Abstract
Immunotherapy might provide an effective treatment for Alzheimer’s disease (AD). A unique feature of AD immunotherapies is that an immune response against a self-antigen needs to be elicited without causing adverse autoimmune reactions. Current research is focused on two possible targets in this regard. One is the inhibition of accumulation and deposition of amyloid beta 1–42 (Aβ42), which is one of the major peptides found in senile plaques, and the second target is hyperphosphorylated tau, which forms neurofibrillary tangles inside the nerve cell and shows association with the progression of dementia. Mouse models have shown that immunotherapy targeting Aβ42 as well as tau with the respective anti-Aβ or anti-tau antibodies can provide significant improvements in these mice. While anti-Aβ immunotherapy (active and passive immunizations) is already in several stages of clinical trials, tau-based immunizations have been analyzed only in mouse models. Recently, as a significant correlation of progression of dementia and levels of phosphorylated tau have been found, high interest has again focused on further development of tau-based therapies. While Aβ immunotherapy might delay the onset of AD, immunotherapy targeting tau might provide benefits in later stages of this disease. Last but not least, targeting Aβ and tau simultaneously with immunotherapy might provide additional therapeutic effects, as these two pathologies are likely synergistic; this is an approach that has not been tested yet. In this review, we will summarize animal models used to test possible therapies for AD, some of the facts about Aβ42 and tau biology, and present an overview on halted, ongoing, and upcoming clinical trials together with ongoing preclinical studies targeting tau or Aβ42.
Introduction
Alzheimer’s disease (AD) is the sixth-leading cause of death in the US, and until now there has been no effective treatment available.Citation1 Current treatment options are only symptomatic and do not affect disease progression. Immunotherapy in which antibodies directed against the two pathophysiological hallmarks of AD – amyloid plaques with amyloid beta 1–42 (Aβ42) as their major component and neurofibrillary tangles (NFTs) that are formed by hyperphosphorylated tau – are possible targets for an immunotherapeutic intervention. Both pathophysiological features are formed from aberrant self-proteins: a major component of the amyloid plaques is Aβ42, which is a small proteolytic fragment from the amyloid precursor protein (APP) with the tendency to aggregate into insoluble fibrils, and the NFTs are derived from hyperphosphorylation of the protein tau, which makes this protein insoluble and leads to aggregate formation and malfunction.Citation2 Other changes in AD brain include inflammation and oxidative stress.Citation3 All of these features lead to severe dysfunction, neurodegeneration, and neuron loss, and end-stage brain from AD patients shows a substantial loss in weight compared to brains of age- and sex-matched nondemented individuals. The most neurotoxic form of Aβ is not the solid plaques but the oligomeric forms of Aβ42. In rodents, a dodecameric form of Aβ42, which was isolated from brains of APP transgenic mice, leads directly to impairment of synaptic plasticity and memory.Citation4 In humans, the same feature was described for an Aβ42 dimer, which had been isolated from human AD brains.Citation5 A recent study showed that all of these Aβ42 oligomers, dimers, trimers, and dodecamers (Aβ56) can be isolated from human brain and cerebrospinal fluid (CSF) and Aβ56 levels correlated positively with levels on soluble tau protein in brain from AD patients.Citation6 The description of Aβ42 aggregation as being outside the nerve cells and tau aggregates being found only inside neurons may not be as strict as proposed. There is evidence that Aβ accumulation occurs first inside the nerve cell and accumulates outside the neurons as disease progresses, and for tau it has also been shown in in vitro and in vivo models that extracellular tau aggregates lead to uptake of these aggregates by the nerve cells in which they induce intracellular tau aggregation, and that the injection of insoluble tau, and to a lesser extent the injection of soluble tau, into mouse brains leads to spreading of tau pathology.Citation7–Citation11
Animal models
In familial AD (FAD), which accounts for less than 5% of all AD cases, mutations were discovered within the APP gene or in genes encoding enzymes involved in the proteolytic degradation of APP, presenilin (PS)-1 and -2, which increase the amyloidogenic processing of APP, and are thus leading to increased Aβ42 levels.Citation12 Based on these mutations, transgenic mouse models were developed that recapitulate some of the features seen in humans. Even though not a perfect replica for the human disease, the mouse models were helpful in discovering mechanisms by which Aβ oligomers and tau oligomers were directly leading to dysfunction and toxicity.Citation13 These mouse models also showed that immunotherapy can modify the development of disease.Citation14 In experiments for Aβ42 immunotherapy, most often used is a double transgenic mouse, which carries a chimeric mouse/human amyloid precursor protein gene (Mo/HuAPP695 Swe) and the gene for mutant human PS1 (PS1-dE9), which are expressed under regulation from different strong promoter sequences, such as the prion protein, platelet-derived growth factor, or the Thy-1 promoter. These transgenic mice develop Aβ42 deposits and senile plaques in the brain by 6–7 months of age.Citation15 While there have been no tau mutations observed in AD, a mutated human tau gene (P301S or P301L) has been associated with forms of frontotemporal dementia with tangle pathology. Mice transgenic for this particular human gene provide tools to study tau pathology and tangle-related neurodegeneration, as well as the evaluation of potential therapies. These mice show the age-dependent development of NFTs, and develop progressive motor dysfunctions correlating with the loss of motor neurons in later stages.Citation16 A triple-transgenic mouse model, which combines Aβ and Tau pathology carrying the mutated human APP and mutated human microtubule-associated protein tau (MAPT) gene, and even a quintuple-transgenic mouse model (5 × FAD) that combines three APP and two PS1 FAD mutations has been generated in an effort to make this model more complete.Citation17–Citation20 An important new animal model was recently described in the rat. Rats transgenic for human APPs and the mutant human PS1 (PS1-dE9) genes developed many of the characteristic features of AD: amyloid plaques, tau tangles, and memory deficits, as well as loss of neurons and neurodegeneration.Citation21 Different from the mouse models, rats developed the NFTs “naturally,” which is in strong support of the order of the pathophysiological findings: Aβ accumulation first, followed by tangle formation. The explanation for this important difference from the mouse models is that the tau proteome is much closer to the human tau proteome than mouse tau. While the mouse expresses only three different tau isoforms in the brain, which is due to the lack of exon 10 splicing, the rat expresses the same six tau protein isoforms that are found in human brain.Citation22,Citation23
Amyloid precursor protein
APP is a type 1 transmembrane protein with a large extracellular domain and a short intracellular segment that is expressed in many tissues, with the highest expression level in the brain. A role for APP in neural tissue is synaptic formation and repair; APP expression is upregulated during differentiation and after neural injury.Citation24,Citation25 Depending on whether APP is processed via the α-secretase or the β-secretase pathway, the products are nonamyloidogenic or amyloidogenic, respectively.Citation26 The aforementioned mutations within the APP gene, which cause FAD, have been found to dramatically increase production of Aβ42.Citation12 Recently, a new mutation within the APP gene was described that showed protection against AD. The APP substitution A673T is adjacent to the β-secretase cleavage site, leading to a 40% reduction in overall Aβ42 levels.Citation27 These findings strongly support the amyloid-cascade hypothesis, which was postulated more than 20 years ago that posits that Aβ accumulation, while it may not be the initial event, plays a central role in the multifactorial pathogenesis of AD.Citation28–Citation31
Clinical human studies: anti-Aβ immunization
Following the observations that Aβ42 accumulation in brain is strongly associated with the development of AD, immunizations against Aβ were tested in AD mouse models. Results showed that this treatment can indeed lead to reduction of total amyloid levels in brain, as well as removal of the senile plaques. Most importantly, a significant effect from the immunotherapy had been shown on mouse memory and performance in behavioral tests.Citation14,Citation32–Citation34
The first clinical trial, AN1792, in which AD patients received Aβ42 peptide injections to induce an antibody immune response, was stopped when 6% of the treated patients developed meningoencephalitis.Citation35–Citation37 But besides the negative side effect, it appeared that Aβ42 immunotherapy had worked in regard to reduction of overall Aβ42 and plaque counts in the brains of immunized patients, even though it did not stop the progression of dementia.Citation38 As immunotherapy has great potential as a disease-modifying intervention in contrast to the currently available symptom-only treatment options, major efforts are in progress to make this therapy for AD safe and effective, and a number of active and passive immunotherapies targeting Aβ peptides are currently in clinical trials.
Based on the observed negative side effect from AN1792 with autoimmune T-cell responses, all of the active immunization trials are now concentrated on the B-cell epitope (Aβ1–6 or Aβ1–15) to produce antibodies while avoiding a possible inflammatory T-cell response. Three of these epitope peptide vaccines for active immunizations – CAD106, (Novartis, Basel, Switzerland), ACC-001 (Elan Corporation, Dublin, Ireland), and Affitope (Affiris AG, Vienna, Austria) – are currently in phase II clinical trials. In the CAD106 vaccine, Aβ1–6 is coupled to a heterologous carrier protein to optimize an immune response, and in the Affitope vaccine, a peptide is used that mimics the Aβ B-cell epitope but has no sequence similarities.Citation39–Citation41 Positive antibody titers and no adverse autoimmune inflammation has been found in clinical trials using these new epitope vaccines. New results were also recently reported from mouse studies using very similar constructed epitope peptide vaccines for potential use in AD patients.Citation42,Citation43
The most promising approach for AD immunotherapy is currently passive immunization with humanized anti-Aβ antibodies. In this approach, preformed anti-Aβ42 antibodies are injected intravenously (IV) with the goal that these antibodies will help to reduce amyloid burden in the brain of AD patients by several possible mechanisms, such as facilitation of phagocytosis of amyloid by microglia, inhibition of amyloid aggregation, or binding of antibodies to amyloid in blood causing a concentration gradient with net efflux of Aβ42 from brain. Observed complications from the injection with some of the monoclonal antibodies (mAbs) is that they show a tendency to cause vasogenic edema and brain microhemorrhage, which have been reported also in mouse models.Citation44–Citation46 Three of the monoclonal antibody therapies – solanezumab from Eli Lilly and Company (Indianapolis, IN, USA), crenezumab from Genentech, (San Francisco, CA, USA), and gantenerumab from Hoffmann-La Roche (Basel, Switzerland) – are in phase II and III clinical trials and ongoing.Citation47–Citation52 A fourth antibody trial, investigating bapineuzumab from Pfizer, (New York, NY, USA), which was completed in 2012, was discontinued when the results obtained did not meet the predicted results.Citation53
Solanezumab and crenezumab are humanized mouse mAbs detecting a mid-region Aβ epitope, Aβ13–28, and Aβ12–23. Solanezumab has a good safety profile, and showed in patients with mild AD the slowing of cognitive decline compared with placebo by one-third.Citation49 An increase of Aβ42 levels in CSF might indicate that this antibody, which binds preferentially soluble forms of Aβ, has the ability to mobilize Aβ from brain amyloid depositions.Citation50 The mAb crenezumab was further modified to carry a certain human immunoglobulin (Ig)-G isoform, IgG4, which is a Th2 antibody isotype carrying noninflammatory features such as reduced Fc-receptor binding on other immune cells. Indeed, results from a phase I clinical trial showed that patients treated with crenezumab showed less brain microhemorrhage and vasogenic edema compared to published observations from other antibody immunotherapy studies.Citation47,Citation51 Gantenerumab is a fully human monoclonal IgG1 antibody detecting two separate epitopes in Aβ42 (Aβ3–11 and Aβ19–28), and it has been reported that this antibody does not bind soluble Aβ but only the fibrillar forms of Aβ. In vitro studies showed that gantenerumab can induce phagocytosis of Aβ fibrils by brain microglia.Citation48 In patients, a decrease of brain amyloid was found in an antibody dose-dependent manner by up to 30%, as shown by positron emission tomography (PET) scans with the fibrillar Aβ-specific Pittsburgh B compound.Citation52
Another drug to treat AD with anti-Aβ antibodies, Gammagard from Baxter International (Deerfield, IL, USA), which is in phase III clinical trials and had shown favorable results to some extent, has failed to meet primary end points such as the slowing of cognitive and functional decline in treated AD patients, and is thus under debate whether trials of it will continue.Citation53 In this study, patients with mild-to-moderate AD received injections of concentrated Ig (IVIg) from healthy persons to utilize naturally occurring autoantibodies that specifically recognize and block the toxic effects of Aβ (nAbs-Aβ). The level of anti-Aβ antibodies in the serum from AD patients increased in proportion to the IVIg dose administered, and CSF Aβ decreased significantly at 6 months of continued treatment, then returned to baseline when treatment was stopped, and decreased again with continuous IVIg treatment, indicating that the naturally occurring anti-Aβ antibodies mobilized Aβ from brain. Mini-mental state scores increased an average of 2.5 points with 6 months of treatment and remained stable with treatment was continued.Citation54–Citation57 In a phase II dose-finding clinical study performed in the US and Germany in which AD patients also received IVIg injections (Octagam®, Octapharma, Toronto, ON, Canada), the main focus was on the safety profile of this therapy.Citation58 In the 6-month treatment period, 14% of patients showed brain microbleeds and one patient had an ischemic stroke, both of which are known side effects of IVIg therapy. The conclusion from this trial was that IVIg has tolerable safety and that further studies with larger patient cohorts and longer treatment times are needed to draw decisive conclusions.
New AD-prevention trials
One of the main arguments for the lack of more definitive positive results from clinical trials in AD is that the treatment was started too late.Citation59–Citation61 It has been shown that Aβ42 concentrations in CSF decline 25 years before the onset of clinical symptoms, which indicates Aβ deposition in brain, and 15 years before clinical symptoms are noticeable; these fibrillar Aβ deposits are visible in PET scans with fibrillar Aβ-specific Pittsburgh compound B.Citation60 Thus, a likely effective prevention and/or intervention have to start much earlier and in patients which do not already show symptoms for AD.
Three major prevention trials are slated to start in 2013: (1) the DIAN (Dominantly Inherited Alzheimer Network) study, (2) the Alzheimer’s Prevention Initiative (API) study, and (3) the Anti-Amyloid Treatment of Asymptomatic Alzheimer’s Disease (A4) study. These studies will focus on therapy in patient cohorts before the onset of clinical symptoms of AD.
The DIAN study will be undertaken in patients who are highly likely to develop AD at an early age, as these patients are carriers of genetic mutations that cause FAD. Treatment methods will be immunotherapy with two anti-Aβ mAbs, solanezumab and gantenerumab, and as a different treatment option a β-secretase inhibitor (LY2886721 from Lilly) will be used, which reduces Aβ deposition, as this reagent blocks the β-secretase enzyme involved in APP turnover on the cell surface.Citation60,Citation62 Also the API study will be undertaken in FAD patients: a large group of FAD carriers in Colombia which will develop AD early with end-stage dementia around the age of 50 years, which is decades earlier than the typical sporadic AD case. In the API study, the patients will receive passive immunizations with the mAb crenezumab.Citation51,Citation63 The A4 study will focus on the most often found form of AD, which is sporadic AD. Enrollment groups for this trial are older patients who are not genetic carriers, but already show early stages of Aβ deposition in the brain as measured by PET scan. In this study, passive immunotherapy will be done with the mAb solanezumab, which binds soluble Aβ, and the hope is that this early treatment will clearly show that treatment before the occurrence of clinical symptoms will lead to better benefits by blocking Aβ accumulation and delay the onset of AD.Citation64
Other alternatives for active Aβ immunotherapy
DNA immunizations differ in many ways from peptide immunizations, and in the search for alternative active immunization therapies, many groups, including ourselves, are investigating this vaccination route.Citation65–Citation72 In DNA immunizations, the DNA encoding the respective antigen is injected into skin or muscle. The DNA is then transcribed and expressed at the injection site. Local dendritic cells will take up the antigen, migrate to local lymph nodes, and present the protein to circulating lymphocytes, thereby initiating a general immune response. Our reports on the effectiveness of DNA Aβ42 immunization in the AD transgenic mouse model were the first to show that Aβ42 levels in the brain were reduced by 41% and Aβ42-containing plaques reduced by 50%.Citation66,Citation67 Similar findings were described by others in later studies.Citation71
shows the results from two groups of DNA Aβ42 trimer-immunized APP/PS1 double-transgenic mice and the respective control DNA (luciferase [Luc])-immunized mice (our group, unpublished results). These mice had been immunized eleven times with DNA Aβ42 trimer or Luc DNA, respectively, via gene gun starting at 4 months of age. The mice in group A had been analyzed 14 days following the final immunization, while the mice in group B were analyzed 4 months following the final immunization. The graphs show a comparison of anti-Aβ42 antibody levels () and total Aβ42 peptide levels in the brains from these mice (). Data shown were obtained by enzyme-linked immunosorbent assay (ELISA) experiments (plates coated with Aβ42 peptide for detection of the anti-Aβ42 antibodies or coated with an anti-Aβ42 antibody for detection of Aβ42 in a sandwich ELISA protocol). In both groups, the Aβ42 levels in brain from the DNA Aβ42 trimer-immunized mice were significantly reduced in comparison to Luc DNA-immunized control animals. In , the Aβ42 levels were reduced 60% compared to the parallel Luc-immunized control mice (9.795 ± 1.455 µg Aβ42 peptide per gram of brain tissue in control mice was reduced to 3.418 ± 0.418 µg Aβ42 peptide in DNA Aβ42-immunized mice), while in the Aβ42 levels were reduced by 25% (48.16 ± 2.914 µg Aβ42 peptide per gram of brain tissue in control mice was reduced to 36.55 ± 1.964 µg Aβ42 peptide in DNA Aβ42-immunized mice). Both these findings were highly significant, with P-values of 0.0006 and 0.0071, respectively. An explanation for this difference is in the time intervals between the final immunizations and the respective analyses of Aβ brain levels. While the mice in group A were still actively producing new antibodies (Aβ42 antibody levels in plasma and Aβ42 peptide levels in brain were shown 14 days after final immunization), in group B, Aβ42 reduction in brain was analyzed 4 months after the final immunization. Consistent with this, total Aβ42 levels in brains were much higher in group B with mean values of 48.2 µg/g wet brain tissue in the control mice compared to 9.7 µg/g wet brain tissue in the control mice of group A, in line with the marked increase of Aβ pathology with an age difference of 4 months (group A was 12 months of age, group B 16 months of age). The anti-Aβ antibody plasma levels were markedly reduced after a 4-month period without booster immunizations in direct comparison to the antibody levels 14 days postimmunization (, P = 0.0125), but even with the reduced antibody levels in plasma, the reduction of Aβ42 brain levels was still significantly different from the Luc DNA-immunized control mice (), and this was shown with the much higher levels of total Aβ42 in brain (compare 10 µg/g in 12-month-old mice [] and 48.2 µg/g wet brain tissue in 16-month-old Luc immunized control mice []) showing that DNA Aβ42 immunotherapy is effective.
Figure 1 (A–E) Effective amyloid beta (Aβ) immunotherapy in an Alzheimer’s disease mouse model with active DNA Aβ42 trimer immunization. Results from two groups of DNA Aβ42 trimer-immunized APP/PS1 double transgenic mice and the respective control DNA-immunized mice are shown (n on the x-axis indicates the number of mice used in this particular experiment). Immunization was started in both groups in 4-month-old mice and was continued for eleven immunizations until the mice were 12 months old. Group A was killed for final analyses (plasma antibody levels, brain Aβ histology, and biochemistry) 14 days following the last immunization, while mice in group B were killed four months after the eleventh immunization. Anti-Aβ42 IgG antibody levels were shown in (A) for group A, and in (C and D) for group B. The comparison of plasma anti-Aβ42 levels of DNA Aβ42-immunized mice and control mice that had received DNA luciferase (Luc), immunizations showed in both groups the presence of Aβ42-specific antibodies in the DNA Aβ42-immunized mice (P = 0.0092 [A] and 0.0305 [D]). In both groups, a significant reduction of Aβ42 levels in brain was found in the DNA Aβ42-immunized mice in comparison to the respective control groups. Mice in group A showed an amyloid reduction of 60% (B), while mice in group B showed a reduction of Aβ42 brain levels of 25% (E). This difference might be due to the time differences in the two groups between final immunizations and brain level analyses, as well as the marked differences in total Aβ42 levels in brain due to the 4-month age difference between the analyses for mice in groups A and B. Symbols used in the diagrams are as follows: in (A, B, D, and E), the grey circles show values from Luc-immunized control mice, and the black circles show values from DNA Aβ42-immunized mice. In (C), the antibody levels were compared in the same mouse group (Group B) 14 days after the final immunization (divided black and white circles) and 4 months after the final immunization (black circles). For statistics (unpaired t-test with two-tailed P-values). P-values of #0.05 were considered significant.
![Figure 1 (A–E) Effective amyloid beta (Aβ) immunotherapy in an Alzheimer’s disease mouse model with active DNA Aβ42 trimer immunization. Results from two groups of DNA Aβ42 trimer-immunized APP/PS1 double transgenic mice and the respective control DNA-immunized mice are shown (n on the x-axis indicates the number of mice used in this particular experiment). Immunization was started in both groups in 4-month-old mice and was continued for eleven immunizations until the mice were 12 months old. Group A was killed for final analyses (plasma antibody levels, brain Aβ histology, and biochemistry) 14 days following the last immunization, while mice in group B were killed four months after the eleventh immunization. Anti-Aβ42 IgG antibody levels were shown in (A) for group A, and in (C and D) for group B. The comparison of plasma anti-Aβ42 levels of DNA Aβ42-immunized mice and control mice that had received DNA luciferase (Luc), immunizations showed in both groups the presence of Aβ42-specific antibodies in the DNA Aβ42-immunized mice (P = 0.0092 [A] and 0.0305 [D]). In both groups, a significant reduction of Aβ42 levels in brain was found in the DNA Aβ42-immunized mice in comparison to the respective control groups. Mice in group A showed an amyloid reduction of 60% (B), while mice in group B showed a reduction of Aβ42 brain levels of 25% (E). This difference might be due to the time differences in the two groups between final immunizations and brain level analyses, as well as the marked differences in total Aβ42 levels in brain due to the 4-month age difference between the analyses for mice in groups A and B. Symbols used in the diagrams are as follows: in (A, B, D, and E), the grey circles show values from Luc-immunized control mice, and the black circles show values from DNA Aβ42-immunized mice. In (C), the antibody levels were compared in the same mouse group (Group B) 14 days after the final immunization (divided black and white circles) and 4 months after the final immunization (black circles). For statistics (unpaired t-test with two-tailed P-values). P-values of #0.05 were considered significant.](/cms/asset/1c9cacd3-9920-4ad5-8556-a8a2320ca4a2/ditt_a_31428_f0001_b.jpg)
DNA immunization differs quantitatively and qualitatively from peptide immunizations, and we and others have shown that Aβ42 DNA vaccination using a gene-gun approach results in a polarized Th2 immune response.Citation65,Citation68–Citation72 In our comparisons of DNA and peptide immunizations, we found that in vitro cell proliferation of potentially inflammatory Aβ42-specific T cells was absent in full-length DNA Aβ42 trimer-immunized mice, making this approach effective and safe for possible immunotherapy in AD patients.Citation73,Citation74
Biological role of tau protein
Tau is a highly soluble cytoplasmic protein that is primarily found in the brain and functions in neurite outgrowth, axonal transport, and microtubule assembly and stability. Neuronal development requires dynamic microtubules with axonal elongation and shortening, while in differentiated neurons the microtubules are relatively stable, and tau participates in these processes by microtubule binding. Alternative splicing and tau phosphorylation allow all these different functions from a single gene. Six isoforms as the result of alternative splicing of the 13 exons encoded by the MAPT gene are found in the human brain, and all of them are likely to have a specific role as they are differently expressed during development. Tau proteins differ by having three (3R) or four (4R) microtubule-binding repeats of 31–32 amino acids each, and possessing one, two, or no amino terminal inserts of 29 amino acids each (36.8–45.9 kDa). Alternative splicing of exon 10 results in the 3R and 4R isoforms. Tau phosphorylation on specific sites modulates function and intracellular localization. Hyperphosphorylated tau protein dissociates from the neuronal microtubule cytoskeleton, leading to microtubule destabilization and the formation of paired helical filaments (PHFs), which precipitate and become visible tangles.Citation75 Furthermore, nonfunctional tau sequesters normally phosphorylated tau proteins, preventing them from binding to microtubules and leading to more dysfunction. While certain sites are phosphorylated early, such as Ser202 (detected with monoclonal antibody mAb AT8) and Ser235 (mAb AT180), other sites, Ser422 and Ser396/S404 (mAb PHF1), were phosphorylated later in more advanced stages of the disease.Citation76
Anti-tau immunizations in mouse models
NFTs are associated with two neurodegenerative diseases: AD and frontotemporal dementia. In AD, NFTs form later in the disease following Aβ42 accumulation, while in frontotemporal dementia no Aβ42 accumulation is present.
Passive and active immunizations against tau have been analyzed in mice using several different mouse strains, as well as different phospho-tau peptides for active immunizations and anti-tau antibodies for passive immunotherapy.Citation77–Citation85 In the first report on results from immunizations with a 30-amino acid-long phosphorylated tau peptide, an effect on the ratios of soluble and insoluble tau, reduction of tangle formation in the immunized mice, and functional benefits observed in behavior testing for these mice were shown.Citation77 These findings were confirmed in later studies from the same laboratory.Citation78,Citation79 Since the mice carrying the P301L mutation develop severe motor impairments as tau pathology advances, it is not possible to analyze memory improvement in these mice, as behavior tests commonly used for memory and cognition, like the Morris water maze, radial arm maze, or the T-maze require extensive motor movement. In a new double-transgenic mouse model, htau/PS1, which shows spatial memory deficits earlier by 12 months of age, it was possible to show that active tau-peptide immunization can prevent cognitive impairments, which was tested by two different maze tests and object recognition.Citation79
There has been only one report presenting an adverse effect of tau immunization in mice, in which the immunized mice developed a late form of experimentally induced autoimmune encephalitis similar to the autoimmune pathogenesis found in mice that had been immunized with myelin oligodendrocyte glycoprotein or myelin basic protein as myelin self-antigens. In this study, the mice had been immunized with full-length tau together with two strong inflammation-inducing substrates – complete Freund’s adjuvant (CFA) and pertussis toxin (PT)Citation80 – because the researchers were interested to see the effects of maximal immune-system activation after immunization with a self-antigen. In a second study, this group used a phosphorylated tau peptide with the same strong adjuvant combination, CFA and PT, and did not observe inflammatory or neurotoxic side effects, but found positive effects in increased microglial activity in brains from immunized mice and reduction of NFTs due to generation of anti-phospho-tau antibodies, which did not cross-react with full-length unhyperphosphorylated tau (the antigen used before), indicative of the importance of choosing the right target for the development of immunotherapeutic strategies.Citation81
Passive immunization with well-characterized anti-tau antibodies, mAbs PHF, which react with phosphorylated Ser396 and Ser404 of the hyperphoshorylated tau protein as an early pathologic conformational epitope on tau confirmed the results seen in active immunization studies. Mice treated with these antibodies showed marked reductions in tau pathology, which was measured with biochemical methods and histology, as well as a significant delay in loss of motor-function decline which was assessed in behavioral testings.Citation82,Citation83
Similar to Aβ immunotherapy, there are several possibilities for antibodies to inhibit or slow the progression of disease. Antibodies might pass the blood–brain barrier and then enter neurons as well to modulate phosphorylation and/or degrade tau directly. It has been shown that tau-targeted immunization reduces the degree of tau phosphorylation in both young and aged mice,Citation77,Citation84 thereby reducing soluble hyperphosphorylated tau species that are toxic. Tau-targeted immunization may clear tau species that are involved in intercellular spreading of tau pathology and may prevent the initiation of tau aggregation.Citation11 Anti-tau antibodies may help to support clearing functions by astrocytes as these were found to be activated in mice with high NFT burden following active immunization.Citation84 Similar to the proposed peripheral sink mechanism for Aβ immunotherapy, anti-tau antibodies may facilitate tau clearance from the brain into the periphery, as an increase in tau concentrations was observed in blood from tau-immunized mice.Citation85 These common antibody-action mechanisms are shown in .
Figure 2 Common features of anti-amyloid beta (Aβ) and anti-tau antibodies. Both pathophysiological hallmarks of Alzheimer’s disease (AD) are caused by overproduction, aggregation, and misfolding of brain self-antigens. Active and passive immunotherapy and the respective anti-Aβ and anti-tau antibodies share common features of antibody actions.
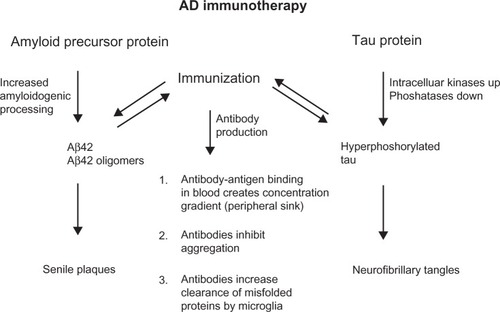
Links between these two pathologies
From pathophysiological analysis in mice and AD patients, it has been shown that Aβ accumulation precedes the formation of NFTs. It has also become clear that Aβ influences tau pathology and tau can influence Aβ pathology. Clearance of Aβ in humans or mice with immunotherapy had effects on tau pathology, and analysis of CSF levels for tau showed a reduction in patients that were positive-antibody responders in the stopped AN-1792 trial.Citation36,Citation86–Citation89 In cell cultures of differentiated rat hippocampal neuronal cells, it was shown that Aβ42-oligomer administration directly led to tau phosphorylation at sites that discriminate among AD and non-AD subjects, which are Ser404, Thr231, Thr181, Ser202, and Thr205. This was found not only for synthetic Aβ oligomers but also for soluble extracts from AD brain containing the Aβ oligomers.Citation90 A reduction of normal endogenous tau has been shown to ameliorate Aβ-induced dysfunction and early mortality in transgenic mice, and tau reduction may thus be an option to treat AD symptoms. Epileptic seizures occur spontaneously in transgenic mice that overexpress APP at synapses, and it has been shown that the reduction of tau can decrease the incidence and severity of pharmacologically induced seizures without changes in Aβ levels. Thus, tau somehow enables Aβ-induced neuronal dysfunction.Citation91–Citation93 In a recent study, it was shown how oligomeric Aβ induces the phosphorylation of tau at specific sites, which drives neurons aberrantly into cell cycles without division (ectopic cell-cycle reentry), leading to cell death and neuron loss. The authors conclude that this might be one reason for the substantial neuronal cell loss in AD.Citation94
While the ongoing and planned clinical trials are all concentrated on reduction of Aβ brain levels, future studies might be needed to test whether targeting Aβ and tau simultaneously can further improve the therapeutic efficacy of immunotherapy for AD.
Acknowledgments
This study was funded by grants from the NIH/NIA Alzheimer’s Disease Center (P30AG12300-17), the Rudman Partnership, and the McCune Foundation.
Disclosure
DLW declares no conflicts of interest. RNR has received clinical trial research grants from Janssen Inc, Novartis, and Pfizer. He holds a US Patent for amyloid beta gene vaccines. He is also on the editorial board of the Journal of the American Medical Association (JAMA) and the editor of JAMA Neurology.
References
- Alzheimer’s Association. [homepage on the Internet] Available at http://www.alz.org/Accessed July 24, 2013
- SelkoeDJAlzheimer’s disease: genes, proteins, and therapyPhysiol Rev200181274176611274343
- Wyss-CorayTInflammation in Alzheimer disease: driving force, bystander or beneficial response?Nat Med20061291005101516960575
- LesnéSKohMTKotilinekLA specific amyloid-beta protein assembly in the brain impairs memoryNature2006440708235235716541076
- ShankarGMLiSMehtaTHAmyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memoryNat Med200814883784218568035
- LesnéSEShermanMAGrantMBrain amyloid-β oligomers in ageing and Alzheimer’s diseaseBrain2013136Pt 51383139823576130
- BillingsLMOddoSGreenKNMcGaughJLLaFerlaFMIntraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic miceNeuron200545567568815748844
- OddoSCaccamoASmithIFGreenKNLaFerlaFMA dynamic relationship between intracellular and extracellular pools of AbetaAm J Pathol2006168118419416400022
- LaFerlaFMGreenKNOddoSIntracellular amyloid-beta in Alzheimer’s diseaseNat Rev Neurosci20078749950917551515
- ClavagueraFBolmontTCrowtherRATransmission and spreading of tauopathy in transgenic mouse brainNat Cell Biol200911790991319503072
- FrostBJacksRLDiamondMIPropagation of tau misfolding from the outside to the inside of a cellJ Biol Chem200928419128451285219282288
- CitronMOltersdorfTHaassCMutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein productionNature199236064056726741465129
- AsheKHZahsKRProbing the biology of Alzheimer’s disease in miceNeuron201066563164520547123
- SchenkDBarbourRDunnWImmunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouseNature1999400674017317710408445
- JankowskyJLSluntHHRatovitskiTJenkinsNACopelandNGBorcheltDRCo-expression of multiple transgenes in mouse CNS: a comparison of strategiesBiomol Eng200117615716511337275
- LewisJMcGowanERockwoodJNeurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau proteinNat Genet200025440240510932182
- OddoSCaccamoAShepherdJDTriple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunctionNeuron200339340942112895417
- OddoSCaccamoAKitazawaMTsengBPLaFerlaFMAmyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s diseaseNeurobiol Aging20032481063107014643377
- FilaliMLalondeRTheriaultPJulienCCalonFPlanelECognitive and non-cognitive behaviors in the triple transgenic mouse model of Alzheimer’s disease expressing mutated APP, PS1, and Mapt (3xTg-AD)Behav Brain Res2012234233434222796601
- OakleyHColeSLLoganSIntraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formationJ Neurosci20062640101291014017021169
- CohenRMRezai-ZadehKWeitzTMA transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric Aβ, and frank neuronal lossJ Neurosci201333156245625623575824
- McMillanPKorvatskaEPoorkajPTau isoform regulation is region- and cell-specific in mouse brainJ Comp Neurol2008511678880318925637
- HanesJZilkaNBartkovaMCaletkovaMDobrotaDNovakMRat tau proteome consists of six tau isoforms: implication for animal models of human tauopathiesJ Neurochem200910851167117619141083
- PrillerCBauerTMittereggerGKrebsBKretzschmarHAHermsJSynapse formation and function is modulated by the amyloid precursor proteinJ Neurosci200626277212722116822978
- WestmarkCJWhat’s happening at synapses? The role of amyloid β-protein precursor and β-amyloid in neurological disordersMol Psychiatry201318442543422925831
- WilquetVDe StrooperBAmyloid-beta precursor protein processing in neurodegenerationCurr Opin Neurobiol200414558258815464891
- JonssonTAtwalJKSteinbergSA mutation in APP protects against Alzheimer’s disease and age-related cognitive declineNature20124887409969922801501
- HardyJNew insights into the genetics of Alzheimer’s diseaseAnn Med19962832552588811169
- HardyJSelkoeDJThe amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeuticsScience2002297558035335612130773
- SelkoeDJAmyloid beta-protein and the genetics of Alzheimer’s diseaseJ Biol Chem19962713118295182988756120
- HardyJAlzheimer’s disease: the amyloid cascade hypothesis: an update and reappraisalJ Alzheimers Dis20069Suppl 315115316914853
- GamesDAdamsDAlessandriniRAlzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor proteinNature199537365145235277845465
- JanusCPearsonJMcLaurinJA beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s diseaseNature2000408681597998211140685
- MorganDDiamondDMGottschallPEA beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s diseaseNature2000408681598298511140686
- OrgogozoJMGilmanSDartiguesJFSubacute meningoen-cephalitis in a subset of patients with AD after Abeta42 immunizationNeurology2003611465412847155
- FoxNCBlackRSGilmanSEffects of Abeta immunization (AN1792) on MRI measures of cerebral volume in Alzheimer diseaseNeurology20056491563157215883317
- GilmanSKollerMBlackRSClinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trialNeurology20056491553156215883316
- HolmesCBocheDWilkinsonDLong-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trialLancet2008372963421622318640458
- RyanJMGrundmanMAnti-amyloid-beta immunotherapy in Alzheimer’s disease: ACC-001 clinical trials are ongoingJ Alzheimers Dis200917224319502708
- SchneebergerAMandlerMOtawaOZaunerWMattnerFSchmidtWDevelopment of AFFITOPE vaccines for Alzheimer’s disease (AD) – from concept to clinical testingJ Nutr Health Aging200913326426719262965
- WinbladBAndreasenNMinthonLSafety, tolerability, and antibody response of active Aβ immunotherapy with CAD106 in patients with Alzheimer’s disease: randomised, double-blind, placebo-controlled, first-in-human studyLancet Neurol201211759760422677258
- DavtyanHGhochikyanAPetrushinaIImmunogenicity, efficacy, safety, and mechanism of action of epitope vaccine (Lu AF20513) for Alzheimer’s disease: prelude to a clinical trialJ Neurosci201333114923493423486963
- LiuBFrostJLSunJMER5101, a novel Aβ1–15:DT conjugate vaccine, generates a robust anti-Aβ antibody response and attenuates Aβ pathology and cognitive deficits in APPswe/PS1∆E9 transgenic miceJ Neurosci201333167027703723595760
- PfeiferMBoncristianoSBondolfiLCerebral hemorrhage after passive anti-Abeta immunotherapyScience20022985597137912434053
- SallowaySSperlingRGilmanSA phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer diseaseNeurology200973242061207019923550
- RoherAECribbsDHKimRCBapineuzumab alters aβ composition: implications for the amyloid cascade hypothesis and anti-amyloid immunotherapyPLoS One201383e5973523555764
- AdolfssonOPihlgrenMToniNAn effector-reduced anti-β-amyloid (Aβ) antibody with unique aβ binding properties promotes neuroprotection and glial engulfment of AβJ Neurosci201232289677968922787053
- BohrmannBBaumannKBenzJGantenerumab: a novel human anti-Aβ antibody demonstrates sustained cerebral amyloid-β binding and elicits cell-mediated removal of human amyloid-βJ Alzheimers Dis2012281496921955818
- LainoCNews from The American Neurological Association Annual Meeeting: Anti-Amyloid-Beta Drug Modestly Slows Cognitive Decline in Mild to Moderate ADNeurology today201212213438
- FarlowMArnoldSEvan DyckCHSafety and biomarker effects of solanezumab in patients with Alzheimer’s diseaseAlzheimers Dement20128426127122672770
- GarberKGenentech’s Alzheimer’s antibody trial to study disease preventionNat Biotechnol201230873173222871696
- OstrowitzkiSDeptulaDThurfjellLMechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumabArch Neurol201269219820721987394
- Alzheimer Research ForumNetworking for a cure. [webpage on the Internet] Available from http://www.alzforum.org/new/detail.asp?id=3485Accessed July 31, 2013
- BlennowKZetterbergHRinneJOEffect of immunotherapy with bapineuzumab on cerebrospinal fluid biomarker levels in patients with mild to moderate Alzheimer diseaseArch Neurol20116981002101022473769
- RelkinNRSzaboPAdamiakB18-Month study of intravenous immunoglobulin for treatment of mild Alzheimer diseaseNeurobiol Aging200930111728173618294736
- RelkinNBettgerLTsakanikasDRavdinLThree-year follow-up on the IVIG for Alzheimer’s phase II studyAlzheimers Dement20128Suppl 43381
- ShayanGAdamiakBRelkinNRLeeKHLongitudinal analysis of novel Alzheimer’s disease proteomic cerebrospinal fluid biomarkers during intravenous immunoglobulin therapyElectrophoresis201233131975197922806462
- DodelRRomingerABartensteinPIntravenous immunoglobulin for treatment of mild-to-moderate Alzheimer’s disease: a phase 2, randomised, double-blind, placebo-controlled, dose-finding trialLancet Neurol201312323324323375965
- RosenbergRNTreat Alzheimer disease before it is symptomaticArch Neurol201168101237123821670383
- BatemanRJXiongCBenzingerTLClinical and biomarker changes in dominantly inherited Alzheimer’s diseaseN Engl J Med2012367979580422784036
- MillerGStopping Alzheimer’s before it startsScience2012337609679079222903991
- MorrisJCAisenPSBatemanRJDeveloping an international network for Alzheimer research: the Dominantly Inherited Alzheimer NetworkClin Investig (Lond)2012210975984
- ReimanEMLangbaumJBFleisherASAlzheimer’s Prevention Initiative: a plan to accelerate the evaluation of presymptomatic treatmentsJ Alzheimers Dis201126Suppl 332132921971471
- CarrilloMCBrashearHRLogovinskyVCan we prevent Alzheimer’s disease? Secondary “prevention” trials in Alzheimer’s diseaseAlzheimers Dement20139212313123411394
- QuBRosenbergRNLiLBoyerPJJohnstonSAGene vaccination to bias the immune response to amyloid-beta peptide as therapy for Alzheimer diseaseArch Neurol200461121859186415596606
- QuBBoyerPJJohnstonSAHynanLSRosenbergRNAβ42 gene vaccination reduces brain amyloid plaque burden in transgenic miceJ Neurol Sci20062441–215115816556449
- QuBXXiangQLiLJohnstonSAHynanLSRosenbergRNAβ42 gene vaccine prevents Aβ42 deposition in brain of double transgenic miceJ Neurol Sci20072601–220421317574274
- QuBXLambracht-WashingtonDFuMEagarTNStüveORosenbergRNAnalysis of three plasmid systems for use in DNA A beta 42 immunization as therapy for Alzheimer’s diseaseVaccine201028325280528720562015
- KimHDJinJJMaxwellJAFukuchiKEnhancing Th2 immune responses against amyloid protein by a DNA prime-adenovirus boost regimen for Alzheimer’s diseaseImmunol Lett20071121303817686533
- MovsesyanNGhochikyanAMkrtichyanMReducing AD-like pathology in 3xTg-AD mouse model by DNA epitope vaccine – a novel immunotherapeutic strategyPLoS One200835e212418461171
- DaSilvaKABrownMEMcLaurinJReduced oligomeric and vascular amyloid-beta following immunization of TgCRND8 mice with an Alzheimer’s DNA vaccineVaccine20092791365137619150380
- DavtyanHMkrtichyanMMovsesyanNDNA prime-protein boost increased the titer, avidity and persistence of anti-Abeta antibodies in wild-type miceGene Ther201017226127119865176
- Lambracht-WashingtonDQuBXFuMEagarTNStüveORosenbergRNDNA beta-amyloid (1–42) trimer immunization for Alzheimer disease in a wild-type mouse modelJAMA2009302161796180219861672
- Lambracht-WashingtonDQuBXFuMDNA immunization against amyloid beta 42 has high potential as safe therapy for Alzheimer’s disease as it diminishes antigen-specific Th1 and Th17 cell proliferationCell Mol Neurobiol201131686787421625960
- LeeGLeugersCJTau and tauopathiesProg Mol Biol Transl Sci201210726329322482453
- DetersNIttnerLMGötzJDivergent phosphorylation pattern of tau in P301L tau transgenic miceEur J Neurosci200828113714718662339
- AsuniAABoutajangoutAQuartermainDSigurdssonEMImmunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvementsJ Neurosci200727349115912917715348
- SigurdssonEMTau-focused immunotherapy for Alzheimer’s disease and related tauopathiesCurr Alzheimer Res20096544645019874269
- BoutajangoutAQuartermainDSigurdssonEMImmunotherapy targeting pathological tau prevents cognitive decline in a new tangle mouse modelJ Neurosci20103049165591656621147995
- RosenmannHGrigoriadisNKarussisDTauopathy-like abnormalities and neurologic deficits in mice immunized with neuronal tau proteinArch Neurol200663101459146717030663
- BoimelMGrigoriadisNLourbopoulosAHaberEAbramskyORosenmannHEfficacy and safety of immunization with phosphorylated tau against neurofibrillary tangles in miceExp Neurol2010224247248520546729
- BoutajangoutAIngadottirJDaviesPSigurdssonEMPassive immunization targeting pathological phospho-tau protein in a mouse model reduces functional decline and clears tau aggregates from the brainJ Neurochem2011118465866721644996
- ChaiXWuSMurrayTKPassive immunization with anti-Tau antibodies in two transgenic models: reduction of Tau pathology and delay of disease progressionJ Biol Chem201128639344573446721841002
- BiMIttnerAKeYDGötzJIttnerLMTau-targeted immunization impedes progression of neurofibrillary histopathology in aged P301L tau transgenic micePLoS One2011612e2686022174735
- TroquierLCaillierezRBurnoufSTargeting phospho-Ser422 by active Tau immunotherapy in the THYTau22 mouse model: a suitable therapeutic approachCurr Alzheimer Res20129439740522272619
- OddoSBillingsLKesslakJPCribbsDHLaFerlaFMAβ immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasomeNeuron200443332133215294141
- OddoSVasilevkoVCaccamoAKitazawaMCribbsDHLaFerlaFMReduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tanglesJ Biol Chem200628151394133942317056594
- Serrano-PozoAWilliamCMFerrerIBeneficial effect of human anti-amyloid-β active immunization on neurite morphology and tau pathologyBrain2010133Pt 51312132720360050
- BocheDDonaldJLoveSReduction of aggregated Tau in neuronal processes but not in the cell bodies after Aβ42 immunisation in Alzheimer’s diseaseActa Neuropathol20101201132020532897
- De FeliceFGWuDLambertMPAlzheimer’s disease-type neuronal tau hyperphosphorylation induced by A beta oligomersNeurobiol Aging20082991334134717403556
- RobersonEDScearce-LevieKPalopJJReducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse modelScience2007316582575075417478722
- IttnerLMKeYDDelerueFDendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse modelsCell2010142338739720655099
- RobersonEDHalabiskyBYooJWAmyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s diseaseJ Neurosci201131270071121228179
- SewardMESwansonENorambuenaAAmyloid-β signals through tau to drive ectopic neuronal cell cycle re-entry in Alzheimer’s diseaseJ Cell Sci2013126Pt 51278128623345405