choulaki C, Papadaki G, Repa A, et al. Enhanced activity of NLRP3 inflammasome in peripheral blood cells of patients with active rheumatoid arthritis. Arthritis Res Ther. 2015;17(1):257. doi:10.1186/s13075-015-0775-2
- Zhao C, Gu Y, Zeng X, Wang J. NLRP3 inflammasome regulates Th17 differentiation in rheumatoid arthritis. Clin Immunol. 2018;197:154–160. doi:10.1016/j.clim.2018.09.007
- Wang Y, Sedlacek AL, Pawaria S, Xu H, Scott MJ, Binder RJ. Cutting edge: the heat shock protein gp96 activates inflammasome-signaling platforms in APCs. J Immunol. 2018;201(8):2209–2214. doi:10.4049/jimmunol.1800505
- Dong X, Zheng Z, Lin P, et al. ACPAs promote IL-1β production in rheumatoid arthritis by activating the NLRP3 inflammasome. Cell Mol Immunol. 2020;17:261–271.
- Jäger E, Murthy S, Schmidt C, et al. Calcium-sensing receptor-mediated NLRP3 inflammasome response to calciprotein particles drives inflammation in rheumatoid arthritis. Nat Commun. 2020;11(1):4243. doi:10.1038/s41467-020-17749-6
- Vande Walle L, Van Opdenbosch N, Jacques P, et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature. 2014;512(7512):69–73. doi:10.1038/nature13322
- Spalinger MR, Kasper S, Gottier C, et al. NLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22. J Clin Invest. 2016;126(5):1783–1800. doi:10.1172/JCI83669
- Lee YH, Bae SC. Vitamin D level in rheumatoid arthritis and its correlation with the disease activity: a meta-analysis. Clin Exp Rheumatol. 2016;34(5):827–833.
- Rao Z, Chen X, Wu J, et al. Vitamin D receptor inhibits NLRP3 activation by impeding its BRCC3-mediated deubiquitination. Front Immunol. 2019;10:2783. doi:10.3389/fimmu.2019.02783
- Thwaites RS, Unterberger S, Chamberlain G, et al. Expression of sterile-α and armadillo motif containing protein (SARM) in rheumatoid arthritis monocytes correlates with TLR2-induced IL-1β and disease activity. Rheumatology. 2021. Available form: https://academic.oup.com/rheumatology/advance-article/doi/10.1093/rheumatology/keab162/6144929. Accessed July 23, 2021.
- Liu Y, Wei W, Wang Y, et al. TNF-α/calreticulin dual signaling induced NLRP3 inflammasome activation associated with HuR nucleocytoplasmic shuttling in rheumatoid arthritis. Inflamm Res. 2019;68(7):597–611. doi:10.1007/s00011-019-01244-w
- Ni M, Wei W, Wang Y, et al. Serum levels of calreticulin in correlation with disease activity in patients with rheumatoid arthritis. J Clin Immunol. 2013;33(5):947–953. doi:10.1007/s10875-013-9885-2
- Tarr JM, Winyard PG, Ryan B, et al. Extracellular calreticulin is present in the joints of patients with rheumatoid arthritis and inhibits FasL (CD95L)-mediated apoptosis of T cells. Arthritis Rheum. 2010;62(10):2919–2929. doi:10.1002/art.27602
- Wu XY, Li KT, Yang HX, et al. Complement C1q synergizes with PTX3 in promoting NLRP3 inflammasome over-activation and pyroptosis in rheumatoid arthritis. J Autoimmun. 2020;106:102336. doi:10.1016/j.jaut.2019.102336
- Wang H, Wang Z, Wang L, et al. IL-6 promotes collagen-induced arthritis by activating the NLRP3 inflammasome through the cathepsin B/S100A9-mediated pathway. Int Immunopharmacol. 2020;88:106985. doi:10.1016/j.intimp.2020.106985
- Anwar MA, Shah M, Kim J, Choi S. Recent clinical trends in Toll-like receptor targeting therapeutics. Med Res Rev. 2019;39(3):1053–1090. doi:10.1002/med.21553
- Danto S, Shojaee N, Singh R, et al. Efficacy and safety of the selective interleukin-1 receptor associated kinase 4 inhibitor, PF-06650833, in patients with active rheumatoid arthritis and inadequate response to methotrexate [abstract 2909]. Arthritis Rheumatol. 2019;71(S10).
- Cohen S, Tuckwell K, Kunder R, et al. Efficacy and safety of fenebrutinib, a BTK inhibitor, compared to placebo in rheumatoid arthritis patients with active disease despite TNF inhibitor treatment: randomized, double blind, Phase 2 Study [abstract 929]. Arthritis Rheumatol. 2019;71(Suppl 10).
- Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov. 2018;17(8):588. doi:10.1038/nrd.2018.97
- El-Sharkawy LY, Brough D, Freeman S. Inhibiting the NLRP3 Inflammasome. Molecules. 2020;25(23):5533. doi:10.3390/molecules25235533
- Connolly M, Rooney PR, McGarry T, et al. Acute serum amyloid A is an endogenous TLR2 ligand that mediates inflammatory and angiogenic mechanisms. Ann Rheum Dis. 2016;75(7):1392–1398. doi:10.1136/annrheumdis-2015-207655
- Shi B, Huang Q, Tak PP, et al. SNAPIN: an endogenous toll-like receptor ligand in rheumatoid arthritis. Ann Rheum Dis. 2012;71(8):1411–1417. doi:10.1136/annrheumdis-2011-200899
- Midwood K, Sacre S, Piccinini AM, et al. Tenascin-C is an endogenous activator of toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med. 2009;15(7):774–780. doi:10.1038/nm.1987
- Grevers LC, de Vries TJ, Vogl T, et al. S100A8 enhances osteoclastic bone resorption in vitro through activation of toll-like receptor 4: implications for bone destruction in murine antigen-induced arthritis. Arthritis Rheum. 2011;63(5):1365–1375. doi:10.1002/art.30290
- Barreto G, Soininen A, Ylinen P, et al. Soluble biglycan: a potential mediator of cartilage degradation in osteoarthritis. Arthritis Res Ther. 2015;17:379. doi:10.1186/s13075-015-0902-0
- Roelofs MF, Boelens WC, Joosten LAB, et al. Identification of small heat shock protein B8 (HSP22) as a novel TLR4 ligand and potential involvement in the pathogenesis of rheumatoid arthritis. J Immunol. 2006;176(11):7021–7027. doi:10.4049/jimmunol.176.11.7021
- Guillou C, Fréret M, Fondard E, et al. Soluble alpha-enolase activates monocytes by CD14-dependent TLR4 signalling pathway and exhibits a dual function. Sci Rep. 2016;6(1):23796. doi:10.1038/srep23796
- Veiko NN, Shubaeva NO, Ivanova SM, Speranskii AI, Lyapunova NA, Spitkovskii DM. Blood serum DNA in patients with rheumatoid arthritis is considerably enriched with fragments of ribosomal repeats containing immunostimulatory CpG-motifs. Bull Exp Biol Med. 2006;142(3):313–316. doi:10.1007/s10517-006-0354-2